The Yeast Saccharomyces cerevisiae as a Model for Understanding RAS Proteins and their Role in Human Tumorigenesis

 , ,
, ,  ,
,  and
and
Abstract
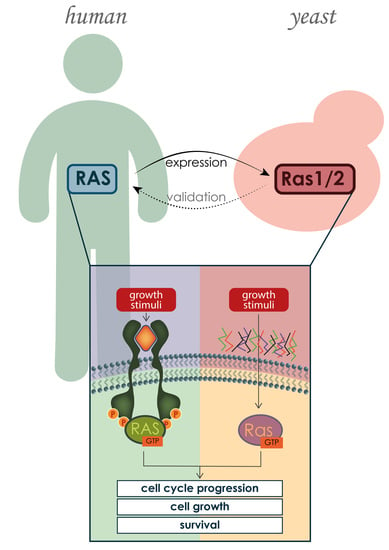
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
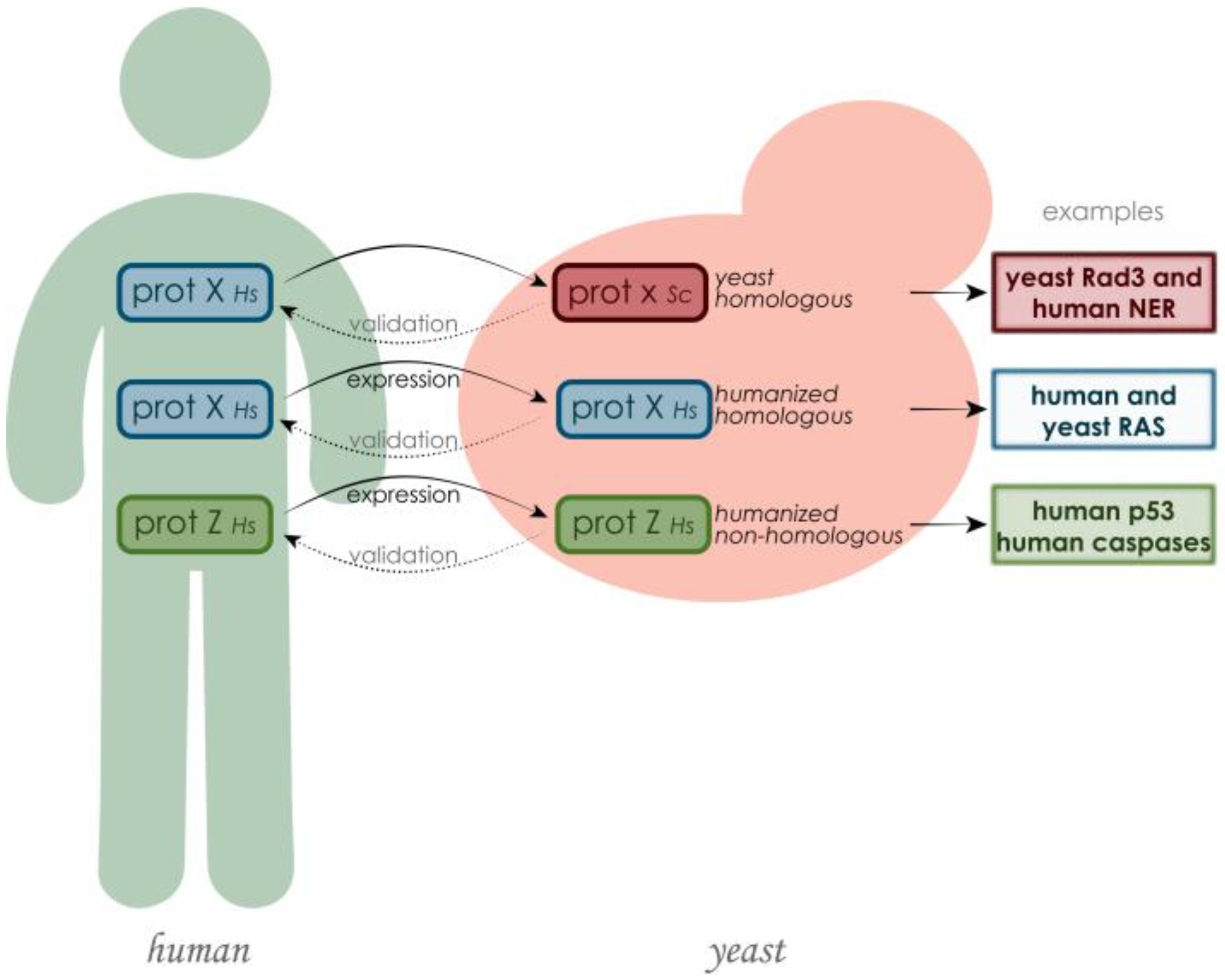
2. S. cerevisiae as a Model Organism for Studying Human Proteins and Molecular Mechanisms Underlying Associated Diseases
S. cerevisiae as a Model Organism for Studying Human RAS Proteins
3. Human and Yeast RAS Proteins: Similarities and Differences
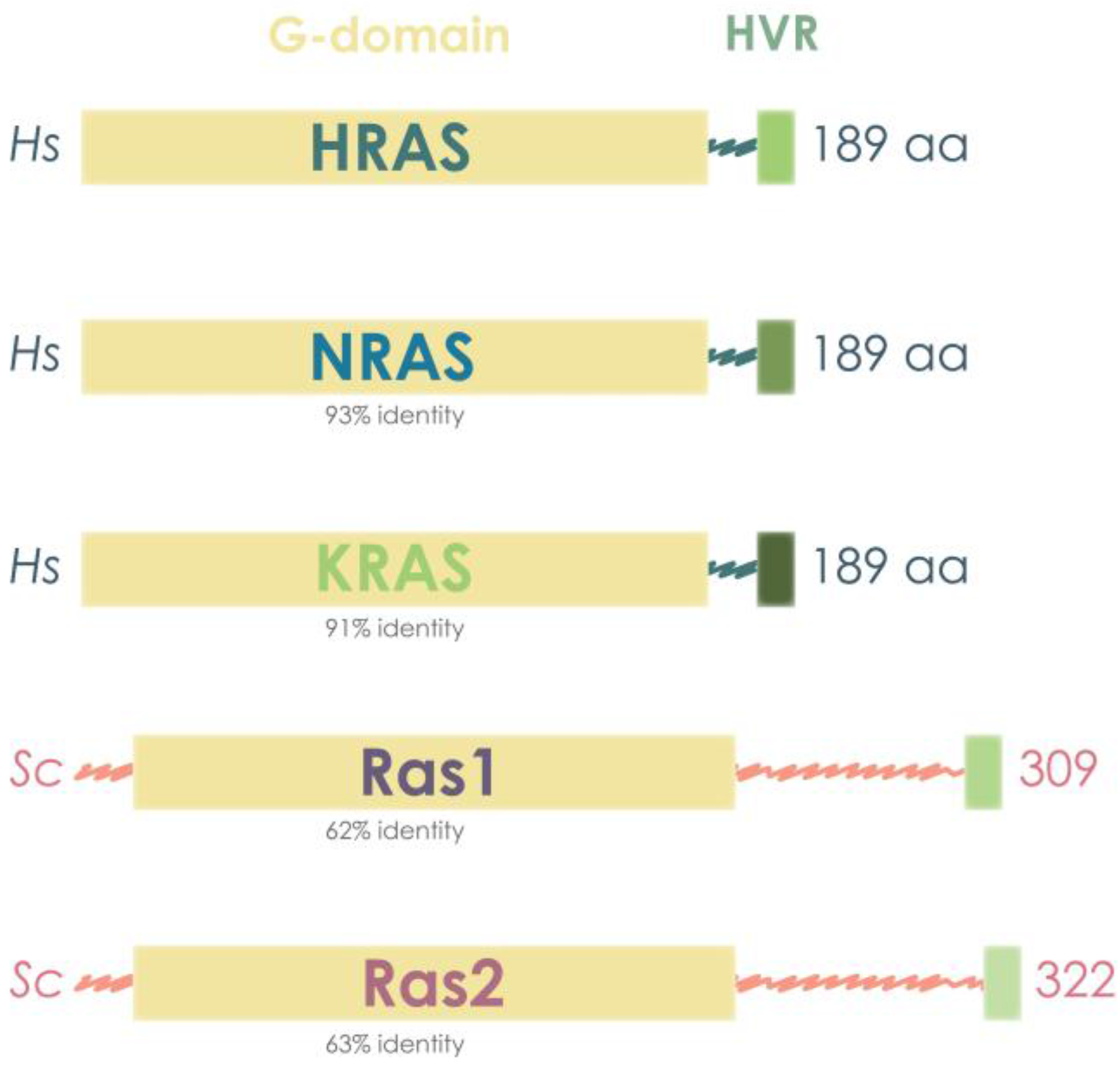
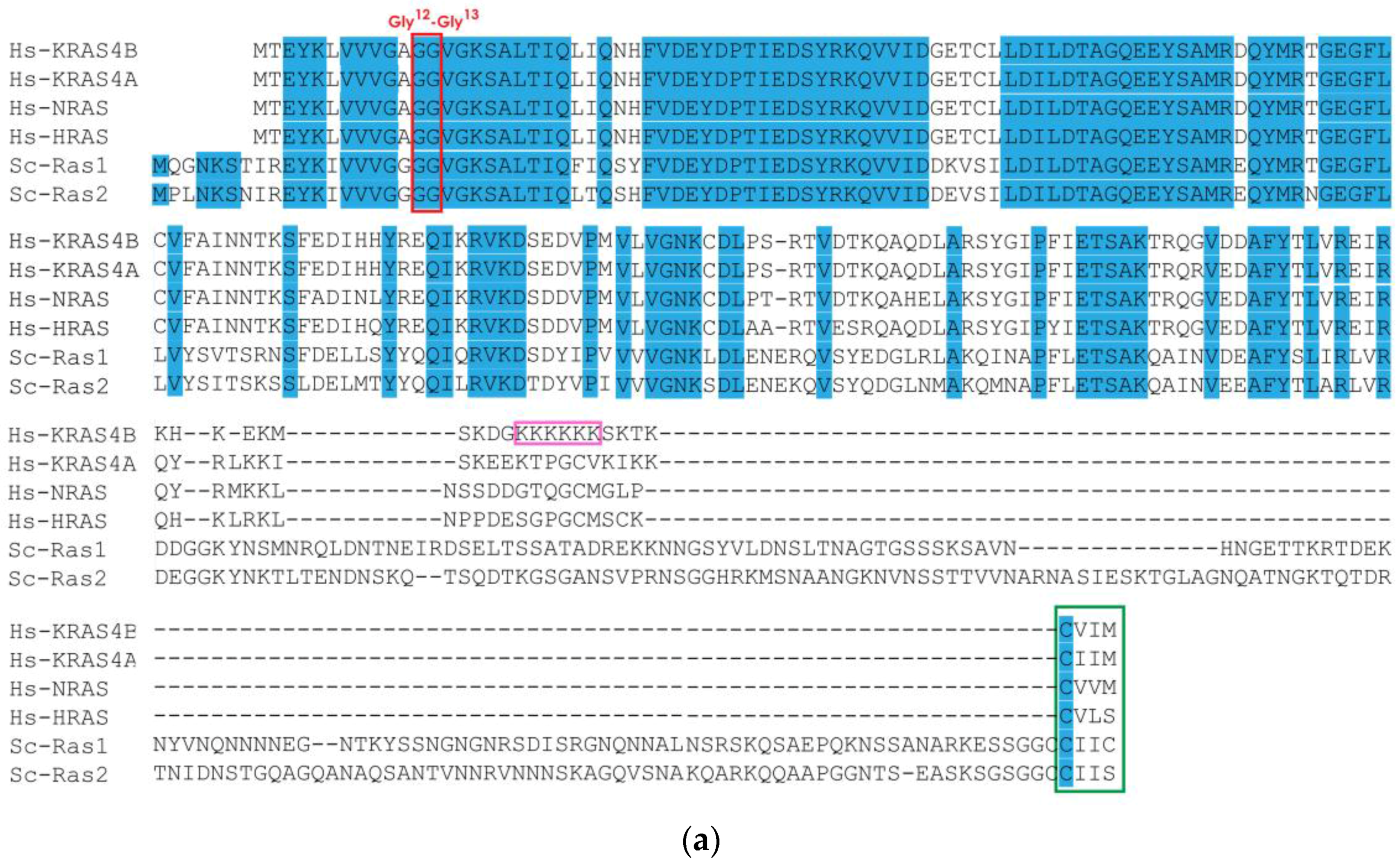
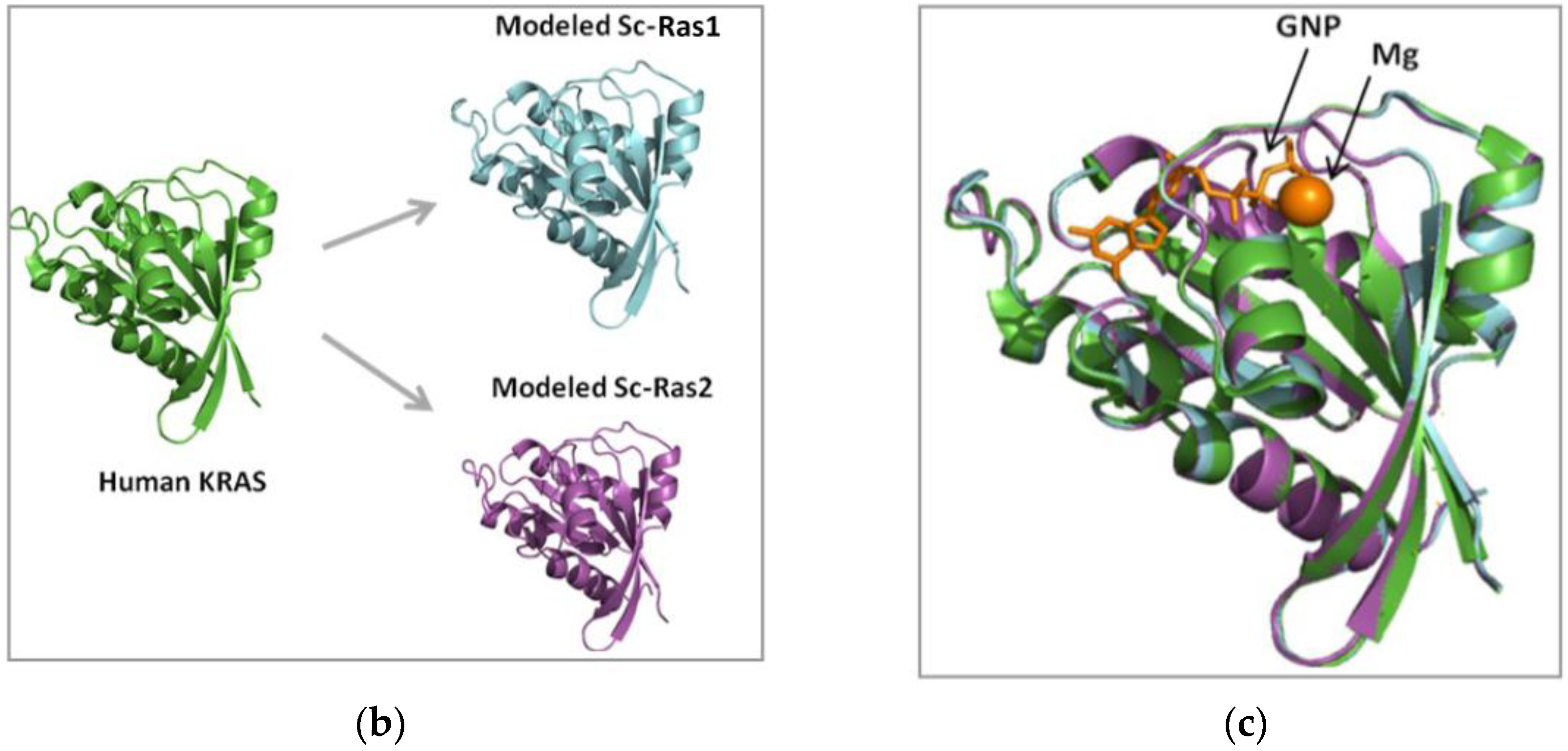
3.1. Human and Yeast RAS Sequence and Structure
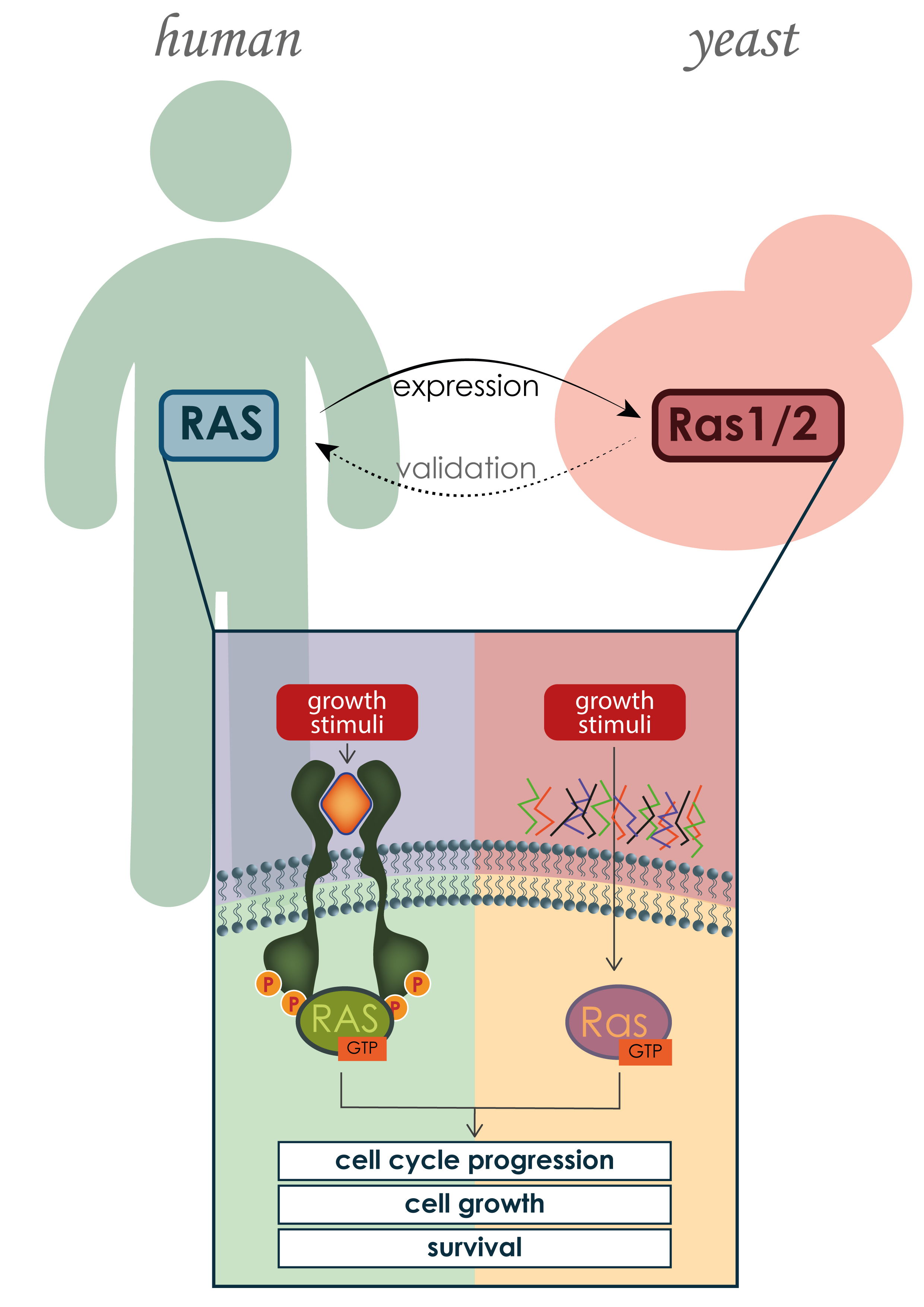
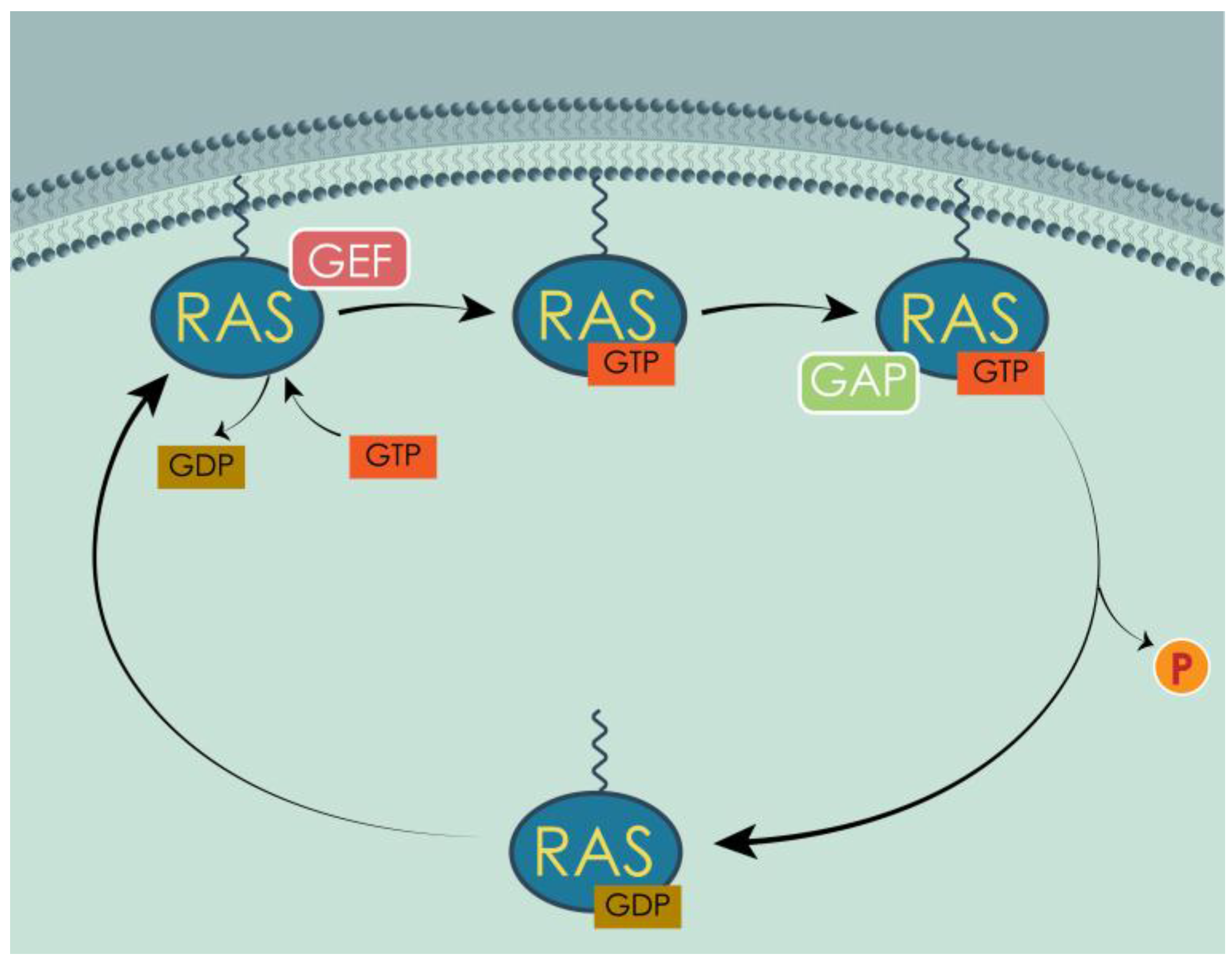
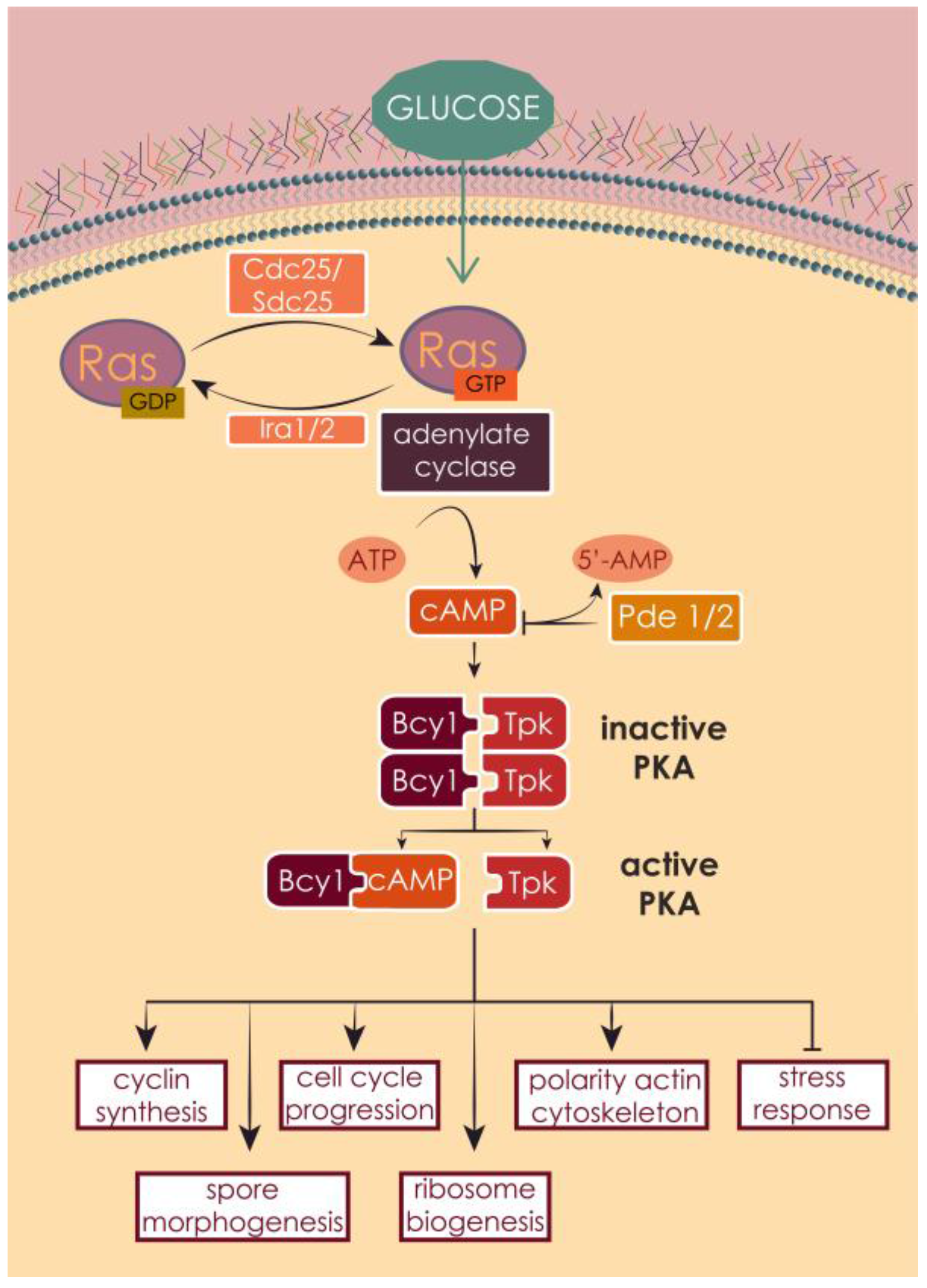
3.2. RAS Mechanism of Action and Regulators
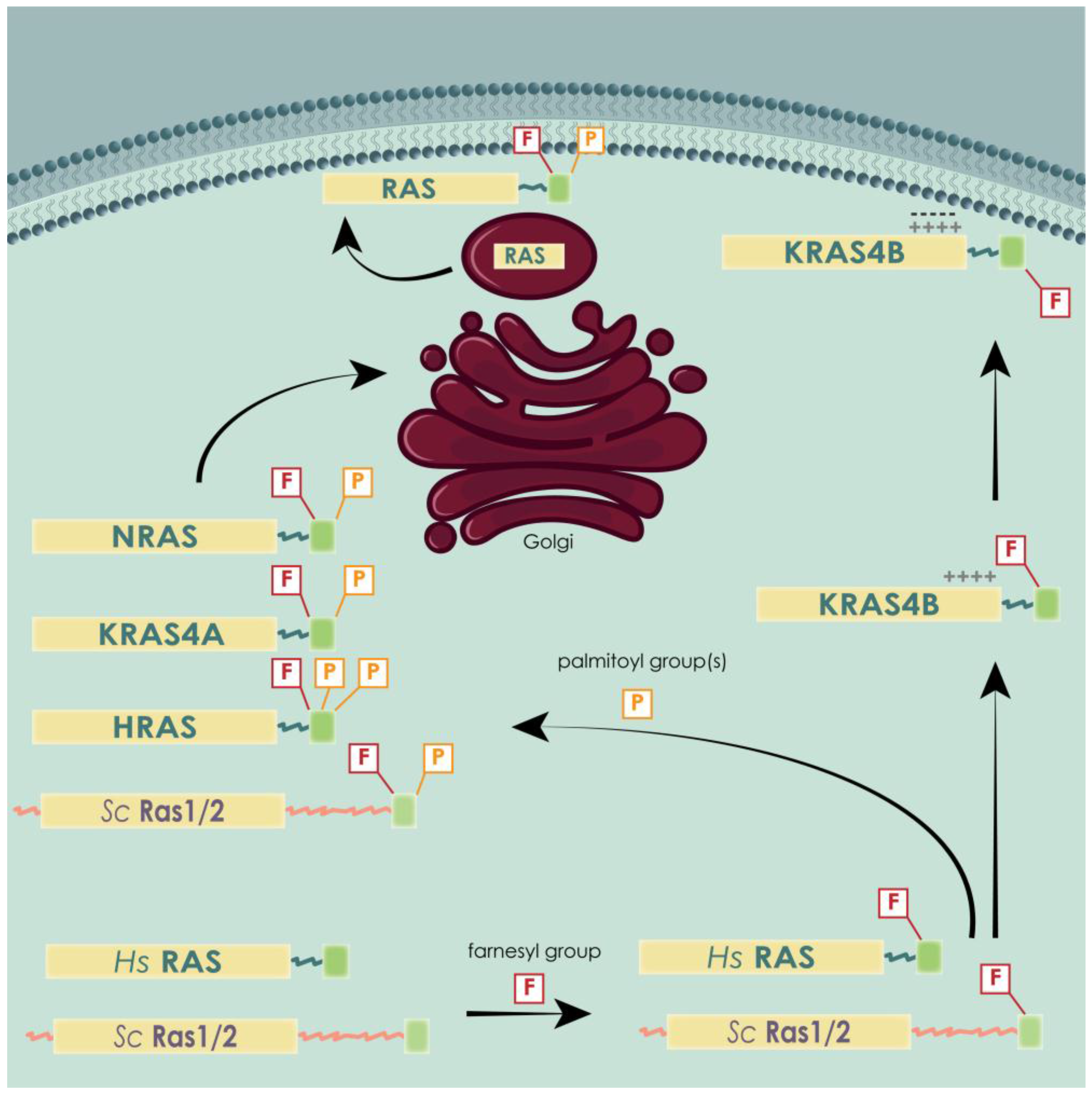
3.3. Post-Translational Modifications of Human and Yeast RAS
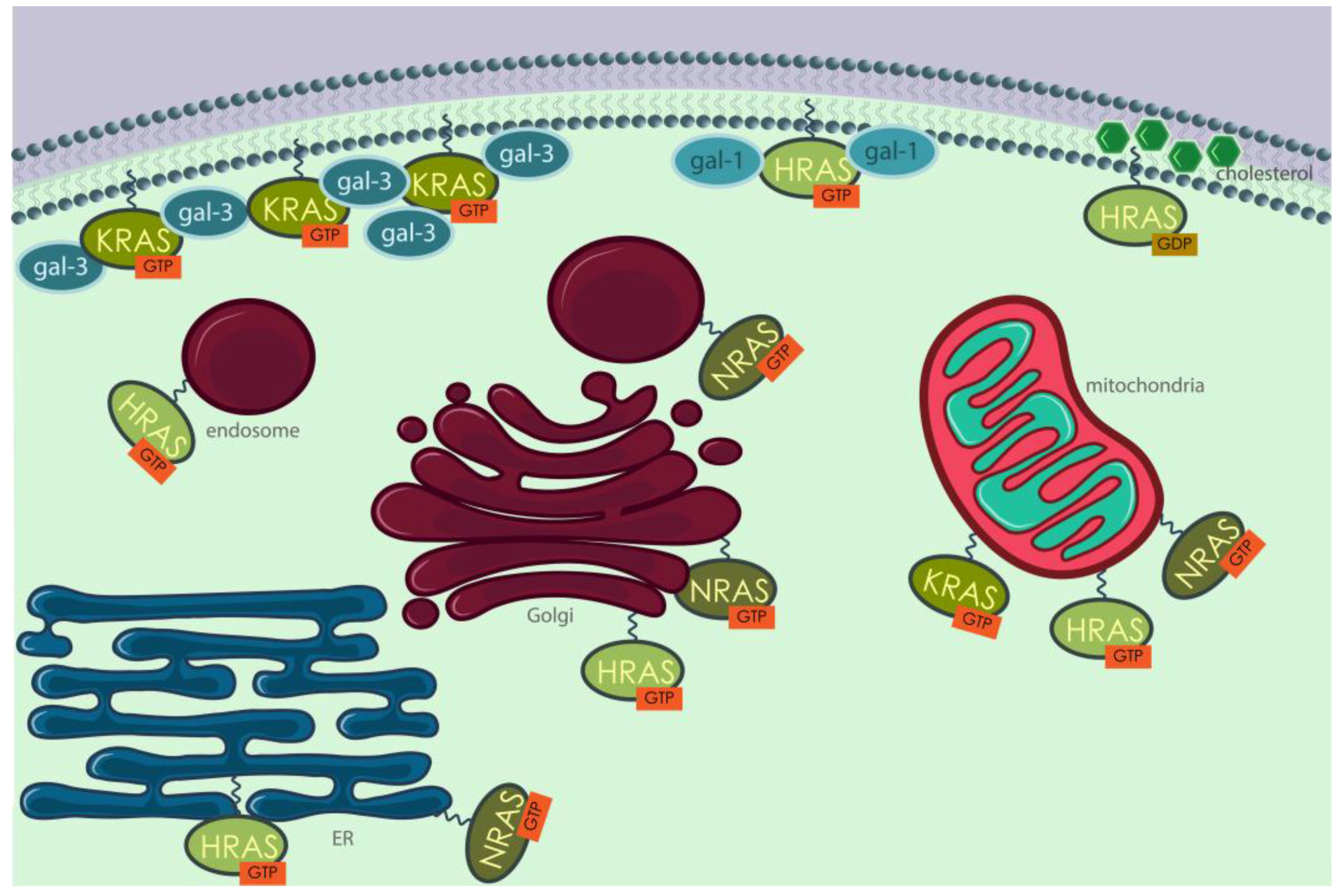
3.4. Differential Localization of Human and Yeast RAS
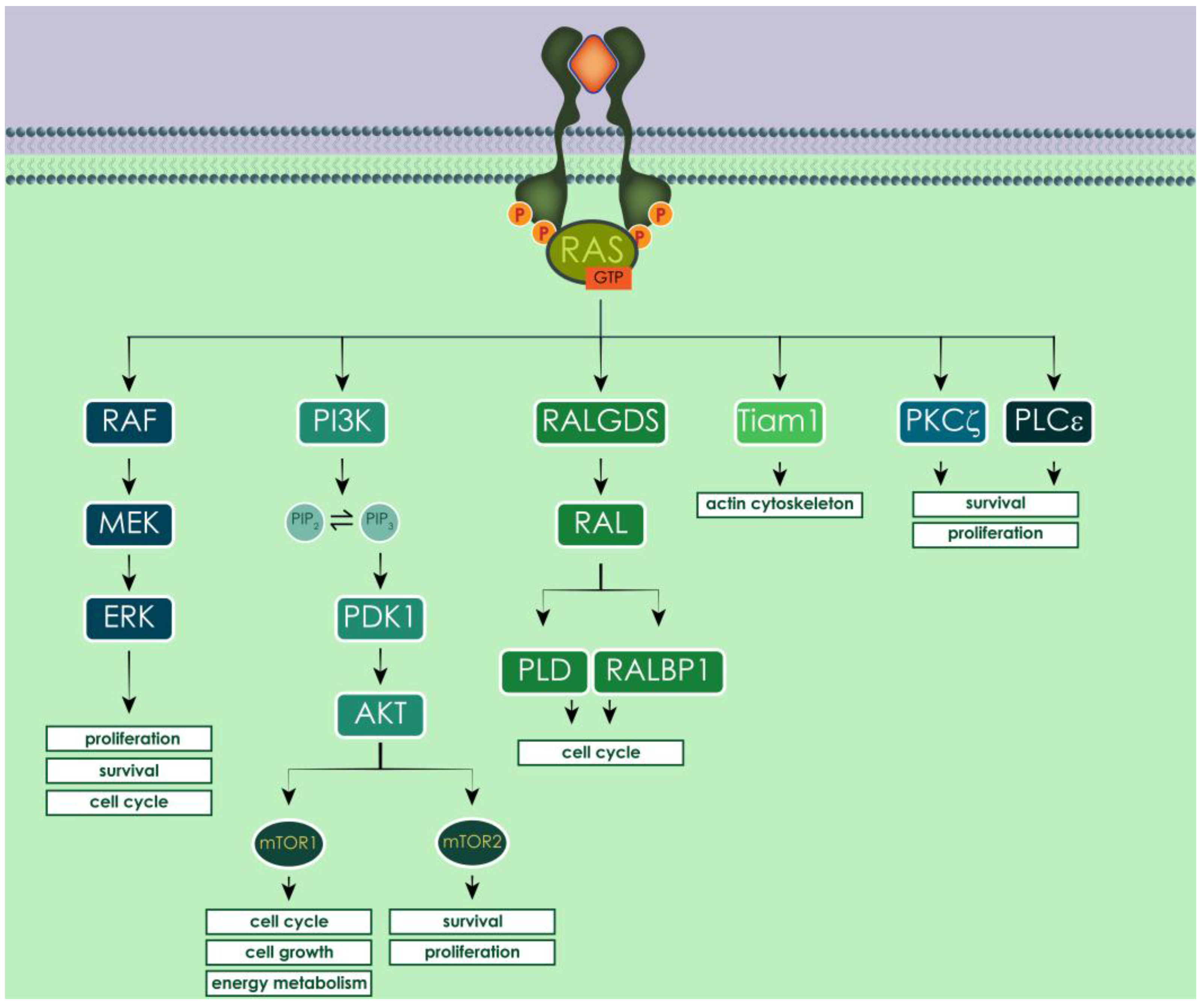
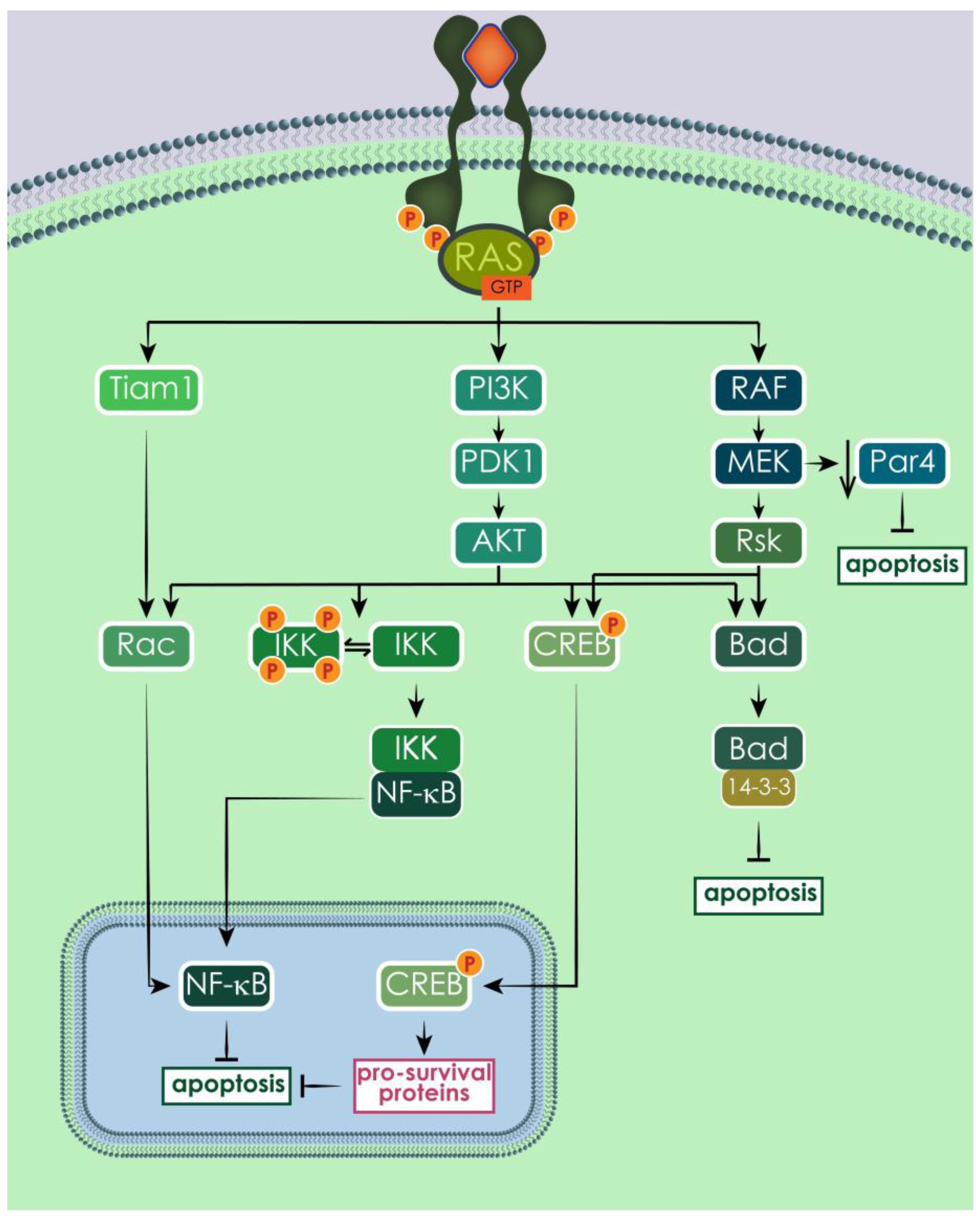
3.5. Human and Yeast RAS Downstream Signaling Pathway
3.6. RAS Effects on Growth, Apoptosis and Autophagy
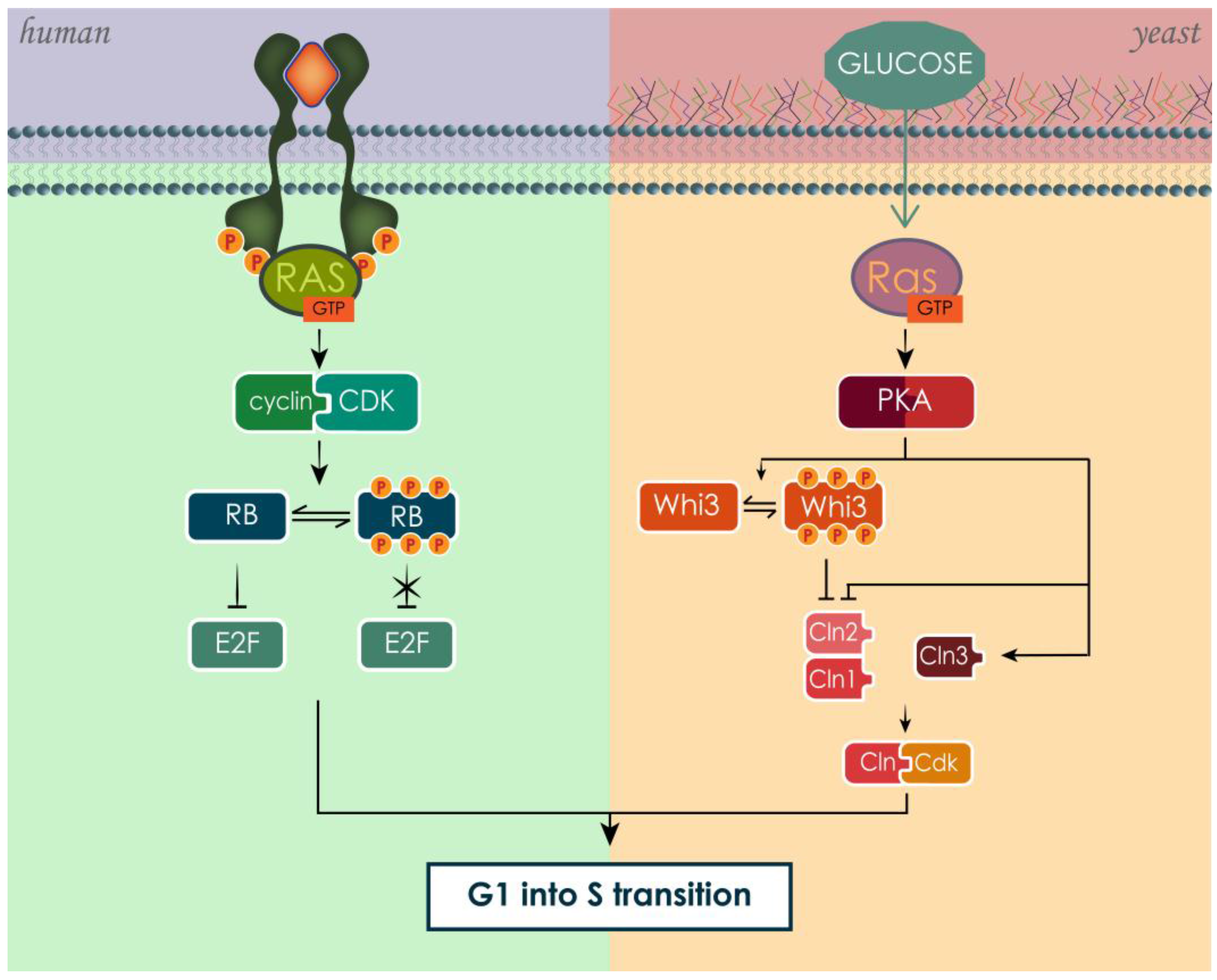
3.6.1. RAS Effects on Cell Cycle in Human and Yeast
3.6.2. RAS Effects on Cell Death in Human and Yeast
3.6.3. RAS Effects on Autophagy in Humans and Yeast
4. Involvement of RAS Proteins in Cancer: Yeast as a Model Organism
4.1. S. cerevisiae as a Model for Studying KRAS-Induced Autophagy in Colorectal Cancer
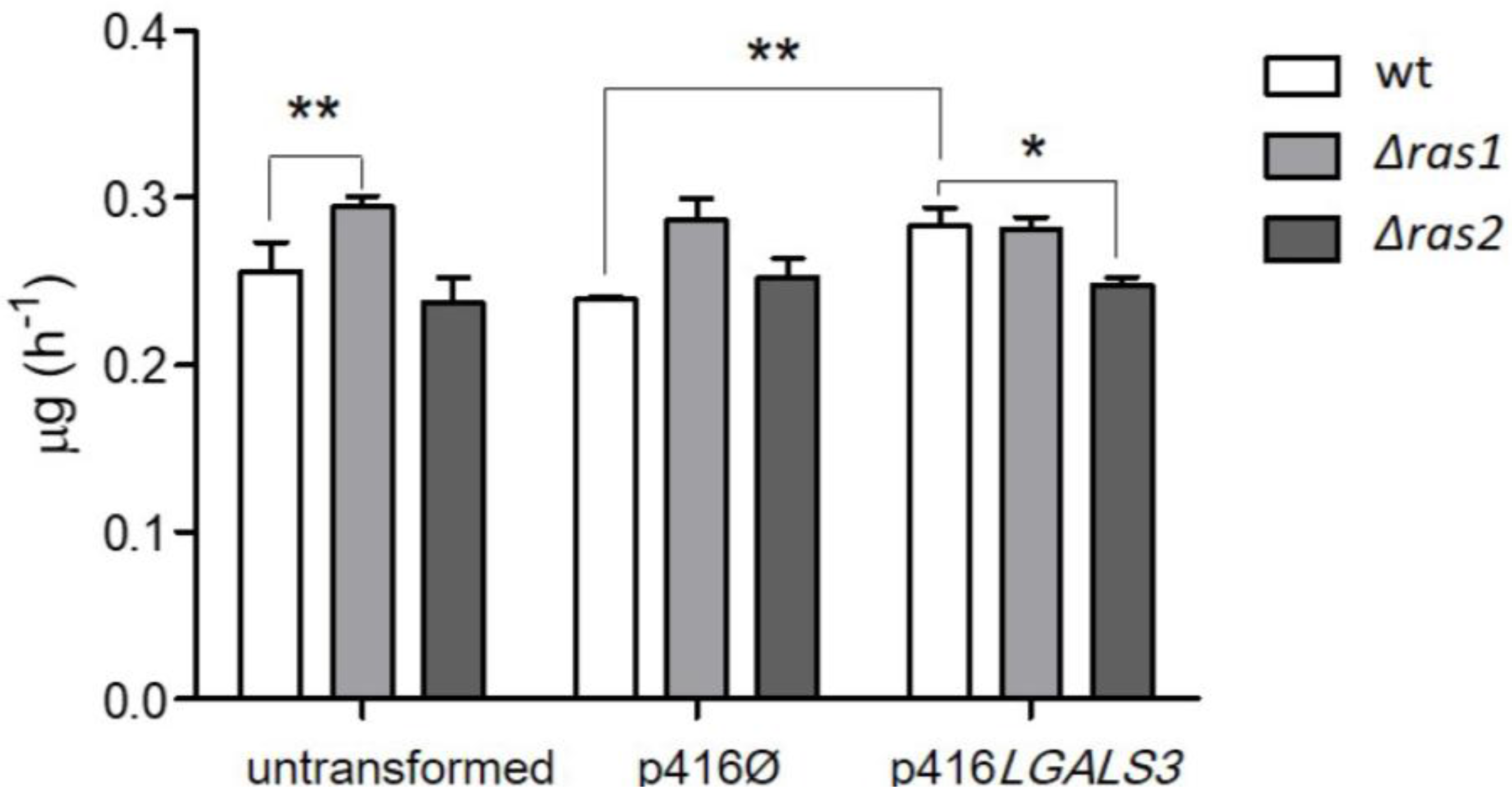
4.2. S. cerevisiae as a Model for Studying KRAS/gal-3 Interaction in Colorectal Cancer
5. Final Remarks
Acknowledgments
Conflicts of Interest
Appendix A
References
- Botstein, D.; Fink, G.R. Yeast: An experimental organism for 21st Century biology. Genetics 2011, 189, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Fields, S.; Johnston, M. Cell biology. Whither model organism research? Science 2005, 307, 1885–1886. [Google Scholar] [CrossRef] [PubMed]
- Mager, W.H.; Winderickx, J. Yeast as a model for medical and medicinal research. Trends Pharmacol. Sci. 2005, 26, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; Fink, G.R. Yeast: An experimental organism for modern biology. Science 1988, 240, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M. Life with 6000 genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.R.; Dietrich, F.S.; Fisk, D.G.; Binkley, G.; Balakrishnan, R.; Costanzo, M.C.; Dwight, S.S.; Hitz, B.C.; Karra, K.; Nash, R.S.; et al. The reference genome sequence of Saccharomyces cerevisiae: Then and now. G3 2014, 4, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Veronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; Andre, B.; et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef] [PubMed]
- DeFeo-Jones, D.; Scolnick, E.M.; Koller, R.; Dhar, R. Ras-related gene sequences identified and isolated from Saccharomyces cerevisiae. Nature 1983, 306, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Dhar, R.; Nieto, A.; Koller, R.; DeFeo-Jones, D.; Scolnick, E.M. Nucleotide sequence of two rasH related-genes isolated from the yeast Saccharomyces cerevisiae. Nucleic Acids Res. 1984, 12, 3611–3618. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.; Kataoka, T.; Fasano, O.; Goldfarb, M.; Strathern, J.; Broach, J.; Wigler, M. Genes in S. cerevisiae encoding proteins with domains homologous to the mammalian ras proteins. Cell 1984, 36, 607–612. [Google Scholar] [CrossRef]
- Ihle, N. Differential Activity of the KRAS Oncogene by Method of Activation: Implications for Signaling and Therapeutic Intervention. Ph.D. Thesis, Faculty of The University of Texas, Houston, TX, USA, 2012. [Google Scholar]
- Wittinghofer, A. Ras Superfamily Small G Proteins: Biology and Mechanisms 1; Springer: Wien, Austria, 2014. [Google Scholar]
- Arrington, A.K.; Heinrich, E.L.; Lee, W.; Duldulao, M.; Patel, S.; Sanchez, J.; Garcia-Aguilar, J.; Kim, J. Prognostic and Predictive Roles of KRAS Mutation in Colorectal Cancer. Int. J. Mol. Sci. 2012, 13, 12153–12168. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jakubowski, M.; Hunt, J.L. KRAS Gene Mutation in Colorectal Cancer Is Correlated with Increased Proliferation and Spontaneous Apoptosis. Am. Soc. Clin. Pathol. 2011, 135, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Bounds, R.; Wolf, H.; Steele, R.J.; Carey, F.A.; Wolf, C.R. Activating K-Ras mutations outwith ‘hotspot’ codons in sporadic colorectal tumours—Implications for personalised cancer medicine. Br. J. Cancer 2010, 102, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Alves, S.; Castro, L.; Fernandes, M.S.; Francisco, R.; Castro, P.; Priault, M.; Chaves, S.R.; Moyer, M.P.; Oliveira, C.; Seruca, R.; et al. Colorectal cancer-related mutant KRAS alleles function as positive regulators of autophagy. Oncotarget 2015, 6, 30787–30802. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.J. An Unidentified Virus Which Causes the Rapid Production of Tumours in Mice. Nature 1964, 204, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Kirsten, W.H.; Mayer, L.A.; Welander, C.W. Infective and noninfective viral murine leukemias. Natl. Cancer Inst. Monogr. 1966, 22, 369–377. [Google Scholar] [PubMed]
- Hall, A.; Marshall, C.J.; Spurr, N.K.; Weiss, R.A. Identification of transforming gene in two human sarcoma cell lines as a new member of the ras gene family located on chromosome 1. Nature 1983, 303, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Papageorge, A.G.; Defeo-Jones, D.; Robinson, P.; Temeles, G.; Scolnick, E.M. Saccharomyces cerevisiae synthesizes proteins related to the p21 gene product of ras genes found in mammals. Mol. Cell. Biol. 1984, 4, 23–29. [Google Scholar] [CrossRef] [PubMed]
- DeFeo-Jones, D.; Tatchell, K.; Robinson, L.C.; Sigal, I.S.; Vass, W.C.; Lowy, D.R.; Scolnick, E.M. Mammalian and yeast ras gene products: Biological function in their heterologous systems. Science 1985, 228, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.; Powers, S.; Cameron, S.; Fasano, O.; Goldfarb, M.; Broach, J.; Wigler, M. Functional homology of mammalian and yeast RAS genes. Cell 1985, 40, 19–26. [Google Scholar] [CrossRef]
- Kataoka, T.; Powers, S.; McGill, C.; Fasano, O.; Strathern, J.; Broach, J.; Wigler, M. Genetic analysis of yeast RAS1 and RAS2 genes. Cell 1984, 37, 437–445. [Google Scholar] [CrossRef]
- Toda, T.; Broek, D.; Field, J.; Michaeli, T.; Cameron, S.; Nikawa, J.; Sass, P.; Birchmeier, C.; Powers, S.; Wigler, M. Exploring the function of RAS oncogenes by studying the yeast Saccharomyces cerevisiae. Princess Takamatsu Symp. 1986, 17, 253–260. [Google Scholar] [PubMed]
- Wigler, M.; Field, J.; Powers, S.; Broek, D.; Toda, T.; Cameron, S.; Nikawa, J.; Michaeli, T.; Colicelli, J.; Ferguson, K. Studies of RAS function in the yeast Saccharomyces cerevisiae. Cold Spring Harb. Symp. Quant. Biol. 1988, 53, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, S.; Huh, W.K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Huh, W.K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nature 2003, 425, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Heinicke, S.; Livstone, M.S.; Lu, C.; Oughtred, R.; Kang, F.; Angiuoli, S.V.; White, O.; Botstein, D.; Dolinski, K. The Princeton Protein Orthology Database (P-POD): A comparative genomics analysis tool for biologists. PLoS ONE 2007, 2, e766. [Google Scholar] [CrossRef] [PubMed]
- Foury, F. Human genetic diseases: A cross-talk between man and yeast. Gene 1997, 195, 1–10. [Google Scholar] [CrossRef]
- Zabrocki, P.; Pellens, K.; Vanhelmont, T.; Vandebroek, T.; Griffioen, G.; Wera, S.; Van Leuven, F.; Winderickx, J. Characterization of a-synuclein aggregation and synergistic toxicity with protein tau in yeast. FEBS J. 2005, 272, 1386–1400. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Franssens, V.; Winderickx, J.; Outeiro, T.F. Yeast models of Parkinson’s disease-associated molecular pathologies. Curr. Opin. Genet. Dev. 2017, 44, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Laurent, J.M.; Young, J.H.; Kachroo, A.H.; Marcotte, E.M. Efforts to make and apply humanized yeast. Brief. Funct. Genom. 2016, 15, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Lindquist, S. Yeast cells provide insight into a-synuclein biology and pathobiology. Science 2003, 302, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Muchowski, P.J. Molecular genetics approaches in yeast to study amyloid diseases. J. Mol. Neurosci. 2004, 23, 49–60. [Google Scholar] [CrossRef]
- Willingham, S.; Outeiro, T.F.; DeVit, M.J.; Lindquist, S.L.; Muchowski, P.J. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or a-synuclein. Science 2003, 302, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Fruhmann, G.; Seynnaeve, D.; Zheng, J.; Ven, K.; Molenberghs, S.; Wilms, T.; Liu, B.; Winderickx, J.; Franssens, V. Yeast buddies helping to unravel the complexity of neurodegenerative disorders. Mech. Ageing Dev. 2017, 161, 288–305. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Coutinho, I.; Soares, J.; Bessa, C.; Leao, M.; Saraiva, L. New insights into cancer-related proteins provided by the yeast model. FEBS J. 2012, 279, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L.; Heitman, J. Yeast as a Tool in Cancer Research; Springer: Dordrecht, The Netherlands, 2007; p. 433. [Google Scholar]
- Matuo, R.; Sousa, F.G.; Soares, D.G.; Bonatto, D.; Saffi, J.; Escargueil, A.E.; Larsen, A.K.; Henriques, J.A. Saccharomyces cerevisiae as a model system to study the response to anticancer agents. Cancer Chemother. Pharmacol. 2012, 70, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Bedalov, A. Yeast as a model system for anticancer drug discovery. Nat. Rev. Cancer 2004, 4, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Menacho-Marquez, M.; Murguia, J.R. Yeast on drugs: Saccharomyces cerevisiae as a tool for anticancer drug research. Clin. Transl. Oncol. 2007, 9, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Guaragnella, N.; Palermo, V.; Galli, A.; Moro, L.; Mazzoni, C.; Giannattasio, S. The expanding role of yeast in cancer research and diagnosis: Insights into the function of the oncosuppressors p53 and BRCA1/2. FEMS Yeast Res. 2014, 14, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Birrell, G.W.; Giaever, G.; Chu, A.M.; Davis, R.W.; Brown, J.M. A genome-wide screen in Saccharomyces cerevisiae for genes affecting UV radiation sensitivity. Proc. Natl. Acad. Sci. USA 2001, 98, 12608–12613. [Google Scholar] [CrossRef] [PubMed]
- Aouida, M.; Page, N.; Leduc, A.; Peter, M.; Ramotar, D. A genome-wide screen in Saccharomyces cerevisiae reveals altered transport as a mechanism of resistance to the anticancer drug bleomycin. Cancer Res. 2004, 64, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.J.; Schaber, M.D.; Srinivasula, S.M.; Alnemri, E.S.; Litwack, G.; Hall, D.J.; Bjornsti, M.A. Cascades of mammalian caspase activation in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1999, 274, 3189–3198. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.E.; Han, D.K.; Hockenbery, D.M. Caspase-3 and inhibitor of apoptosis protein(s) interactions in Saccharomyces cerevisiae and mammalian cells. FEBS Lett. 2000, 481, 13–18. [Google Scholar] [CrossRef]
- Hayashi, H.; Cuddy, M.; Shu, V.C.; Yip, K.W.; Madiraju, C.; Diaz, P.; Matsuyama, T.; Kaibara, M.; Taniyama, K.; Vasile, S.; et al. Versatile assays for high throughput screening for activators or inhibitors of intracellular proteases and their cellular regulators. PLoS ONE 2009, 4, e7655. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Reed, J.C. Bax inhibitor-1, a mammalian apoptosis suppressor identified by functional screening in yeast. Mol. Cell 1998, 1, 337–346. [Google Scholar] [CrossRef]
- Zhang, H.; Cowan-Jacob, S.W.; Simonen, M.; Greenhalf, W.; Heim, J.; Meyhack, B. Structural basis of BFL-1 for its interaction with BAX and its anti-apoptotic action in mammalian and yeast cells. J. Biol. Chem. 2000, 275, 11092–11099. [Google Scholar] [CrossRef] [PubMed]
- Brezniceanu, M.L.; Volp, K.; Bosser, S.; Solbach, C.; Lichter, P.; Joos, S.; Zornig, M. HMGB1 inhibits cell death in yeast and mammalian cells and is abundantly expressed in human breast carcinoma. FASEB J. 2003, 17, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Torgler, C.N.; de Tiani, M.; Raven, T.; Aubry, J.P.; Brown, R.; Meldrum, E. Expression of bak in S. pombe results in a lethality mediated through interaction with the calnexin homologue Cnx1. Cell Death Differ. 1997, 4, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, Q.; Krajewski, S.; Krajewska, M.; Xie, Z.; Fuess, S.; Kitada, S.; Pawlowski, K.; Godzik, A.; Reed, J.C. BAR: An apoptosis regulator at the intersection of caspases and Bcl-2 family proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 2597–2602. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Hanada, M.; Bodrug, S.; Irie, S.; Iwama, N.; Boise, L.H.; Thompson, C.B.; Golemis, E.; Fong, L.; Wang, H.G.; et al. Interactions among members of the Bcl-2 protein family analyzed with a yeast two-hybrid system. Proc. Natl. Acad. Sci. USA 1994, 91, 9238–9242. [Google Scholar] [CrossRef] [PubMed]
- Cheok, C.F.; Verma, C.S.; Baselga, J.; Lane, D.P. Translating p53 into the clinic. Nat. Rev. Clin. Oncol. 2011, 8, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Yousef, A.F.; Xu, G.W.; Mendez, M.; Brandl, C.J.; Mymryk, J.S. Coactivator requirements for p53-dependent transcription in the yeast Saccharomyces cerevisiae. Int. J. Cancer 2008, 122, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Fields, S.; Jang, S.K. Presence of a potent transcription activating sequence in the p53 protein. Science 1990, 249, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Scharer, E.; Iggo, R. Mammalian p53 can function as a transcription factor in yeast. Nucleic Acids Res. 1992, 20, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Nigro, J.M.; Sikorski, R.; Reed, S.I.; Vogelstein, B. Human p53 and CDC2Hs genes combine to inhibit the proliferation of Saccharomyces cerevisiae. Mol. Cell. Biol. 1992, 12, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Mokdad-Gargouri, R.; Belhadj, K.; Gargouri, A. Translational control of human p53 expression in yeast mediated by 5’-UTR-ORF structural interaction. Nucleic Acids Res. 2001, 29, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.D.; Merrill, G.F. Deletion of the Saccharomyces cerevisiae TRR1 gene encoding thioredoxin reductase inhibits p53-dependent reporter gene expression. J. Biol. Chem. 1998, 273, 5431–5434. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Ma, X.; Lindner, D.J.; Karra, S.; Hofmann, E.R.; Reddy, S.P.; Kalvakolanu, D.V. Modulation of p53 dependent gene expression and cell death through thioredoxin-thioredoxin reductase by the Interferon-Retinoid combination. Oncogene 2001, 20, 4235–4248. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, I.; Pereira, C.; Pereira, G.; Goncalves, J.; Corte-Real, M.; Saraiva, L. Distinct regulation of p53-mediated apoptosis by protein kinase Ca, d, e and z: Evidence in yeast for transcription-dependent and -independent p53 apoptotic mechanisms. Exp. Cell Res. 2011, 317, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Robert, V.; Michel, P.; Flaman, J.M.; Chiron, A.; Martin, C.; Charbonnier, F.; Paillot, B.; Frebourg, T. High frequency in esophageal cancers of p53 alterations inactivating the regulation of genes involved in cell cycle and apoptosis. Carcinogenesis 2000, 21, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Di Como, C.J.; Prives, C. Human tumor-derived p53 proteins exhibit binding site selectivity and temperature sensitivity for transactivation in a yeast-based assay. Oncogene 1998, 16, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.G.; McGrath, J.P.; Levinson, A.D. Expression of normal and activated human Ha-ras cDNAs in Saccharomyces cerevisiae. Mol. Cell. Biol. 1985, 5, 2746–2752. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, A.; Matsumoto, K.; Tamanoi, F. A novel yeast mutant defective in the processing of ras proteins: Assessment of the effect of the mutation on processing steps. EMBO J. 1987, 6, 223–228. [Google Scholar] [PubMed]
- Toda, T.; Uno, I.; Ishikawa, T.; Powers, S.; Kataoka, T.; Broek, D.; Cameron, S.; Broach, J.; Matsumoto, K.; Wigler, M. In yeast, RAS proteins are controlling elements of adenylate cyclase. Cell 1985, 40, 27–36. [Google Scholar] [CrossRef]
- Robinson, L.C.; Gibbs, J.B.; Marshall, M.S.; Sigal, I.S.; Tatchell, K. CDC25: A component of the RAS-adenylate cyclase pathway in Saccharomyces cerevisiae. Science 1987, 235, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Broek, D.; Toda, T.; Michaeli, T.; Levin, L.; Birchmeier, C.; Zoller, M.; Powers, S.; Wigler, M. The S. cerevisiae CDC25 gene product regulates the RAS/adenylate cyclase pathway. Cell 1987, 48, 789–799. [Google Scholar] [CrossRef]
- Wei, W.; Mosteller, R.D.; Sanyal, P.; Gonzales, E.; McKinney, D.; Dasgupta, C.; Li, P.; Liu, B.X.; Broek, D. Identification of a mammalian gene structurally and functionally related to the CDC25 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1992, 89, 7100–7104. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D.; Fu, P.; Simon, M.; Senior, P. Identification of murine homologues of the Drosophila son of sevenless gene: Potential activators of ras. Proc. Natl. Acad. Sci. USA 1992, 89, 6511–6515. [Google Scholar] [CrossRef] [PubMed]
- Shou, C.; Farnsworth, C.L.; Neel, B.G.; Feig, L.A. Molecular cloning of cDNAs encoding a guanine-nucleotide-releasing factor for Ras p21. Nature 1992, 358, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.F.; Lin, B.; Tanaka, K.; Dunn, D.; Wood, D.; Gesteland, R.; White, R.; Weiss, R.; Tamanoi, F. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 1990, 63, 835–841. [Google Scholar] [CrossRef]
- Gibbs, J.B.; Schaber, M.D.; Allard, W.J.; Sigal, I.S.; Scolnick, E.M. Purification of ras GTPase activating protein from bovine brain. Proc. Natl. Acad. Sci. USA 1988, 85, 5026–5030. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; You, M.; Wang, Y. Alternative splicing of the K-ras gene in mouse tissues and cell lines. Exp. Lung Res. 2001, 27, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Arozarena, I.; Berciano, M.T.; Aaronson, D.S.; Pellicer, A.; Lafarga, M.; Crespo, P. Differences on the inhibitory specificities of H-Ras, K-Ras, and N-Ras (N17) dominant negative mutants are related to their membrane microlocalization. J. Biol. Chem. 2003, 278, 4572–4581. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Calvo, F.; Crespo, P. Ras, an actor on many stages: Posttranslational modifications, localization, and site-specified events. Genes Cancer 2011, 2, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Tatchell, K.; Chaleff, D.T.; DeFeo-Jones, D.; Scolnick, E.M. Requirement of either of a pair of ras-related genes of Saccharomyces cerevisiae for spore viability. Nature 1984, 309, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, J.; Goody, R.S.; Wittinghofer, A. Preparation and characterization of nucleotide-free and metal ion-free p21 “apoprotein”. J. Biol. Chem. 1987, 262, 8455–8458. [Google Scholar] [PubMed]
- Breviario, D.; Hinnebusch, A.; Cannon, J.; Tatchell, K.; Dhar, R. Carbon source regulation of RAS1 expression in Saccharomyces cerevisiae and the phenotypes of ras2-cells. Proc. Natl. Acad. Sci. USA 1986, 83, 4152–4156. [Google Scholar] [CrossRef] [PubMed]
- Breviario, D.; Hinnebusch, A.G.; Dhar, R. Multiple regulatory mechanisms control the expression of the RAS1 and RAS2 genes of Saccharomyces cerevisiae. EMBO J. 1988, 7, 1805–1813. [Google Scholar] [PubMed]
- Fraenkel, D.G. On ras gene function in yeast. Proc. Natl. Acad. Sci. USA 1985, 82, 4740–4744. [Google Scholar] [CrossRef] [PubMed]
- Tatchell, K.; Robinson, L.C.; Breitenbach, M. RAS2 of Saccharomyces cerevisiae is required for gluconeogenic growth and proper response to nutrient limitation. Proc. Natl. Acad. Sci. USA 1985, 82, 3785–3789. [Google Scholar] [CrossRef] [PubMed]
- Marshall, M.S.; Gibbs, J.B.; Scolnick, E.M.; Sigal, I.S. Regulatory function of the Saccharomyces cerevisiae RAS C-terminus. Mol. Cell. Biol. 1987, 7, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, A.; Tamanoi, F. Processing and fatty acid acylation of RAS1 and RAS2 proteins in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1986, 83, 1266–1270. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, L.; Carneiro, J.; van Asch, B.; Moleirinho, A.; Pereira, F.; Amorim, A. Epistatic interactions modulate the evolution of mammalian mitochondrial respiratory complex components. BMC Genom. 2009, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, J.; Duarte-Pereira, S.; Azevedo, L.; Castro, L.F.; Aguiar, P.; Moreira, I.S.; Amorim, A.; Silva, R.M. The evolutionary portrait of metazoan NAD salvage. PLoS ONE 2013, 8, e64674. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.B.; Marshall, M.S. The ras oncogene--an important regulatory element in lower eucaryotic organisms. Microbiol. Rev. 1989, 53, 171–185. [Google Scholar] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmuller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Acharya, A.; Prakash, B. Structural plasticity mediates distinct GAP-dependent GTP hydrolysis mechanisms in Rab33 and Rab5. FEBS J. 2017, 284, 4358–4375. [Google Scholar] [CrossRef] [PubMed]
- Temeles, G.L.; Gibbs, J.B.; D’Alonzo, J.S.; Sigal, I.S.; Scolnick, E.M. Yeast and mammalian ras proteins have conserved biochemical properties. Nature 1985, 313, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef] [PubMed]
- Spaargaren, M.; Bischoff, J.R. Identification of the guanine nucleotide dissociation stimulator for Ral as a putative effector molecule of R-ras, H-ras, K-ras, and Rap. Proc. Natl. Acad. Sci. USA 1994, 91, 12609–12613. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Demo, S.D.; Ye, Z.H.; Chen, Y.W.; Williams, L.T. ralGDS family members interact with the effector loop of ras p21. Mol. Cell. Biol. 1994, 14, 7483–7491. [Google Scholar] [CrossRef] [PubMed]
- Hofer, F.; Fields, S.; Schneider, C.; Martin, G.S. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc. Natl. Acad. Sci. USA 1994, 91, 11089–11093. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Milburn, M.V.; Tong, L.; de Vos, A.M.; Brunger, A.; Yamaizumi, Z.; Nishimura, S.; Kim, S.H. Molecular switch for signal transduction: Structural differences between active and inactive forms of protooncogenic ras proteins. Science 1990, 247, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Schlichting, I.; Almo, S.C.; Rapp, G.; Wilson, K.; Petratos, K.; Lentfer, A.; Wittinghofer, A.; Kabsch, W.; Pai, E.F.; Petsko, G.A.; et al. Time-resolved X-ray crystallographic study of the conformational change in Ha-Ras p21 protein on GTP hydrolysis. Nature 1990, 345, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Scheffzek, K.; Lautwein, A.; Kabsch, W.; Ahmadian, M.R.; Wittinghofer, A. Crystal structure of the GTPase-activating domain of human p120GAP and implications for the interaction with Ras. Nature 1996, 384, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Repasky, G.A.; Chenette, E.J.; Der, C.J. Renewing the conspiracy theory debate: Does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004, 14, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Trahey, M.; McCormick, F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 1987, 238, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Bernards, A.; Settleman, J. GAP control: Regulating the regulators of small GTPases. Trends Cell Biol. 2004, 14, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Hall, A. Guanine nucleotide exchange factors for Rho GTPases: Turning on the switch. Genes Dev. 2002, 16, 1587–1609. [Google Scholar] [CrossRef] [PubMed]
- Martegani, E.; Vanoni, M.; Zippel, R.; Coccetti, P.; Brambilla, R.; Ferrari, C.; Sturani, E.; Alberghina, L. Cloning by functional complementation of a mouse cDNA encoding a homologue of CDC25, a Saccharomyces cerevisiae RAS activator. EMBO J. 1992, 11, 2151–2157. [Google Scholar] [PubMed]
- Segal, M.; Marbach, I.; Engelberg, D.; Simchen, G.; Levitzki, A. Interaction between the Saccharomyces cerevisiae CDC25 gene product and mammalian ras. J. Biol. Chem. 1992, 267, 22747–22751. [Google Scholar] [PubMed]
- Ballester, R.; Marchuk, D.; Boguski, M.; Saulino, A.; Letcher, R.; Wigler, M.; Collins, F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990, 63, 851–859. [Google Scholar] [CrossRef]
- Goodman, L.E.; Judd, S.R.; Farnsworth, C.C.; Powers, S.; Gelb, M.H.; Glomset, J.A.; Tamanoi, F. Mutants of Saccharomyces cerevisiae defective in the farnesylation of Ras proteins. Proc. Natl. Acad. Sci. USA 1990, 87, 9665–9669. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, A.; Tamanoi, F. RAS2 protein of Saccharomyces cerevisiae undergoes removal of methionine at N terminus and removal of three amino acids at C terminus. J. Biol. Chem. 1990, 265, 3362–3368. [Google Scholar] [PubMed]
- Fujiyama, A.; Tsunasawa, S.; Tamanoi, F.; Sakiyama, F. S-farnesylation and methyl esterification of C-terminal domain of yeast RAS2 protein prior to fatty acid acylation. J. Biol. Chem. 1991, 266, 17926–17931. [Google Scholar] [PubMed]
- Shih, T.Y.; Weeks, M.O.; Young, H.A.; Scholnick, E.M. Identification of a sarcoma virus-coded phosphoprotein in nonproducer cells transformed by Kirsten or Harvey murine sarcoma virus. Virology 1979, 96, 64–79. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. Ras family signaling: Therapeutic targeting. Cancer Biol. Ther. 2002, 1, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Schafer, W.R.; Kim, R.; Sterne, R.; Thorner, J.; Kim, S.H.; Rine, J. Genetic and pharmacological suppression of oncogenic mutations in ras genes of yeast and humans. Science 1989, 245, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F.; Magee, A.I.; Childs, J.E.; Marshall, C.J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989, 57, 1167–1177. [Google Scholar] [CrossRef]
- Casey, P.J.; Solski, P.A.; Der, C.J.; Buss, J.E. p21ras is modified by a farnesyl isoprenoid. Proc. Natl. Acad. Sci. USA 1989, 86, 8323–8327. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F.; Cadwallader, K.; Paterson, H.; Marshall, C.J. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 1991, 10, 4033–4039. [Google Scholar] [CrossRef]
- Rowell, C.A.; Kowalczyk, J.J.; Lewis, M.D.; Garcia, A.M. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J. Biol. Chem. 1997, 272, 14093–14097. [Google Scholar] [CrossRef] [PubMed]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, A.; Prior, I.A.; Lindsay, M.; Parton, R.G.; Hancock, J.F. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol. Cell. Biol. 2000, 20, 2475–2487. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.; Chiu, V.K.; Silletti, J.; Feoktistov, M.; Morimoto, T.; Michaelson, D.; Ivanov, I.E.; Philips, M.R. Endomembrane trafficking of ras: The CAAX motif targets proteins to the ER and Golgi. Cell 1999, 98, 69–80. [Google Scholar] [CrossRef]
- Roy, M.O.; Leventis, R.; Silvius, J.R. Mutational and biochemical analysis of plasma membrane targeting mediated by the farnesylated, polybasic carboxy terminus of K-ras4B. Biochemistry 2000, 39, 8298–8307. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F.; Paterson, H.; Marshall, C.J. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 1990, 63, 133–139. [Google Scholar] [CrossRef]
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, S. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta 2007, 1773, 1177–1195. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F.; Cadwallader, K.; Marshall, C.J. Methylation and proteolysis are essential for efficient membrane binding of prenylated p21K-ras(B). EMBO J. 1991, 10, 641–646. [Google Scholar] [PubMed]
- Agudo-Ibanez, L.; Crespo, P.; Casar, B. Analysis of Ras/ERK Compartmentalization by Subcellular Fractionation. Methods Mol. Biol. 2017, 1487, 151–162. [Google Scholar] [PubMed]
- Ahearn, I.M.; Tsai, F.D.; Court, H.; Zhou, M.; Jennings, B.C.; Ahmed, M.; Fehrenbacher, N.; Linder, M.E.; Philips, M.R. FKBP12 binds to acylated H-ras and promotes depalmitoylation. Mol. Cell 2011, 41, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Harding, A.; Yan, J.; Sluimer, J.; Parton, R.G.; Hancock, J.F. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat. Cell Biol. 2001, 3, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Luetterforst, R.; Harding, A.; Apolloni, A.; Etheridge, M.; Stang, E.; Rolls, B.; Hancock, J.F.; Parton, R.G. Dominant-negative caveolin inhibits H-Ras function by disrupting cholesterol-rich plasma membrane domains. Nat. Cell Biol. 1999, 1, 98–105. [Google Scholar] [PubMed]
- Ashery, U.; Yizhar, O.; Rotblat, B.; Elad-Sfadia, G.; Barkan, B.; Haklai, R.; Kloog, Y. Spatiotemporal organization of Ras signaling: Rasosomes and the galectin switch. Cell. Mol. Neurobiol. 2006, 26, 471–495. [Google Scholar] [CrossRef] [PubMed]
- Carozzi, A.J.; Roy, S.; Morrow, I.C.; Pol, A.; Wyse, B.; Clyde-Smith, J.; Prior, I.A.; Nixon, S.J.; Hancock, J.F.; Parton, R.G. Inhibition of lipid raft-dependent signaling by a dystrophy-associated mutant of caveolin-3. J. Biol. Chem. 2002, 277, 17944–17949. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Muncke, C.; Parton, R.G.; Hancock, J.F. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 2003, 160, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, V.K.; Bivona, T.; Hach, A.; Sajous, J.B.; Silletti, J.; Wiener, H.; Johnson, R.L., 2nd; Cox, A.D.; Philips, M.R. Ras signalling on the endoplasmic reticulum and the Golgi. Nat. Cell Biol. 2002, 4, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sorkin, A. Coordinated traffic of Grb2 and Ras during epidermal growth factor receptor endocytosis visualized in living cells. Mol. Biol. Cell 2002, 13, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; McClure, M.; Huang, F.; Carter, R. Interaction of EGF receptor and grb2 in living cells visualized by fluorescence resonance energy transfer (FRET) microscopy. Curr. Biol. 2000, 10, 1395–1398. [Google Scholar] [CrossRef]
- Mochizuki, N.; Yamashita, S.; Kurokawa, K.; Ohba, Y.; Nagai, T.; Miyawaki, A.; Matsuda, M. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature 2001, 411, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.T.; Carracedo, A.; Locasale, J.W.; Anastasiou, D.; Takeuchi, K.; Kahoud, E.R.; Haviv, S.; Asara, J.M.; Pandolfi, P.P.; Cantley, L.C. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci. Signal. 2011, 4, ra13. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Tebar, F.; Alvarez-Moya, B.; Lopez-Alcala, C.; Calvo, M.; Enrich, C.; Agell, N.; Nakamura, T.; Matsuda, M.; Bachs, O. A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J. Cell Biol. 2009, 184, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, A.; Perez-Sala, D.; Martinez, A.C. Bcl-2 differentially targets K-, N-, and H-Ras to mitochondria in IL-2 supplemented or deprived cells: Implications in prevention of apoptosis. Oncogene 1999, 18, 4930–4939. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hancock, J.F. Ras trafficking, localization and compartmentalized signalling. Semin. Cell Dev. Biol. 2012, 23, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Wittinghofer, A.; Franken, S.M.; Scheidig, A.J.; Rensland, H.; Lautwein, A.; Pai, E.F.; Goody, R.S. Three-dimensional structure and properties of wild-type and mutant H-ras-encoded p21. Ciba Found. Symp. 1993, 176, 6–21, discussion 21–27. [Google Scholar] [PubMed]
- Elad-Sfadia, G.; Haklai, R.; Balan, E.; Kloog, Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J. Biol. Chem. 2004, 279, 34922–34930. [Google Scholar] [CrossRef] [PubMed]
- Shalom-Feuerstein, R.; Plowman, S.J.; Rotblat, B.; Ariotti, N.; Tian, T.; Hancock, J.F.; Kloog, Y. K-ras nanoclustering is subverted by overexpression of the scaffold protein galectin-3. Cancer Res. 2008, 68, 6608–6616. [Google Scholar] [CrossRef] [PubMed]
- Paz, A.; Haklai, R.; Elad-Sfadia, G.; Ballan, E.; Kloog, Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 2001, 20, 7486–7493. [Google Scholar] [CrossRef] [PubMed]
- Belotti, F.; Tisi, R.; Paiardi, C.; Rigamonti, M.; Groppi, S.; Martegani, E. Localization of Ras signaling complex in budding yeast. Biochim. Biophys. Acta 2012, 1823, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Sobering, A.K.; Romeo, M.J.; Vay, H.A.; Levin, D.E. A novel Ras inhibitor, Eri1, engages yeast Ras at the endoplasmic reticulum. Mol. Cell. Biol. 2003, 23, 4983–4990. [Google Scholar] [CrossRef] [PubMed]
- Broggi, S.; Martegani, E.; Colombo, S. Nuclear Ras2-GTP Controls Invasive Growth in Saccharomyces cerevisiae. PLoS ONE 2013, 8, e79274. [Google Scholar] [CrossRef] [PubMed]
- Tisi, R.; Belotti, F.; Martegani, E. Yeast as a model for Ras signalling. Methods Mol. Biol. 2014, 1120, 359–390. [Google Scholar] [PubMed]
- Cheng, C.-M.; Chang, E.C. Cell Cycle. In Busy Traveling Ras; Bioscience, L., Ed.; Landes Bioscience: Houston, TX, USA, 2011; Volume 10, pp. 1180–1181. [Google Scholar]
- Saif, M.W. Colorectal cancer in review: The role of the EGFR pathway. Expert Opin. Investig. Drugs 2010, 19, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; De Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Tejpar, S.; Morgillo, F.; De Luca, A.; Van Cutsem, E.; Ciardiello, F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol. 2009, 6, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Schulze, W.X.; Deng, L.; Mann, M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol. Syst. Biol. 2005, 1. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Shields, J.M.; Pruitt, K.; McFall, A.; Shaub, A.; Der, C.J. Understanding Ras: ‘it ain’t over ’til it’s over’. Trends Cell Biol. 2000, 10, 147–154. [Google Scholar] [CrossRef]
- Herrmann, C. Ras-effector interactions: After one decade. Curr. Opin. Struct. Biol. 2003, 13, 122–129. [Google Scholar] [CrossRef]
- Efferth, T. Signal transduction pathways of the epidermal growth factor receptor in colorectal cancer and their inhibition by small molecules. Curr. Med. Chem. 2012, 19, 5735–5744. [Google Scholar] [CrossRef] [PubMed]
- Scheid, M.P.; Woodgett, J.R. Phosphatidylinositol 3’ kinase signaling in mammary tumorigenesis. J. Mammary Gland Biol. Neoplasia 2001, 6, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Scheid, M.P.; Woodgett, J.R. PKB/AKT: Functional insights from genetic models. Nat. Rev. Mol. Cell Biol. 2001, 2, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Roy, S.; Apolloni, A.; Lane, A.; Hancock, J.F. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J. Biol. Chem. 1998, 273, 24052–24056. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Zhang, Y.J. Targeting mTOR network in colorectal cancer therapy. World J. Gastroenterol. 2014, 20, 4178–4188. [Google Scholar] [CrossRef] [PubMed]
- Busti, S.; Coccetti, P.; Alberghina, L.; Vanoni, M. Glucose signaling-mediated coordination of cell growth and cell cycle in Saccharomyces cerevisiae. Sensors 2010, 10, 6195–6240. [Google Scholar] [CrossRef] [PubMed]
- Tamanoi, F. Ras signaling in yeast. Genes Cancer 2011, 2, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Francois, J.; Hers, H.G. The control of glycogen metabolism in yeast. 2. A kinetic study of the two forms of glycogen synthase and of glycogen phosphorylase and an investigation of their interconversion in a cell-free extract. Eur. J. Biochem. 1988, 174, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Francois, J.; Van Schaftingen, E.; Hers, H.G. The mechanism by which glucose increases fructose 2,6-bisphosphate concentration in Saccharomyces cerevisiae. A cyclic-AMP-dependent activation of phosphofructokinase 2. Eur. J. Biochem. 1984, 145, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Boado, Y.S.; Herrero, P.; Gascon, S.; Moreno, F. Catabolite inactivation of isocitrate lyase from Saccharomyces cerevisiae. Arch. Microbiol. 1987, 147, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Bandlow, W. cAMP-dependent protein kinase activity in yeast mitochondria. Z. Naturforsch. C 1987, 42, 1291–1302. [Google Scholar] [PubMed]
- Ortiz, C.H.; Maia, J.C.; Tenan, M.N.; Braz-Padrao, G.R.; Mattoon, J.R.; Panek, A.D. Regulation of yeast trehalase by a monocyclic, cyclic AMP-dependent phosphorylation-dephosphorylation cascade system. J. Bacteriol. 1983, 153, 644–651. [Google Scholar] [PubMed]
- Pohlig, G.; Holzer, H. Phosphorylation and inactivation of yeast fructose-1,6-bisphosphatase by cyclic AMP-dependent protein kinase from yeast. J. Biol. Chem. 1985, 260, 13818–13823. [Google Scholar] [PubMed]
- Rittenhouse, J.; Moberly, L.; Marcus, F. Phosphorylation in vivo of yeast (Saccharomyces cerevisiae) fructose-1,6-bisphosphatase at the cyclic AMP-dependent site. J. Biol. Chem. 1987, 262, 10114–10119. [Google Scholar] [PubMed]
- Rödel, G.; Muller, G.; Bandlow, W. Cyclic AMP receptor protein from yeast mitochondria: Submitochondrial localization and preliminary characterization. J. Bacteriol. 1985, 161, 7–12. [Google Scholar] [PubMed]
- Uno, I.; Matsumoto, K.; Adachi, K.; Ishikawa, T. Genetic and biochemical evidence that trehalase is a substrate of cAMP-dependent protein kinase in yeast. J. Biol. Chem. 1983, 258, 10867–10872. [Google Scholar] [PubMed]
- Cameron, S.; Levin, L.; Zoller, M.; Wigler, M. cAMP-independent control of sporulation, glycogen metabolism, and heat shock resistance in S. cerevisiae. Cell 1988, 53, 555–566. [Google Scholar] [CrossRef]
- Uno, I.; Matsumoto, K.; Hirata, A.; Ishikawa, T. Outer plaque assembly and spore encapsulation are defective during sporulation of adenylate cyclase-deficient mutants of Saccharomyces cerevisiae. J. Cell Biol. 1985, 100, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Uno, I.; Oshima, Y.; Ishikawa, T. Isolation and characterization of yeast mutants deficient in adenylate cyclase and cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1982, 79, 2355–2359. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Uno, I.; Ishikawa, T. Control of cell division in Saccharomyces cerevisiae mutants defective in adenylate cyclase and cAMP-dependent protein kinase. Exp. Cell Res. 1983, 146, 151–161. [Google Scholar] [CrossRef]
- Matsumoto, K.; Uno, I.; Ishikawa, T. Initiation of meiosis in yeast mutants defective in adenylate cyclase and cyclic AMP-dependent protein kinase. Cell 1983, 32, 417–423. [Google Scholar] [CrossRef]
- Kido, M.; Shima, F.; Satoh, T.; Asato, T.; Kariya, K.; Kataoka, T. Critical function of the Ras-associating domain as a primary Ras-binding site for regulation of Saccharomyces cerevisiae adenylyl cyclase. J. Biol. Chem. 2002, 277, 3117–3123. [Google Scholar] [CrossRef] [PubMed]
- Thevelein, J.M.; de Winde, J.H. Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 1999, 33, 904–918. [Google Scholar] [CrossRef] [PubMed]
- Broach, J.R.; Deschenes, R.J. The function of ras genes in Saccharomyces cerevisiae. Adv. Cancer Res. 1990, 54, 79–139. [Google Scholar] [PubMed]
- Thevelein, J.M. The RAS-adenylate cyclase pathway and cell cycle control in Saccharomyces cerevisiae. Antonie van Leeuwenhoek 1992, 62, 109–130. [Google Scholar] [CrossRef] [PubMed]
- Thevelein, J.M. Signal transduction in yeast. Yeast 1994, 10, 1753–1790. [Google Scholar] [CrossRef] [PubMed]
- Tatchell, T. RAS genes in the budding yeast Saccharomyces cerevisiae. In Signal Transduction: Prokaryotic and Simple Eukaryotic Systems; Kurjan, J., Taylor, B.L., Eds.; Academic Press: San Diego, CA, USA, 1993; pp. 147–188. [Google Scholar]
- Toda, T.; Cameron, S.; Sass, P.; Zoller, M.; Scott, J.D.; McMullen, B.; Hurwitz, M.; Krebs, E.G.; Wigler, M. Cloning and characterization of BCY1, a locus encoding a regulatory subunit of the cyclic AMP-dependent protein kinase in Saccharomyces cerevisiae. Mol. Cell. Biol. 1987, 7, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Cameron, S.; Sass, P.; Zoller, M.; Wigler, M. Three different genes in S. cerevisiae encode the catalytic subunits of the cAMP-dependent protein kinase. Cell 1987, 50, 277–287. [Google Scholar] [CrossRef]
- Nikawa, J.; Cameron, S.; Toda, T.; Ferguson, K.M.; Wigler, M. Rigorous feedback control of cAMP levels in Saccharomyces cerevisiae. Genes Dev. 1987, 1, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Mbonyi, K.; van Aelst, L.; Arguelles, J.C.; Jans, A.W.; Thevelein, J.M. Glucose-induced hyperaccumulation of cyclic AMP and defective glucose repression in yeast strains with reduced activity of cyclic AMP-dependent protein kinase. Mol. Cell. Biol. 1990, 10, 4518–4523. [Google Scholar] [CrossRef] [PubMed]
- Zaman, S.; Lippman, S.I.; Zhao, X.; Broach, J.R. How Saccharomyces responds to nutrients. Annu. Rev. Genet. 2008, 42, 27–81. [Google Scholar] [CrossRef] [PubMed]
- Gancedo, J.M. The early steps of glucose signalling in yeast. FEMS Microbiol. Rev. 2008, 32, 673–704. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Bretscher, A. Ras regulates the polarity of the yeast actin cytoskeleton through the stress response pathway. Mol. Biol. Cell 2001, 12, 1541–1555. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Wagner, M.; Dunham, M.J.; Shin, M.E.; Ahmed, N.T.; Winter, E. The Ras/cAMP pathway and the CDK-like kinase Ime2 regulate the MAPK Smk1 and spore morphogenesis in Saccharomyces cerevisiae. Genetics 2009, 181, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Hubler, L.; Bradshaw-Rouse, J.; Heideman, W. Connections between the Ras-cyclic AMP pathway and G1 cyclin expression in the budding yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 1993, 13, 6274–6282. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Kale, S.P.; Childress, A.M.; Pinswasdi, C.; Jazwinski, S.M. Divergent roles of RAS1 and RAS2 in yeast longevity. J. Biol. Chem. 1994, 269, 18638–18645. [Google Scholar] [PubMed]
- Peeper, D.S.; Upton, T.M.; Ladha, M.H.; Neuman, E.; Zalvide, J.; Bernards, R.; DeCaprio, J.A.; Ewen, M.E. Ras signalling linked to the cell-cycle machinery by the retinoblastoma protein. Nature 1997, 386, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.; Der, C.J. Ras and Rho regulation of the cell cycle and oncogenesis. Cancer Lett. 2001, 171, 1–10. [Google Scholar] [CrossRef]
- Coleman, M.L.; Marshall, C.J.; Olson, M.F. RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat. Rev. Mol. Cell Biol. 2004, 5, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Cell cycle: Routine role for Ras. Curr. Biol. 1997, 7, R258–R260. [Google Scholar] [CrossRef]
- Mittnacht, S.; Paterson, H.; Olson, M.F.; Marshall, C.J. Ras signalling is required for inactivation of the tumour suppressor pRb cell-cycle control protein. Curr. Biol. 1997, 7, 219–221. [Google Scholar] [CrossRef]
- Yang, J.J.; Kang, J.S.; Krauss, R.S. Transformation-restoring factor: A low molecular weight secreted factor required for anchorage-independent growth of oncogene-resistant mutant cell lines. Oncogene 1995, 10, 1291–1299. [Google Scholar] [PubMed]
- Kang, J.S.; Krauss, R.S. Ras induces anchorage-independent growth by subverting multiple adhesion-regulated cell cycle events. Mol. Cell. Biol. 1996, 16, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.; Pestell, R.G.; Der, C.J. Ras inactivation of the retinoblastoma pathway by distinct mechanisms in NIH 3T3 fibroblast and RIE-1 epithelial cells. J. Biol. Chem. 2000, 275, 40916–40924. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.S.; Smith, M.R.; Stacey, D.W. Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature 1985, 313, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Feramisco, J.R.; Gross, M.; Kamata, T.; Rosenberg, M.; Sweet, R.W. Microinjection of the oncogene form of the human H-ras (T-24) protein results in rapid proliferation of quiescent cells. Cell 1984, 38, 109–117. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Unger, M.W. Unequal division in Saccharomyces cerevisiae and its implications for the control of cell division. J. Cell Biol. 1977, 75, 422–435. [Google Scholar] [CrossRef] [PubMed]
- Vanoni, M.; Vai, M.; Popolo, L.; Alberghina, L. Structural heterogeneity in populations of the budding yeast Saccharomyces cerevisiae. J. Bacteriol. 1983, 156, 1282–1291. [Google Scholar] [PubMed]
- Johnston, G.C.; Ehrhardt, C.W.; Lorincz, A.; Carter, B.L. Regulation of cell size in the yeast Saccharomyces cerevisiae. J. Bacteriol. 1979, 137, 1–5. [Google Scholar] [PubMed]
- Lord, P.G.; Wheals, A.E. Rate of cell cycle initiation of yeast cells when cell size is not a rate-determining factor. J. Cell Sci. 1983, 59, 183–201. [Google Scholar] [PubMed]
- Mitchison, J.M. The Biology of the Cell Cycle; Cambridge University Press: Cambridge, UK, 1971. [Google Scholar]
- Baroni, M.D.; Monti, P.; Alberghina, L. Repression of growth-regulated G1 cyclin expression by cyclic AMP in budding yeast. Nature 1994, 371, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Tokiwa, G.; Tyers, M.; Volpe, T.; Futcher, B. Inhibition of G1 cyclin activity by the Ras/cAMP pathway in yeast. Nature 1994, 371, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Mizunuma, M.; Tsubakiyama, R.; Ogawa, T.; Shitamukai, A.; Kobayashi, Y.; Inai, T.; Kume, K.; Hirata, D. Ras/cAMP-dependent protein kinase (PKA) regulates multiple aspects of cellular events by phosphorylating the Whi3 cell cycle regulator in budding yeast. J. Biol. Chem. 2013, 288, 10558–10566. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Ras signalling and apoptosis. Curr. Opin. Genet. Dev. 1998, 8, 49–54. [Google Scholar] [CrossRef]
- Chang, F.; Steelman, L.S.; Shelton, J.G.; Lee, J.T.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.; McCubrey, J.A. Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway. Int. J. Oncol. 2003, 22, 469–480. [Google Scholar] [PubMed]
- Cox, A.D.; Der, C.J. The dark side of Ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Sulciner, D.J.; Irani, K.; Yu, Z.X.; Ferrans, V.J.; Goldschmidt-Clermont, P.; Finkel, T. rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kB activation. Mol. Cell. Biol. 1996, 16, 7115–7121. [Google Scholar] [CrossRef] [PubMed]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef] [PubMed]
- Joneson, T.; Bar-Sagi, D. Suppression of Ras-induced apoptosis by the Rac GTPase. Mol. Cell. Biol. 1999, 19, 5892–5901. [Google Scholar] [CrossRef] [PubMed]
- Mayo, M.W.; Baldwin, A.S. The transcription factor NF-kB: Control of oncogenesis and cancer therapy resistance. Biochim. Biophys. Acta 2000, 1470, M55–M62. [Google Scholar] [PubMed]
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Romashkova, J.A.; Makarov, S.S. NF-kB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Janknecht, R.; Hunter, T. The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of Bad on Ser136. Curr. Biol. 1998, 8, 779–782. [Google Scholar] [CrossRef]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yu, S.; Eder, A.; Mao, M.; Bast, R.C., Jr.; Boyd, D.; Mills, G.B. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene 1999, 18, 6635–6640. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ruan, H.; Demeter, M.R.; Comb, M.J. p90(RSK) blocks bad-mediated cell death via a protein kinase C-dependent pathway. J. Biol. Chem. 1999, 274, 34859–34867. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Montminy, M. CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 1998, 273, 32377–32379. [Google Scholar] [CrossRef] [PubMed]
- Nalca, A.; Qiu, S.G.; El-Guendy, N.; Krishnan, S.; Rangnekar, V.M. Oncogenic Ras sensitizes cells to apoptosis by Par-4. J. Biol. Chem. 1999, 274, 29976–29983. [Google Scholar] [CrossRef] [PubMed]
- Ries, S.; Biederer, C.; Woods, D.; Shifman, O.; Shirasawa, S.; Sasazuki, T.; McMahon, M.; Oren, M.; McCormick, F. Opposing effects of Ras on p53: Transcriptional activation of mdm2 and induction of p19ARF. Cell 2000, 103, 321–330. [Google Scholar] [CrossRef]
- Tsuchida, T.; Kijima, H.; Hori, S.; Oshika, Y.; Tokunaga, T.; Kawai, K.; Yamazaki, H.; Ueyama, Y.; Scanlon, K.J.; Tamaoki, N.; et al. Adenovirus-mediated anti-K-ras ribozyme induces apoptosis and growth suppression of human pancreatic carcinoma. Cancer Gene Ther. 2000, 7, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Navarro, P.; Valverde, A.M.; Benito, M.; Lorenzo, M. Activated Ha-ras induces apoptosis by association with phosphorylated Bcl-2 in a mitogen-activated protein kinase-independent manner. J. Biol. Chem. 1999, 274, 18857–18863. [Google Scholar] [CrossRef] [PubMed]
- Gourlay, C.W.; Du, W.; Ayscough, K.R. Apoptosis in yeast–mechanisms and benefits to a unicellular organism. Mol. Microbiol. 2006, 62, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, K.U.; Fussi, H.; Ruckenstuhl, C. Yeast apoptosis—From genes to pathways. Semin. Cancer Biol. 2007, 17, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Gutierrez, D.; Eisenberg, T.; Buttner, S.; Meisinger, C.; Kroemer, G.; Madeo, F. Apoptosis in yeast: Triggers, pathways, subroutines. Cell Death Differ. 2010, 17, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.J.; Crowe, J.D.; Ramsdale, M. Ras pathway signaling accelerates programmed cell death in the pathogenic fungus Candida albicans. Proc. Natl. Acad. Sci. USA 2006, 103, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Gourlay, C.W.; Ayscough, K.R. Actin-induced hyperactivation of the Ras signaling pathway leads to apoptosis in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006, 26, 6487–6501. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, M.L.; Damsz, B.; Coca, M.A.; Ibeas, J.I.; Yun, D.J.; Pardo, J.M.; Hasegawa, P.M.; Bressan, R.A. A plant defense response effector induces microbial apoptosis. Mol. Cell 2001, 8, 921–930. [Google Scholar] [CrossRef]
- Santos, J.; Sousa, M.J.; Leao, C. Ammonium is toxic for aging yeast cells, inducing death and shortening of the chronological lifespan. PLoS ONE 2012, 7, e37090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.; Leao, C.; Sousa, M.J. Ammonium-dependent shortening of CLS in yeast cells starved for essential amino acids is determined by the specific amino acid deprived, through different signaling pathways. Oxid. Med. Cell. Longev. 2013, 2013, 161986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeo, F.; Frohlich, E.; Ligr, M.; Grey, M.; Sigrist, S.J.; Wolf, D.H.; Frohlich, K.U. Oxygen stress: A regulator of apoptosis in yeast. J. Cell Biol. 1999, 145, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Ludovico, P.; Sousa, M.J.; Silva, M.T.; Leao, C.; Corte-Real, M. Saccharomyces cerevisiae commits to a programmed cell death process in response to acetic acid. Microbiology 2001, 147, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.D.; Sotoca, R.; Johansson, B.; Ludovico, P.; Sansonetty, F.; Silva, M.T.; Peinado, J.M.; Corte-Real, M. Hyperosmotic stress induces metacaspase- and mitochondria-dependent apoptosis in Saccharomyces cerevisiae. Mol. Microbiol. 2005, 58, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsui, K.; Nakagawa, D.; Nakamura, M.; Okamoto, T.; Tsurugi, K. Valproic acid induces apoptosis dependent of Yca1p at concentrations that mildly affect the proliferation of yeast. FEBS Lett. 2005, 579, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, M.; Ramachandran, L.; Feng, L.; Sharma, K.; Sun, X.; Marchetti, M.; Huberman, J.A.; Burhans, W.C. Apoptosis in budding yeast caused by defects in initiation of DNA replication. J. Cell Sci. 2005, 118, 3543–3553. [Google Scholar] [CrossRef] [PubMed]
- Gourlay, C.W.; Ayscough, K.R. Identification of an upstream regulatory pathway controlling actin-mediated apoptosis in yeast. J. Cell Sci. 2005, 118, 2119–2132. [Google Scholar] [CrossRef] [PubMed]
- Lastauskiene, E.; Zinkeviciene, A.; Citavicius, D. Ras/PKA signal transduction pathway participates in the regulation of Saccharomyces cerevisiae cell apoptosis in an acidic environment. Biotechnol. Appl. Biochem. 2014, 61, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Mollapour, M.; Phelan, J.P.; Millson, S.H.; Piper, P.W.; Cooke, F.T. Weak acid and alkali stress regulate phosphatidylinositol bisphosphate synthesis in Saccharomyces cerevisiae. Biochem. J. 2006, 395, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy. Curr. Biol. 2005, 15, R282–R283. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.; Kiffin, R.; Cuervo, A.M. Pathophysiology of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2420–2434. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Levine, B.; Deretic, V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007, 7, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vicencio, J.M.; Kepp, O.; Tasdemir, E.; Maiuri, M.C.; Kroemer, G. To die or not to die: That is the autophagic question. Curr. Mol. Med. 2008, 8, 78–91. [Google Scholar] [PubMed]
- Kroemer, G.; Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Autophagy: Regulation by energy sensing. Curr. Biol. 2011, 21, R227–R229. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Bauvy, C.; Codogno, P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J. Biol. Chem. 2003, 278, 16667–16674. [Google Scholar] [CrossRef] [PubMed]
- Furuta, S.; Hidaka, E.; Ogata, A.; Yokota, S.; Kamata, T. Ras is involved in the negative control of autophagy through the class I PI3-kinase. Oncogene 2004, 23, 3898–3904. [Google Scholar] [CrossRef] [PubMed]
- Elgendy, M.; Sheridan, C.; Brumatti, G.; Martin, S.J. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell 2011, 42, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Lan, S.H.; Cheng, D.E.; Chen, W.K.; Shen, C.H.; Lee, Y.R.; Zuchini, R.; Liu, H.S. Ras-related tumorigenesis is suppressed by BNIP3-mediated autophagy through inhibition of cell proliferation. Neoplasia 2011, 13, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.Y.; Lee, Y.K.; Yoon, Y.S.; Xu, W.G.; Yoon, J.K.; Choi, S.E.; Ko, Y.G.; Kim, M.J.; Lee, S.J.; et al. Involvement of mitophagy in oncogenic K-Ras-induced transformation: Overcoming a cellular energy deficit from glucose deficiency. Autophagy 2011, 7, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Woo, S.J.; Yoon, C.H.; Lee, J.S.; An, S.; Choi, Y.H.; Hwang, S.G.; Yoon, G.; Lee, S.J. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 2011, 286, 12924–12932. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Cebollero, E.; Reggiori, F. Regulation of autophagy in yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2009, 1793, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy—Unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Stephan, J.S.; Yeh, Y.Y.; Ramachandran, V.; Deminoff, S.J.; Herman, P.K. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc. Natl. Acad. Sci. USA 2009, 106, 17049–17054. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Schothorst, J.; Kankipati, H.N.; Van Zeebroeck, G.; Rubio-Texeira, M.; Thevelein, J.M. Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2014, 38, 254–299. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Fabrizio, P. Chronological aging in Saccharomyces cerevisiae. Subcell. Biochem. 2012, 57, 101–121. [Google Scholar] [PubMed]
- Jacinto, E.; Lorberg, A. TOR regulation of AGC kinases in yeast and mammals. Biochem. J. 2008, 410, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.; Soulard, A.; Huber, A.; Lippman, S.; Mukhopadhyay, D.; Deloche, O.; Wanke, V.; Anrather, D.; Ammerer, G.; Riezman, H.; et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 2007, 26, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Zaman, S.; Broach, J.R.; Klionsky, D.J. Protein kinase A and Sch9 cooperatively regulate induction of autophagy in Saccharomyces cerevisiae. Mol. Biol. Cell 2007, 18, 4180–4189. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Pendergast, A.M.; Quilliam, L.A.; Cripe, L.D.; Bassing, C.H.; Dai, Z.; Li, N.; Batzer, A.; Rabun, K.M.; Der, C.J.; Schlessinger, J.; et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell 1993, 75, 175–185. [Google Scholar] [CrossRef]
- Donovan, S.; Shannon, K.M.; Bollag, G. GTPase activating proteins: Critical regulators of intracellular signaling. Biochim. Biophys. Acta 2002, 1602, 23–45. [Google Scholar] [CrossRef]
- Yarden, Y. The EGFR family and its ligands in human cancer. Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37 (Suppl. 4), S3–S8. [Google Scholar] [CrossRef]
- Barault, L.; Veyrie, N.; Jooste, V.; Lecorre, D.; Chapusot, C.; Ferraz, J.M.; Lievre, A.; Cortet, M.; Bouvier, A.M.; Rat, P.; et al. Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. Int. J. Cancer 2008, 122, 2255–2259. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Velho, S.; Moutinho, C.; Ferreira, A.; Preto, A.; Domingo, E.; Capelinha, A.F.; Duval, A.; Hamelin, R.; Machado, J.C.; et al. KRAS and BRAF oncogenic mutations in MSS colorectal carcinoma progression. Oncogene 2007, 26, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Velculescu, V.E. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 2004, 3, 1221–1224. [Google Scholar] [CrossRef] [PubMed]
- Broek, D.; Samiy, N.; Fasano, O.; Fujiyama, A.; Tamanoi, F.; Northup, J.; Wigler, M. Differential activation of yeast adenylate cyclase by wild-type and mutant RAS proteins. Cell 1985, 41, 763–769. [Google Scholar] [CrossRef]
- Cazzanelli, G. Study the Roles of Human Galectin-3 Using the Yeast Saccharomyces cerevisiae and Colorectal Cancer Cells as Eukaryotic Models. Ph.D. Thesis, the University of Minho, Braga, Portugal, 2017. [Google Scholar]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Karantza, V. Autophagy as a therapeutic target in cancer. Cancer Biol. Ther. 2011, 11, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Shalom-Feuerstein, R.; Cooks, T.; Raz, A.; Kloog, Y. Galectin-3 regulates a molecular switch from N-Ras to K-Ras usage in human breast carcinoma cells. Cancer Res. 2005, 65, 7292–7300. [Google Scholar] [CrossRef] [PubMed]
- Abankwa, D.; Gorfe, A.A.; Inder, K.; Hancock, J.F. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. USA 2010, 107, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Bhagatji, P.; Leventis, R.; Rich, R.; Lin, C.J.; Silvius, J.R. Multiple cellular proteins modulate the dynamics of K-ras association with the plasma membrane. Biophys. J. 2010, 99, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Grafi-Cohen, M.; Kraiem, Z.; Kloog, Y. Galectin-3 promotes chronic activation of K-Ras and differentiation block in malignant thyroid carcinomas. Mol. Cancer Ther. 2010, 9, 2208–2219. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Ji, B.; Ramachandran, V.; Wang, H.; Hafley, M.; Logsdon, C.; Bresalier, R.S. Overexpressed galectin-3 in pancreatic cancer induces cell proliferation and invasion by binding Ras and activating Ras signaling. PLoS ONE 2012, 7, e42699. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.L.; Huang, E.Y.; Jhu, E.W.; Huang, Y.H.; Su, W.H.; Chuang, P.C.; Yang, K.D. Overexpression of galectin-3 enhances migration of colon cancer cells related to activation of the K-Ras-Raf-Erk1/2 pathway. J. Gastroenterol. 2013, 48, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.I.; Liu, F.T.; Panjwani, N. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J. Exp. Med. 2010, 207, 1981–1993. [Google Scholar] [CrossRef] [PubMed]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin b(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Han, J.W.; McCormick, F.; Macara, I.G. Regulation of Ras-GAP and the neurofibromatosis-1 gene product by eicosanoids. Science 1991, 252, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Rymond, B. Appendix A1: Yeast Nomenclature Systematic Open Reading Frame (ORF) and Other Genetic Designations. In Alternative pre-mRNA Splicing; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 603–607. [Google Scholar]
- Sherman, F. Getting started with yeast. Methods Enzymol. 2002, 350, 3–41. [Google Scholar] [PubMed]













© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cazzanelli, G.; Pereira, F.; Alves, S.; Francisco, R.; Azevedo, L.; Dias Carvalho, P.; Almeida, A.; Côrte-Real, M.; Oliveira, M.J.; Lucas, C.; et al. The Yeast Saccharomyces cerevisiae as a Model for Understanding RAS Proteins and their Role in Human Tumorigenesis. Cells 2018, 7, 14. https://doi.org/10.3390/cells7020014
Cazzanelli G, Pereira F, Alves S, Francisco R, Azevedo L, Dias Carvalho P, Almeida A, Côrte-Real M, Oliveira MJ, Lucas C, et al. The Yeast Saccharomyces cerevisiae as a Model for Understanding RAS Proteins and their Role in Human Tumorigenesis. Cells. 2018; 7(2):14. https://doi.org/10.3390/cells7020014
Chicago/Turabian StyleCazzanelli, Giulia, Flávia Pereira, Sara Alves, Rita Francisco, Luísa Azevedo, Patrícia Dias Carvalho, Ana Almeida, Manuela Côrte-Real, Maria José Oliveira, Cândida Lucas, and et al. 2018. "The Yeast Saccharomyces cerevisiae as a Model for Understanding RAS Proteins and their Role in Human Tumorigenesis" Cells 7, no. 2: 14. https://doi.org/10.3390/cells7020014
APA StyleCazzanelli, G., Pereira, F., Alves, S., Francisco, R., Azevedo, L., Dias Carvalho, P., Almeida, A., Côrte-Real, M., Oliveira, M. J., Lucas, C., Sousa, M. J., & Preto, A. (2018). The Yeast Saccharomyces cerevisiae as a Model for Understanding RAS Proteins and their Role in Human Tumorigenesis. Cells, 7(2), 14. https://doi.org/10.3390/cells7020014