Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors

Abstract
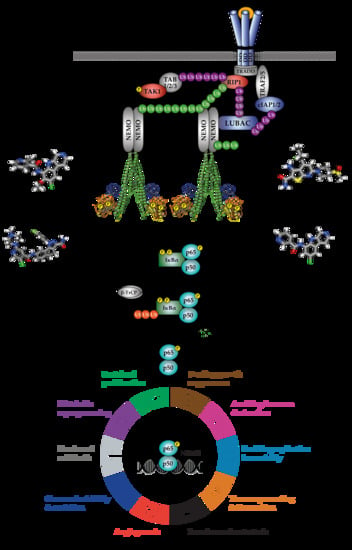
:
1. Introduction
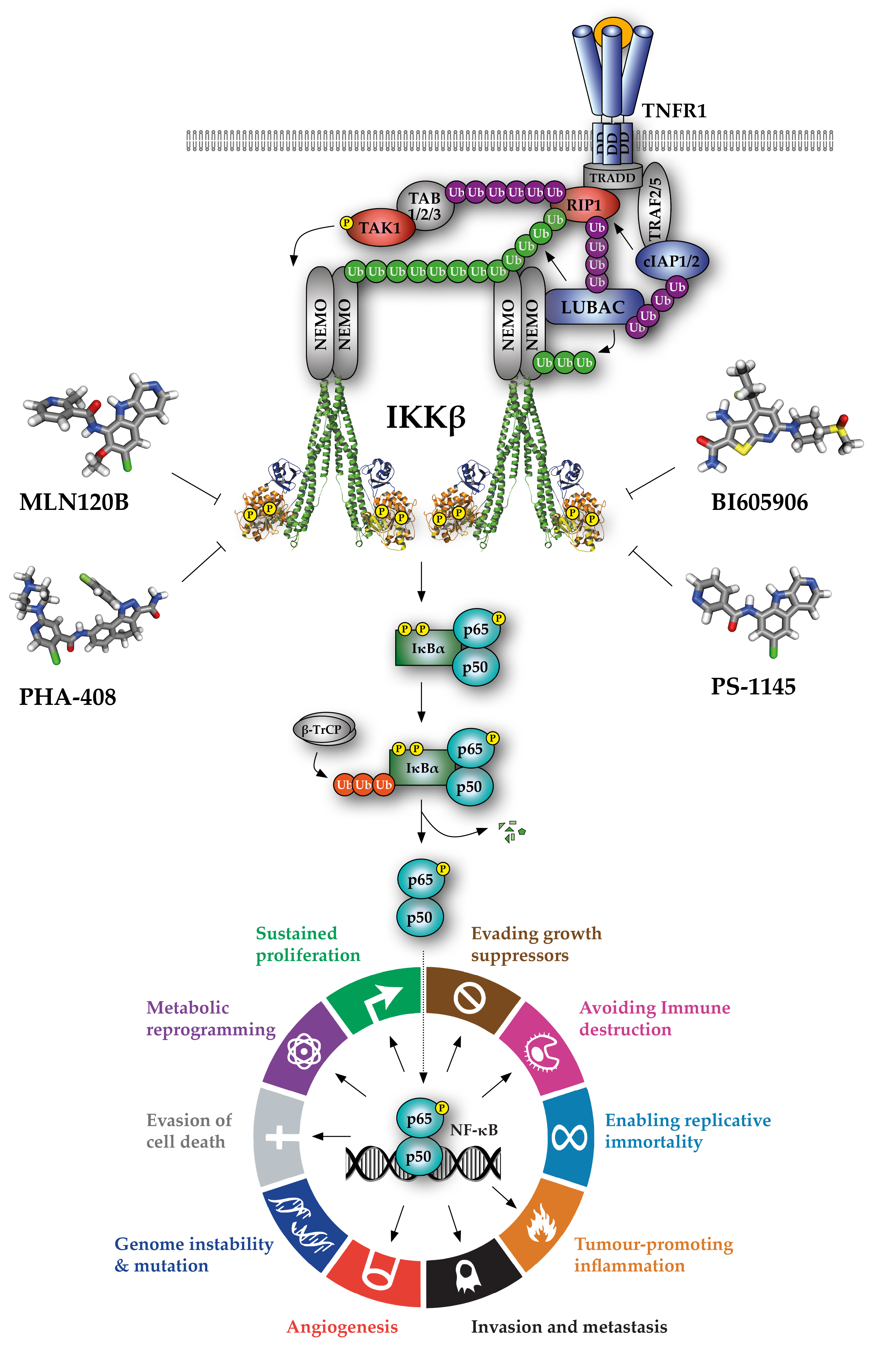
2. IKKβ Inhibitors
3. Pre-clinical Development of IKKβ Inhibitors
4. Potential Reasons for the Lack of Clinical Success of IKKβ Inhibitors
5. Safety Concerns Surrounding the Therapeutic Use of IKKβ Inhibitors
6. Recent Therapeutic Opportunities to Target IKKβ
6.1. Cancers Exhibiting Clear ‘Addiction’ to Canonical NF-κB Signalling
6.2. Use of IKKβ Inhibitors in Combination Therapies to Combat Chemoresistance
6.3. IKKβ Inhibitors as an Adjunct to Cancer Immunotherapies
6.4. Targeted Delivery of IKKβ Inhibitors to Specific Tissues
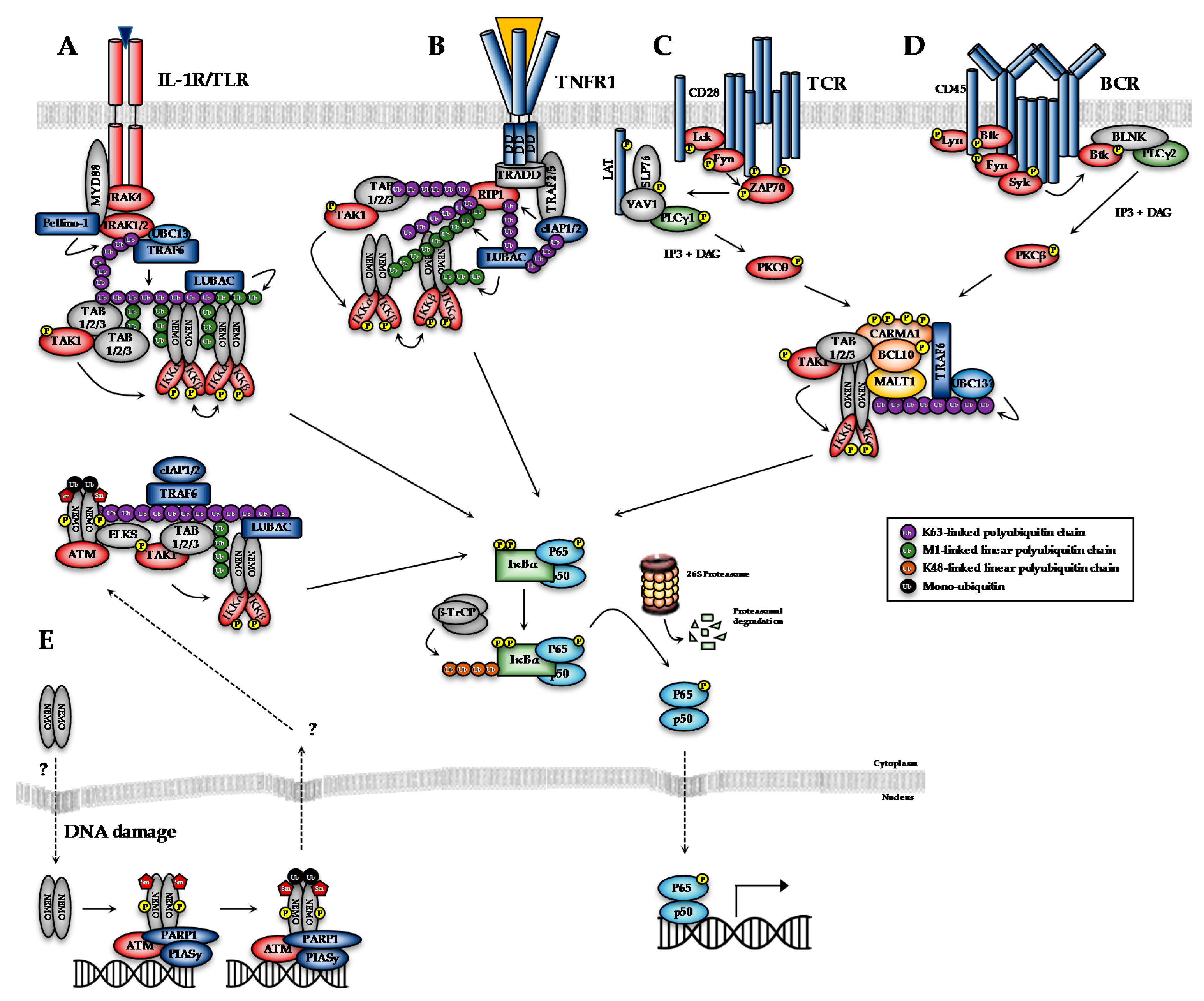
7. Alternative Approaches to Targeting the NF-κB Pathway
7.1. Targeting Upstream NF-κB Signalling Components
7.2. Targeting IKKα or NEMO
7.3. Targeting NF-κB-Independent Functions of the IKKs
7.4. Targeting IκBα Degradation
7.5. Targeting NF-κB Activity Directly
7.6. Targeting Downstream Effectors of NF-κB-Dependent Pathogenesis
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC DLBCL | Activated B cell-like Diffuse large B-cell lymphoma |
| BCR | B-cell receptor |
| BET | Bromodomain and extra-terminal |
| BTK | Burton tyrosine kinase |
| β-TrCP | Beta-transducing repeat-containing protein |
| CAC | Colitis-associated carcinoma |
| CAF | Cancer-associated fibroblast |
| CLL | Chronic lymphomatic leukemia |
| CNV | Choroid neovascularization |
| COPD | Chronic obstructive pulmonary disease |
| CSN5 | COP9 signalosome 5 |
| CTL | Cytotoxic T-lymphocyte |
| DC | Dendritic cell |
| DEN | Diethylnitrosoamine |
| DMF | Dimethyl fumarate |
| DSB | Double-strand break |
| EGFR | Epidermal growth factor receptor |
| HCC | Hepatocellular carcinoma |
| HNSCC | Head and neck squamous cell carcinoma |
| ICD | Immunogenic cell death |
| IKK | IκB kinase |
| IL-1 | Interleukin-1 |
| iNUB | Inhibitor of NEMO-Ubiquitin binding |
| IκB | Inhibitor of kappa B |
| KD | Kinase domain |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LPS | Lipopolysaccharide |
| LUBAC | Linear ubiquitin chain assembly complex |
| MALT | Mucosa-associated lymphoid tissue |
| MCL | Mantle cell lymphoma |
| MM | Multiple myeloma |
| mTORC | Mammalian target of rapamycin complex |
| NBD | NEMO-binding domain |
| NF-κB | nuclear factor- “kappa-light-chain-enhancer” of activated B-cells |
| NSCLC | Non-small-cell lung carcinoma |
| OV | Oncolytic virus |
| PD/PDL | Programmed death/PD-ligand 1 |
| POC | Proof-of-concept |
| RA | Rheumatoid arthritis |
| RHD | Rel homology domain |
| SCF | S phase kinase-associated protein 1 (SKP1)-cullin 1-F-box protein |
| SCID | Severe combined immunodeficient |
| SDD | Scaffold/dimerization domain |
| SH2 | Src Homology 2 |
| TAM | Tumour-associated macrophage |
| TCR | T-cell receptor |
| TLR | Toll-like receptor |
| TME | Tumour microenvironment |
| TNFα | Tumor necrosis factor-alpha |
| Treg | Regulatory T cells |
| UBC | Ubiquitin-conjugating enzyme |
| ULD | Ubiquitin-like domain |
References
- Napetschnig, J.; Wu, H. Molecular basis of NK-kappaB signaling. Annu. Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-canonical NK-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NK-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xia, Y.; Parker, A.S.; Verma, I.M. IKK biology. Immunol. Rev. 2012, 246, 239–253. [Google Scholar] [CrossRef] [PubMed]
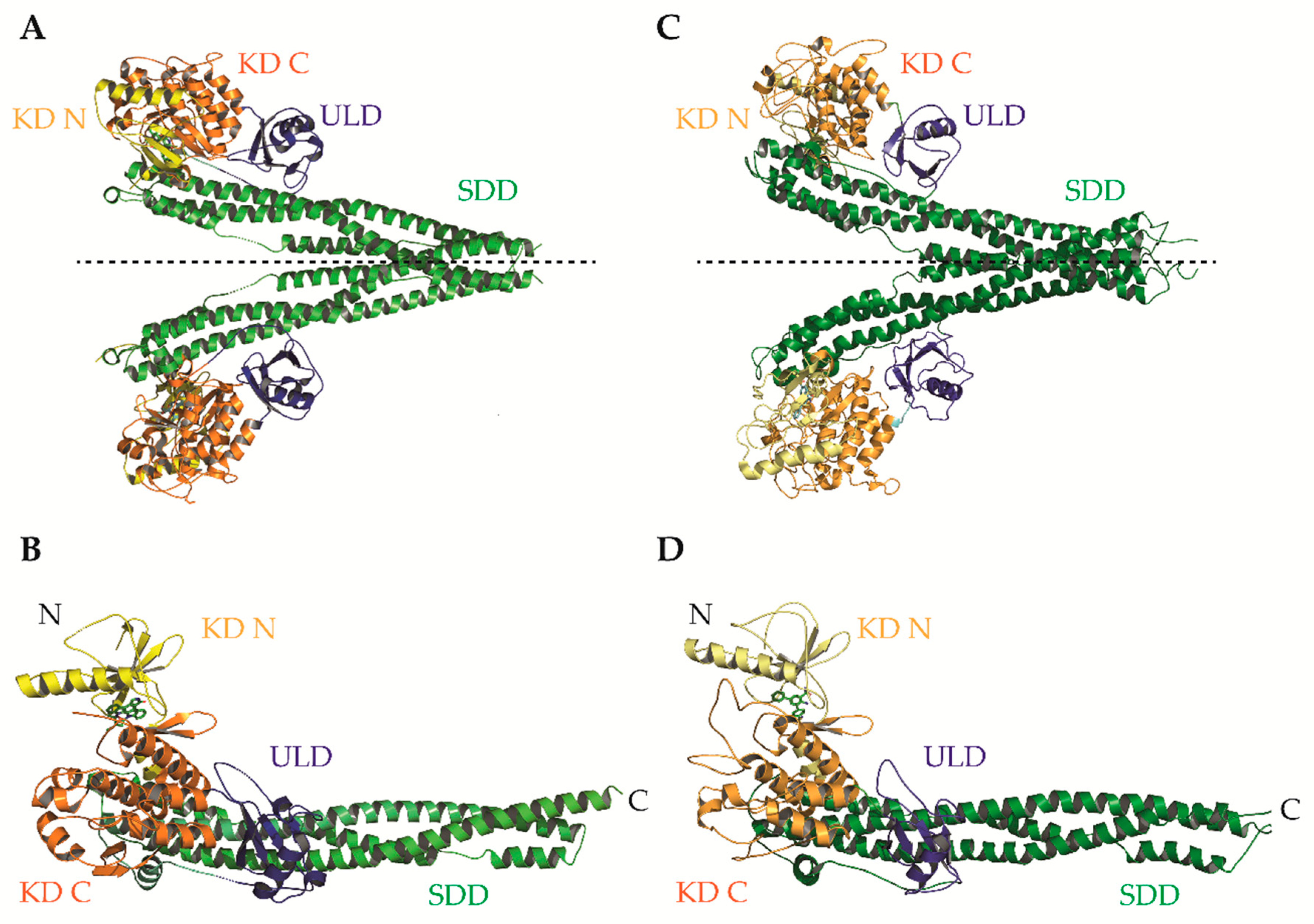
- Liu, S.; Misquitta, Y.R.; Olland, A.; Johnson, M.A.; Kelleher, K.S.; Kriz, R.; Lin, L.L.; Stahl, M.; Mosyak, L. Crystal structure of a human IkappaB kinase beta asymmetric dimer. J. Biol. Chem. 2013, 288, 22758–22767. [Google Scholar] [CrossRef] [PubMed]
- Polley, S.; Huang, D.B.; Hauenstein, A.V.; Fusco, A.J.; Zhong, X.; Vu, D.; Schrofelbauer, B.; Kim, Y.; Hoffmann, A.; Verma, I.M.; et al. A structural basis for IkappaB kinase 2 activation via oligomerization-dependent trans auto-phosphorylation. PLoS Biol. 2013, 11, e1001581. [Google Scholar] [CrossRef] [PubMed]
- Polley, S.; Passos, D.O.; Huang, D.B.; Mulero, M.C.; Mazumder, A.; Biswas, T.; Verma, I.M.; Lyumkis, D.; Ghosh, G. Structural basis for the activation of IKK1/alpha. Cell Rep. 2016, 17, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. IKK-1 and IKK-2: Cytokine-activated IkappaB kinases essential for NK-kappaB activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Delhase, M.; Hayakawa, M.; Chen, Y.; Karin, M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science 1999, 284, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Scholefield, J.; Henriques, R.; Savulescu, A.F.; Fontan, E.; Boucharlat, A.; Laplantine, E.; Smahi, A.; Israël, A.; Agou, F.; Mhlanga, M.M. Super-resolution microscopy reveals a preformed nemo lattice structure that is collapsed in incontinentia pigmenti. Nat. Commun. 2016, 7, 12629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Clark, K.; Lawrence, T.; Peggie, M.W.; Cohen, P. An unexpected twist to the activation of IKKβ: TAK1 primes IKKβ for activation by autophosphorylation. Biochem. J. 2014, 461, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, T.; Machleidt, T.; Alkalay, I.; Kronke, M.; Ben-Neriah, Y.; Baeuerle, P.A. Rapid proteolysis of I kappa B-alpha is necessary for activation of transcription factor NK-kappa B. Nature 1993, 365, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Gerstberger, S.; Carlson, L.; Franzoso, G.; Siebenlist, U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 1995, 267, 1485–1488. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hagler, J.; Palombella, V.J.; Melandri, F.; Scherer, D.; Ballard, D.; Maniatis, T. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995, 9, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Baldi, L.; Brown, K.; Franzoso, G.; Siebenlist, U. Critical role for lysines 21 and 22 in signal-induced, ubiquitin-mediated proteolysis of ib. J. Biol. Chem. 1996, 271, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFβ-TRCP–ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 2006, 25, 6717–6730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, F.; Smith, E.L.; Carmody, R.J. The regulation of NK-kappaB subunits by phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NK-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-kappaB addiction and its role in cancer: ‘One size does not fit all’. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-kappaB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Chariot, A. The NK-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 2009, 19, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Lee, D.F.; Xia, W.; Golfman, L.S.; Ou-Yang, F.; Yang, J.Y.; Zou, Y.; Bao, S.; Hanada, N.; Saso, H.; et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 2004, 117, 225–237. [Google Scholar] [CrossRef]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mtor pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.W.; Chu, W.; Hu, Y.; Delhase, M.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J. Exp. Med. 1999, 189, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Van Antwerp, D.; Mercurio, F.; Lee, K.F.; Verma, I.M. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science 1999, 284, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y. Abnormal morphogenesis but intact IKK activation in mice lacking the IKK subunit of ib kinase. Science 1999, 284, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Li, Z.-W.; Baud, V.; Karin, M. IKKβ is essential for protecting T cells from TNFα-induced apoptosis. Immunity 2001, 14, 217–230. [Google Scholar] [CrossRef]
- Kaisho, T.; Takeda, K.; Tsujimura, T.; Kawai, T.; Nomura, F.; Terada, N.; Akira, S. IkappaB kinase alpha is essential for mature B cell development and function. J. Exp. Med. 2001, 193, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, E.; Droin, N.M.; Delhase, M.; Haas, E.; Cao, Y.; Makris, C.; Li, Z.W.; Karin, M.; Ware, C.F.; Green, D.R. The lymphotoxin-beta receptor induces different patterns of gene expression via two NK-kappaB pathways. Immunity 2002, 17, 525–535. [Google Scholar] [CrossRef]
- Bonizzi, G.; Bebien, M.; Otero, D.C.; Johnson-Vroom, K.E.; Cao, Y.; Vu, D.; Jegga, A.G.; Aronow, B.J.; Ghosh, G.; Rickert, R.C.; et al. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:P52 dimers. EMBO J. 2004, 23, 4202–4210. [Google Scholar] [CrossRef] [PubMed]
- Anthony, N.G.; Baiget, J.; Berretta, G.; Boyd, M.; Breen, D.; Edwards, J.; Gamble, C.; Gray, A.I.; Harvey, A.L.; Hatziieremia, S.; et al. Inhibitory kappa B kinase alpha (IKKalpha) inhibitors that recapitulate their selectivity in cells against isoform-related biomarkers. J. Med. Chem. 2017, 60, 7043–7066. [Google Scholar] [CrossRef] [PubMed]
- Kobori, M.; Yang, Z.; Gong, D.; Heissmeyer, V.; Zhu, H.; Jung, Y.K.; Gakidis, M.A.; Rao, A.; Sekine, T.; Ikegami, F.; et al. Wedelolactone suppresses LPS-induced caspase-11 expression by directly inhibiting the IKK complex. Cell Death Differ. 2004, 11, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Nam, N.H. Naturally occurring NK-kappaB inhibitors. Mini Rev. Med. Chem. 2006, 6, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Llona-Minguez, S.; Baiget, J.; Mackay, S.P. Small-molecule inhibitors of IkappaB kinase (IKK) and IKK-related kinases. Pharm. Pat. Anal. 2013, 2, 481–498. [Google Scholar] [CrossRef] [PubMed]
- Arepalli, S.K.; Choi, M.; Jung, J.K.; Lee, H. Novel NK-kappaB inhibitors: A patent review (2011–2014). Expert Opin. Ther. Pat. 2015, 25, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Peggie, M.; Plater, L.; Sorcek, R.J.; Young, E.R.; Madwed, J.B.; Hough, J.; McIver, E.G.; Cohen, P. Novel cross-talk within the IKK family controls innate immunity. Biochem. J. 2011, 434, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, K.; Sasseville, V.G.; Wen, D.; Bielecki, A.; Yang, H.; Simpson, C.; Grant, E.; Hepperle, M.; Harriman, G.; Jaffee, B.; et al. Rapid TNFR1-dependent lymphocyte depletion in vivo with a selective chemical inhibitor of IKKbeta. Blood 2006, 107, 4266–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hideshima, T.; Neri, P.; Tassone, P.; Yasui, H.; Ishitsuka, K.; Raje, N.; Chauhan, D.; Podar, K.; Mitsiades, C.; Dang, L.; et al. MLN120B, a novel IkappaB kinase beta inhibitor, blocks multiple myeloma cell growth in vitro and in vivo. Clin. Cancer Res. 2006, 12, 5887–5894. [Google Scholar] [CrossRef] [PubMed]
- Schopf, L.; Savinainen, A.; Anderson, K.; Kujawa, J.; DuPont, M.; Silva, M.; Siebert, E.; Chandra, S.; Morgan, J.; Gangurde, P.; et al. IKKbeta inhibition protects against bone and cartilage destruction in a rat model of rheumatoid arthritis. Arthritis Rheum. 2006, 54, 3163–3173. [Google Scholar] [CrossRef] [PubMed]
- Mbalaviele, G.; Sommers, C.D.; Bonar, S.L.; Mathialagan, S.; Schindler, J.F.; Guzova, J.A.; Shaffer, A.F.; Melton, M.A.; Christine, L.J.; Tripp, C.S.; et al. A novel, highly selective, tight binding IkappaB kinase-2 (IKK-2) inhibitor: A tool to correlate IKK-2 activity to the fate and functions of the components of the nuclear factor-kappaB pathway in arthritis-relevant cells and animal models. J. Pharmacol. Exp. Ther. 2009, 329, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Chiang, P.C.; Kishore, N.N.; Thompson, D.C. Combined use of pharmacokinetic modeling and a steady-state delivery approach allows early assessment of IkappaB kinase-2 (IKK-2) target safety and efficacy. J. Pharm. Sci. 2010, 99, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Sommers, C.D.; Thompson, J.M.; Guzova, J.A.; Bonar, S.L.; Rader, R.K.; Mathialagan, S.; Venkatraman, N.; Holway, V.W.; Kahn, L.E.; Hu, G.; et al. Novel tight-binding inhibitory factor-kappaB kinase (IKK-2) inhibitors demonstrate target-specific anti-inflammatory activities in cellular assays and following oral and local delivery in an in vivo model of airway inflammation. J. Pharmacol. Exp. Ther. 2009, 330, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Rajendrasozhan, S.; Hwang, J.W.; Yao, H.; Kishore, N.; Rahman, I. Anti-inflammatory effect of a selective IkappaB kinase-beta inhibitor in rat lung in response to LPS and cigarette smoke. Pulm. Pharmacol. Ther. 2010, 23, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Podolin, P.L.; Callahan, J.F.; Bolognese, B.J.; Li, Y.H.; Carlson, K.; Davis, T.G.; Mellor, G.W.; Evans, C.; Roshak, A.K. Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IkappaB kinase 2, TPCA-1 (2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and antigen-induced T cell proliferation. J. Pharmacol. Exp. Ther. 2005, 312, 373–381. [Google Scholar] [PubMed]
- Sachse, F.; Becker, K.; Basel, T.J.; Weiss, D.; Rudack, C. IKK-2 inhibitor TPCA-1 represses nasal epithelial inflammation in vitro. Rhinology 2011, 49, 168–173. [Google Scholar] [PubMed]
- Du, Z.; Whitt, M.A.; Baumann, J.; Garner, J.M.; Morton, C.L.; Davidoff, A.M.; Pfeffer, L.M. Inhibition of type I interferon-mediated antiviral action in human glioma cells by the IKK inhibitors BMS-345541 and TPCA-1. J. Interferon Cytokine Res. 2012, 32, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Nan, J.; Du, Y.; Chen, X.; Bai, Q.; Wang, Y.; Zhang, X.; Zhu, N.; Zhang, J.; Hou, J.; Wang, Q.; et al. TPCA-1 is a direct dual inhibitor of STAT3 and NK-kappaB and regresses mutant EGFR-associated human non-small cell lung cancers. Mol. Cancer Ther. 2014, 13, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Birrell, M.A.; Wong, S.; Hardaker, E.L.; Catley, M.C.; McCluskie, K.; Collins, M.; Haj-Yahia, S.; Belvisi, M.G. IkappaB kinase-2-independent and -dependent inflammation in airway disease models: Relevance of IKK-2 inhibition to the clinic. Mol. Pharmacol. 2006, 69, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Lu, Q.; Gaddipati, S.; Kasetti, R.B.; Wang, W.; Pasparakis, M.; Kaplan, H.J.; Li, Q. IKK2 inhibition attenuates laser-induced choroidal neovascularization. PLoS ONE 2014, 9, e87530. [Google Scholar] [CrossRef] [PubMed]
- Kishore, N.; Sommers, C.; Mathialagan, S.; Guzova, J.; Yao, M.; Hauser, S.; Huynh, K.; Bonar, S.; Mielke, C.; Albee, L.; et al. A selective IKK-2 inhibitor blocks NK-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J. Biol. Chem. 2003, 278, 32861–32871. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Shi, Z.; Liu, Y.; Stack, M.S. Inhibitors of NK-kappaB reverse cellular invasion and target gene upregulation in an experimental model of aggressive oral squamous cell carcinoma. Oral Oncol. 2014, 50, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, H.; Chim, S.M.; Zhou, L.; Zhao, J.; Feng, H.; Wei, Q.; Wang, Q.; Zheng, M.H.; Tan, R.X.; et al. SC-514, a selective inhibitor of IKKbeta attenuates rankl-induced osteoclastogenesis and NK-kappaB activation. Biochem. Pharmacol. 2013, 86, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Negi, G.; Sharma, S.S. Inhibition of IkappaB kinase (IKK) protects against peripheral nerve dysfunction of experimental diabetes. Mol. Neurobiol. 2015, 51, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Lipstein, M.; Rodriguez, R.; Serrano, X.O.; McIntosh, C.; Tsai, W.Y.; Wasmuth, A.S.; Jaken, S.; O’Connor, O.A. The novel IKK2 inhibitor ly2409881 potently synergizes with histone deacetylase inhibitors in preclinical models of lymphoma through the downregulation of NK-kappaB. Clin. Cancer Res. 2015, 21, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.C.; Dang, L.C.; Soucy, F.; Grenier, L.; Mazdiyasni, H.; Hottelet, M.; Parent, L.; Pien, C.; Palombella, V.; Adams, J. Novel IKK inhibitors: Beta-carbolines. Bioorg. Med. Chem. Lett. 2003, 13, 2419–2422. [Google Scholar] [CrossRef]
- Vodanovic-Jankovic, S.; Hari, P.; Jacobs, P.; Komorowski, R.; Drobyski, W.R. NF-kappaB as a target for the prevention of graft-versus-host disease: Comparative efficacy of bortezomib and PS-1145. Blood 2006, 107, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Richardson, P.; Mitsiades, C.; Mitsiades, N.; Hayashi, T.; Munshi, N.; Dang, L.; Castro, A.; Palombella, V.; et al. NK-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002, 277, 16639–16647. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.T.; Davis, R.E.; Pierce, J.; Hepperle, M.; Xu, Y.; Hottelet, M.; Nong, Y.; Wen, D.; Adams, J.; Dang, L.; et al. Small molecule inhibitors of iκb kinase are selectively toxic for subgroups of diffuse large B-cell lymphoma defined by gene expression profiling. Clin. Cancer Res. 2005, 11, 28–40. [Google Scholar] [PubMed]
- Choi, S.I.; Lee, S.Y.; Jung, W.J.; Lee, S.H.; Lee, E.J.; Min, K.H.; Hur, G.Y.; Lee, S.H.; Lee, S.Y.; Kim, J.H.; et al. The effect of an IkappaB-kinase-beta (IKKbeta) inhibitor on tobacco smoke-induced pulmonary inflammation. Exp. Lung Res. 2016, 42, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Ziegelbauer, K.; Gantner, F.; Lukacs, N.W.; Berlin, A.; Fuchikami, K.; Niki, T.; Sakai, K.; Inbe, H.; Takeshita, K.; Ishimori, M.; et al. A selective novel low-molecular-weight inhibitor of IkappaB kinase-beta (IKK-beta) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br. J. Pharmacol. 2005, 145, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Bassères, D.S.; Ebbs, A.; Cogswell, P.C.; Baldwin, A.S. IKK is a therapeutic target in kras-induced lung cancer with disrupted p53 activity. Genes Cancer 2014, 5, 41–55. [Google Scholar] [PubMed]
- Moss, N.C.; Stansfield, W.E.; Willis, M.S.; Tang, R.H.; Selzman, C.H. IKKbeta inhibition attenuates myocardial injury and dysfunction following acute ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2248–H2253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Qian, L.; Flood, P.M.; Shi, J.S.; Hong, J.S.; Gao, H.M. Inhibition of IkappaB kinase-beta protects dopamine neurons against lipopolysaccharide-induced neurotoxicity. J. Pharmacol. Exp. Ther. 2010, 333, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, S.B.; Carlson, C.B.; Riddle, S.M.; Zhao, J.; Vogel, K.W.; Nichols, R.J.; Bi, K. Screening for novel LRRK2 inhibitors using a high-throughput tr-fret cellular assay for LRRK2 SER935 phosphorylation. PLoS ONE 2012, 7, e43580. [Google Scholar] [CrossRef] [PubMed]
- Waelchli, R.; Bollbuck, B.; Bruns, C.; Buhl, T.; Eder, J.; Feifel, R.; Hersperger, R.; Janser, P.; Revesz, L.; Zerwes, H.G.; et al. Design and preparation of 2-benzamido-pyrimidines as inhibitors of IKK. Bioorg. Med. Chem. Lett. 2006, 16, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Sordi, R.; Chiazza, F.; Johnson, F.L.; Patel, N.S.; Brohi, K.; Collino, M.; Thiemermann, C. Inhibition of IkappaB kinase attenuates the organ injury and dysfunction associated with hemorrhagic shock. Mol. Med. 2015, 21, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Coldewey, S.M.; Rogazzo, M.; Collino, M.; Patel, N.S.; Thiemermann, C. Inhibition of IkappaB kinase reduces the multiple organ dysfunction caused by sepsis in the mouse. Dis. Models Mech. 2013, 6, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.S.; Tao, W.; Miao, Q.B.; Zhu, Y.B.; Yang, Y.F. Improvement of ventilation-induced lung injury in a rodent model by inhibition of inhibitory kappaB kinase. J. Trauma Acute Care Surg. 2014, 76, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.L.; Patel, N.S.A.; Purvis, G.S.D.; Chiazza, F.; Chen, J.; Sordi, R.; Hache, G.; Merezhko, V.V.; Collino, M.; Yaqoob, M.M.; et al. Inhibition of IkappaB kinase at 24 hours after acute kidney injury improves recovery of renal function and attenuates fibrosis. J. Am. Heart Assoc. 2017, 6, e005092. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Konno, M.; Muto, S.; Kambe, N.; Morii, E.; Nakahata, T.; Itai, A.; Matsuda, H. A novel NK-kappaB inhibitor, IMD-0354, suppresses neoplastic proliferation of human mast cells with constitutively activated c-kit receptors. Blood 2005, 105, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Muto, S.; Konno, M.; Itai, A.; Matsuda, H. A new IkappaB kinase beta inhibitor prevents human breast cancer progression through negative regulation of cell cycle transition. Cancer Res. 2006, 66, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Kanduri, M.; Tobin, G.; Aleskog, A.; Nilsson, K.; Rosenquist, R. The novel NK-kappaB inhibitor IMD-0354 induces apoptosis in chronic lymphocytic leukemia. Blood Cancer J. 2011, 1, e12. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, T.; Saito, Y.; Saitoh, T.; Dewan, M.Z.; Shioya, A.; Kobayashi, M.; Kawachi, H.; Muto, S.; Itai, A.; Uota, S.; et al. Inhibition of IkappaB kinase beta restrains oncogenic proliferation of pancreatic cancer cells. J. Med. Dent. Sci. 2008, 55, 49–59. [Google Scholar] [PubMed]
- Uota, S.; Zahidunnabi Dewan, M.; Saitoh, Y.; Muto, S.; Itai, A.; Utsunomiya, A.; Watanabe, T.; Yamamoto, N.; Yamaoka, S. An IkappaB kinase 2 inhibitor IMD-0354 suppresses the survival of adult T-cell leukemia cells. Cancer Sci. 2012, 103, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Shimada, M.; Sakakibara, S.; Yoshino, T.; Masuda, T.; Shintani, T.; Sato, H.; Koriyama, Y.; Fukushima, K.; Nunami, N.; et al. Synthesis and structure-activity relationships of novel IKK-beta inhibitors. Part 3: Orally active anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2004, 14, 4019–4022. [Google Scholar] [CrossRef] [PubMed]
- Sanda, T.; Iida, S.; Ogura, H.; Asamitsu, K.; Murata, T.; Bacon, K.B.; Ueda, R.; Okamoto, T. Growth inhibition of multiple myeloma cells by a novel IkappaB kinase inhibitor. Clin. Cancer Res. 2005, 11, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Sanda, T.; Asamitsu, K.; Ogura, H.; Iida, S.; Utsunomiya, A.; Ueda, R.; Okamoto, T. Induction of cell death in adult T-cell leukemia cells by a novel IkappaB kinase inhibitor. Leukemia 2006, 20, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Victoriano, A.F.; Asamitsu, K.; Hibi, Y.; Imai, K.; Barzaga, N.G.; Okamoto, T. Inhibition of human immunodeficiency virus type 1 replication in latently infected cells by a novel IkappaB kinase inhibitor. Antimicrob. Agents Chemother. 2006, 50, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NK-kappa B-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, K.W.; Shuster, D.J.; Gillooly, K.M.; Dambach, D.M.; Pattoli, M.A.; Lu, P.; Zhou, X.D.; Qiu, Y.; Zusi, F.C.; Burke, J.R. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 2003, 48, 2652–2659. [Google Scholar] [CrossRef] [PubMed]
- MacMaster, J.F.; Dambach, D.M.; Lee, D.B.; Berry, K.K.; Qiu, Y.; Zusi, F.C.; Burke, J.R. An inhibitor of IkappaB kinase, BMS-345541, blocks endothelial cell adhesion molecule expression and reduces the severity of dextran sulfate sodium-induced colitis in mice. Inflamm. Res. 2003, 52, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Townsend, R.M.; Postelnek, J.; Susulic, V.; McIntyre, K.W.; Shuster, D.J.; Qiu, Y.; Zusi, F.C.; Burke, J.R. A highly selective inhibitor of IkappaB kinase, BMS-345541, augments graft survival mediated by suboptimal immunosuppression in a murine model of cardiac graft rejection. Transplantation 2004, 77, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Buontempo, F.; Chiarini, F.; Bressanin, D.; Tabellini, G.; Melchionda, F.; Pession, A.; Fini, M.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Activity of the selective IkappaB kinase inhibitor BMS-345541 against T-cell acute lymphoblastic leukemia: Involvement of FOXO3a. Cell Cycle 2012, 11, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- Ping, H.; Yang, F.; Wang, M.; Niu, Y.; Xing, N. IKK inhibitor suppresses epithelial-mesenchymal transition and induces cell death in prostate cancer. Oncol. Rep. 2016, 36, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.S.; Oberley, C.; Hooper, C.P.; Grindle, K.; Wuerzberger-Davis, S.; Wolff, J.; McCool, K.; Rui, L.; Miyamoto, S. Withaferin a disrupts ubiquitin-based nemo reorganization induced by canonical NK-kappaB signaling. Exp. Cell Res. 2015, 331, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Choi, B.Y. Withaferin-A—A natural anticancer agent with pleitropic mechanisms of action. Int. J. Mol. Sci. 2016, 17, 290. [Google Scholar] [CrossRef] [PubMed]
- Kaileh, M.; Vanden Berghe, W.; Heyerick, A.; Horion, J.; Piette, J.; Libert, C.; De Keukeleire, D.; Essawi, T.; Haegeman, G. Withaferin a strongly elicits IkappaB kinase beta hyperphosphorylation concomitant with potent inhibition of its kinase activity. J. Biol. Chem. 2007, 282, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Heyninck, K.; Lahtela-Kakkonen, M.; Van der Veken, P.; Haegeman, G.; Vanden Berghe, W. Withaferin a inhibits NK-kappaB activation by targeting cysteine 179 in IKKbeta. Biochem. Pharmacol. 2014, 91, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, W.; Sabbe, L.; Kaileh, M.; Haegeman, G.; Heyninck, K. Molecular insight in the multifunctional activities of withaferin a. Biochem. Pharmacol. 2012, 84, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Roh, E.; Lee, H.Y.; Lee, I.J.; Ahn, B.; Jung, S.H.; Lee, H.; Han, S.B.; Kim, Y. Benzoxathiole derivative blocks lipopolysaccharide-induced nuclear factor-kappaB activation and nuclear factor-kappaB-regulated gene transcription through inactivating inhibitory kappaB kinase beta. Mol. Pharmacol. 2008, 73, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Li, C.; Wang, X.; Dian, L.; Zhang, X.; Li, L.; Chen, S.; Cao, R.; Li, L.; Huang, N.; et al. Ainsliadimer a selectively inhibits IKKα/β by covalently binding a conserved cysteine. Nat. Commun. 2015, 6, 6522. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.A.; Collins, I. Measuring and interpreting the selectivity of protein kinase inhibitors. J. Chem. Biol. 2009, 2, 131–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, J.W.; Schoenleber, R.; Jesmok, G.; Best, J.; Moore, S.A.; Collins, T.; Gerritsen, M.E. Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effectsin vivo. J.Biol. Chem. 1997, 272, 21096–21103. [Google Scholar] [CrossRef] [PubMed]
- Strickson, S.; Campbell, D.G.; Emmerich, C.H.; Knebel, A.; Plater, L.; Ritorto, M.S.; Shpiro, N.; Cohen, P. The anti-inflammatory drug bay 11-7082 suppresses the myd88-dependent signalling network by targeting the ubiquitin system. Biochem. J. 2013, 451, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gray, N.S. Snapshot: Kinase inhibitors II. Mol. Cell 2015, 58, 710. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Sung, B.; Kunnumakkara, A.B.; Sethi, G.; Chaturvedi, M.M.; Aggarwal, B.B. Berberine modifies cysteine 179 of IkappaBalpha kinase, suppresses nuclear factor-kappaB-regulated antiapoptotic gene products, and potentiates apoptosis. Cancer Res. 2008, 68, 5370–5379. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NK-kappaB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Byun, M.S.; Choi, J.; Jue, D.M. Cysteine-179 of IkappaB kinase beta plays a critical role in enzyme activation by promoting phosphorylation of activation loop serines. Exp. Mol. Med. 2006, 38, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Bernier, M.; Kwon, Y.K.; Pandey, S.K.; Zhu, T.N.; Zhao, R.J.; Maciuk, A.; He, H.J.; Decabo, R.; Kole, S. Binding of manumycin a inhibits IkappaB kinase beta activity. J. Biol. Chem. 2006, 281, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.C.; Bardhan, S.; Li, C.; Pace, E.A.; Porco, J.A., Jr.; Gilmore, T.D. Jesterone dimer, a synthetic derivative of the fungal metabolite jesterone, blocks activation of transcription factor nuclear factor kappaB by inhibiting the inhibitor of kappaB kinase. Mol. Pharmacol. 2003, 64, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.S.; Matthews, D.E.; et al. Dynamic redox control of NK-kappaB through glutaredoxin-regulated s-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef] [PubMed]
- May, M.J.; D’Acquisto, F.; Madge, L.A.; Glockner, J.; Pober, J.S.; Ghosh, S. Selective inhibition of NK-kappaB activation by a peptide that blocks the interaction of nemo with the IkappaB kinase complex. Science 2000, 289, 1550–1554. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, L.; Ai, G.; Spitale, R.C.; Bhat, G.J. Molecular targets of aspirin and cancer prevention. Br. J. Cancer 2014, 111, 61. [Google Scholar] [CrossRef] [PubMed]
- Kopp, E.; Ghosh, S. Inhibition of NK-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef] [PubMed]
- McDade, T.P.; Perugini, R.A.; Vittimberga, F.J., Jr.; Carrigan, R.C.; Callery, M.P. Salicylates inhibit NK-kappaB activation and enhance tnf-alpha-induced apoptosis in human pancreatic cancer cells. J. Surg. Res. 1999, 83, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Yin, M.J.; Lin, K.M.; Gaynor, R.B. Sulindac inhibits activation of the NK-kappaB pathway. J. Biol. Chem. 1999, 274, 27307–27314. [Google Scholar] [CrossRef] [PubMed]
- Kinase Profiling Inhibitor Database, MRC Protein Phosphorylation Unit at the University of Dundee. Available online: http://www.kinase-screen.mrc.ac.uk/screening-compounds/349388?order=field_results_inhibition&sort=asc (accessed on 1 August 2018).
- Schwenger, P.; Alpert, D.; Skolnik, E.Y.; Vilcek, J. Activation of p38 mitogen-activated protein kinase by sodium salicylate leads to inhibition of tumor necrosis factor-induced IkappaB alpha phosphorylation and degradation. Mol. Cell. Biol. 1998, 18, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Alpert, D.; Vilček, J. Inhibition of iκb kinase activity by sodium salicylate in vitro does not reflect its inhibitory mechanism in intact cells. J. Biol. Chem. 2000, 275, 10925–10929. [Google Scholar] [CrossRef] [PubMed]
- Stark, L.A.; Din, F.V.; Zwacka, R.M.; Dunlop, M.G. Aspirin-induced activation of the NK-kappaB signaling pathway: A novel mechanism for aspirin-mediated apoptosis in colon cancer cells. FASEB J. 2001, 15, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Stark, L.A.; Reid, K.; Sansom, O.J.; Din, F.V.; Guichard, S.; Mayer, I.; Jodrell, D.I.; Clarke, A.R.; Dunlop, M.G. Aspirin activates the NK-kappaB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis 2007, 28, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Loveridge, C.J.; MacDonald, A.D.; Thoms, H.C.; Dunlop, M.G.; Stark, L.A. The proapoptotic effects of sulindac, sulindac sulfone and indomethacin are mediated by nucleolar translocation of the RelA(p65) subunit of NK-kappaB. Oncogene 2008, 27, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Mladenova, D.; Pangon, L.; Currey, N.; Ng, I.; Musgrove, E.A.; Grey, S.T.; Kohonen-Corish, M.R. Sulindac activates NK-kappaB signaling in colon cancer cells. Cell Commun. Signal. 2013, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.B.; Yang, X.; Clark, R.; Choi, J.; Baek, S.J.; Lee, S.H. A mechanistic study of the proapoptotic effect of tolfenamic acid: Involvement of NK-kappaB activation. Carcinogenesis 2013, 34, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Stark, L.A. Aspirin prevention of colorectal cancer: Focus on NK-kappaB signalling and the nucleolus. Biomedicines 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, L.; Beyaert, R. Receptor proximal kinases in NK-kappaB signaling as potential therapeutic targets in cancer and inflammation. Biochem. Pharmacol. 2014, 92, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, Y.; Sawada, T.; Nagatani, K.; Komagata, Y.; Inoue, T.; Muto, S.; Itai, A.; Yamamoto, K. Effect of nuclear factor-kappaB inhibition on rheumatoid fibroblast-like synoviocytes and collagen induced arthritis. J. Rheumatol. 2005, 32, 1440–1447. [Google Scholar] [PubMed]
- Onai, Y.; Suzuki, J.; Kakuta, T.; Maejima, Y.; Haraguchi, G.; Fukasawa, H.; Muto, S.; Itai, A.; Isobe, M. Inhibition of IkappaB phosphorylation in cardiomyocytes attenuates myocardial ischemia/reperfusion injury. Cardiovasc. Res. 2004, 63, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Pippione, A.C.; Federico, A.; Ducime, A.; Sainas, S.; Boschi, D.; Barge, A.; Lupino, E.; Piccinini, M.; Kubbutat, M.; Contreras, J.-M.; et al. 4-Hydroxy-n-[3,5-bis(trifluoromethyl)phenyl]-1,2,5-thiadiazole-3-carboxamide: A novel inhibitor of the canonical NF-κB cascade. MedChemComm 2017, 8, 1850–1855. [Google Scholar] [CrossRef] [PubMed]
- Tegeder, I.; Niederberger, E.; Schmidt, R.; Kunz, S.; Guhring, H.; Ritzeler, O.; Michaelis, M.; Geisslinger, G. Specific inhibition of IkappaB kinase reduces hyperalgesia in inflammatory and neuropathic pain models in rats. J. Neurosci. 2004, 24, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Grothe, K.; Flechsenhar, K.; Paehler, T.; Ritzeler, O.; Beninga, J.; Saas, J.; Herrmann, M.; Rudolphi, K. IkappaB kinase inhibition as a potential treatment of osteoarthritis—Results of a clinical proof-of-concept study. Osteoarthr. Cartil. 2017, 25, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Palanki, M.S.S.; Gayo-Fung, L.M.; Shevlin, G.I.; Erdman, P.; Sato, M.; Goldman, M.; Ransone, L.J.; Spooner, C. Structure–activity relationship studies of ethyl 2-[(3-methyl-2,5-dioxo(3-pyrrolinyl))amino]-4-(trifluoromethyl)pyrimidine-5-carboxylate: An inhibitor of ap-1 and NF-κB mediated gene expression. Bioorg. Med. Chem. Lett. 2002, 12, 2573–2577. [Google Scholar] [CrossRef]
- Frelin, C.; Imbert, V.; Griessinger, E.; Loubat, A.; Dreano, M.; Peyron, J.F. AS602868, a pharmacological inhibitor of IKK2, reveals the apoptotic potential of tnf-alpha in jurkat leukemic cells. Oncogene 2003, 22, 8187–8194. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liang, H.; Meng, H.; Deng, X.; Zhang, X.; Lai, L. A novel allosteric inhibitor that prevents IKKβ activation. MedChemComm 2018, 9, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The Pymol Molecular Graphics System, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Schrodinger, LLC. The Jymol Molecular Graphics Development Component, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Adli, M.; Merkhofer, E.; Cogswell, P.; Baldwin, A.S. IKKalpha and IKKbeta each function to regulate NK-kappaB activation in the tnf-induced/canonical pathway. PLoS ONE 2010, 5, e9428. [Google Scholar] [CrossRef] [PubMed]
- Nottingham, L.K.; Yan, C.H.; Yang, X.; Si, H.; Coupar, J.; Bian, Y.; Cheng, T.F.; Allen, C.; Arun, P.; Gius, D.; et al. Aberrant IKKalpha and IKKbeta cooperatively activate NK-kappaB and induce EGFR/ap1 signaling to promote survival and migration of head and neck cancer. Oncogene 2014, 33, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Slotta, C.; Storm, J.; Pfisterer, N.; Henkel, E.; Kleinwachter, S.; Pieper, M.; Ruiz-Perera, L.M.; Greiner, J.F.W.; Kaltschmidt, B.; Kaltschmidt, C. IKK1/2 protect human cells from tnf-mediated ripk1-dependent apoptosis in an NK-kappaB-independent manner. Biochim. Biophys. Acta 2018, 1865, 1025–1033. [Google Scholar] [PubMed]
- Lam, L.T.; Davis, R.E.; Ngo, V.N.; Lenz, G.; Wright, G.; Xu, W.; Zhao, H.; Yu, X.; Dang, L.; Staudt, L.M. Compensatory IKKalpha activation of classical NK-kappaB signaling during IKKbeta inhibition identified by an RNA interference sensitization screen. Proc. Natl. Acad. Sci. USA 2008, 105, 20798–20803. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhang, M.; Sun, S.C. Beta-TRCP binding and processing of NK-kappaB2/p100 involve its phosphorylation at serines 866 and 870. Cell. Signal. 2006, 18, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the rela component of NK-kappa B. Nature 1995, 376, 167–170. [Google Scholar] [PubMed]
- Tanaka, M.; Fuentes, M.E.; Yamaguchi, K.; Durnin, M.H.; Dalrymple, S.A.; Hardy, K.L.; Goeddel, D.V. Embryonic lethality, liver degeneration, and impaired NK-kappa B activation in IKK-beta-deficient mice. Immunity 1999, 10, 421–429. [Google Scholar] [CrossRef]
- Chen, L.W.; Egan, L.; Li, Z.W.; Greten, F.R.; Kagnoff, M.F.; Karin, M. The two faces of IKK and NK-kappaB inhibition: Prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat. Med. 2003, 9, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Pannicke, U.; Baumann, B.; Fuchs, S.; Henneke, P.; Rensing-Ehl, A.; Rizzi, M.; Janda, A.; Hese, K.; Schlesier, M.; Holzmann, K.; et al. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N. Engl. J. Med. 2013, 369, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Burns, S.O.; Plagnol, V.; Gutierrez, B.M.; Al Zahrani, D.; Curtis, J.; Gaspar, M.; Hassan, A.; Jones, A.M.; Malone, M.; Rampling, D.; et al. Immunodeficiency and disseminated mycobacterial infection associated with homozygous nonsense mutation of IKKbeta. J. Allergy Clin. Immunol. 2014, 134, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Mousallem, T.; Yang, J.; Urban, T.J.; Wang, H.; Adeli, M.; Parrott, R.E.; Roberts, J.L.; Goldstein, D.B.; Buckley, R.H.; Zhong, X.P. A nonsense mutation in IKBKB causes combined immunodeficiency. Blood 2014, 124, 2046–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, C.; Jakobsen, M.A.; Larsen, M.J.; Muller, A.C.; Hansen, S.; Lillevang, S.T.; Fisker, N.; Barington, T. Immunodeficiency associated with a nonsense mutation of IKBKB. J. Clin. Immunol. 2014, 34, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NK-kappaB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M. Regulation of tissue homeostasis by NK-kappaB signalling: Implications for inflammatory diseases. Nat. Rev. Immunol. 2009, 9, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Baltimore, D. Constitutive NK-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev. 1995, 9, 2736–2746. [Google Scholar] [CrossRef] [PubMed]
- Klement, J.F.; Rice, N.R.; Car, B.D.; Abbondanzo, S.J.; Powers, G.D.; Bhatt, P.H.; Chen, C.H.; Rosen, C.A.; Stewart, C.L. IkappaBalpha deficiency results in a sustained NK-kappaB response and severe widespread dermatitis in mice. Mol. Cell. Biol. 1996, 16, 2341–2349. [Google Scholar] [CrossRef] [PubMed]
- Courtois, G.; Gilmore, T.D. Mutations in the NK-kappaB signaling pathway: Implications for human disease. Oncogene 2006, 25, 6831–6843. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Splittgerber, R.; Yull, F.E.; Kantrow, S.; Ayers, G.D.; Karin, M.; Richmond, A. Conditional ablation of IKKb inhibits melanoma tumor development in mice. J. Clin. Investig. 2010, 120, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/beta/NK-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. NF-kappaB: Tumor promoter or suppressor? Trends Cell Biol. 2004, 14, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.-C.; Enzler, T.; Seita, J.; Timmer, A.M.; Lee, C.-Y.; Lai, T.-Y.; Yu, G.-Y.; Lai, L.-C.; Temkin, V.; Sinzig, U.; et al. IL-1β-driven neutrophilia preserves antibacterial defense in the absence of the kinase IKKβ. Nat. Immunol. 2010, 12, 144. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- McLoed, A.G.; Sherrill, T.P.; Cheng, D.S.; Han, W.; Saxon, J.A.; Gleaves, L.A.; Wu, P.; Polosukhin, V.V.; Karin, M.; Yull, F.E.; et al. Neutrophil-derived IL-1beta impairs the efficacy of NK-kappaB inhibitors against lung cancer. Cell Rep. 2016, 16, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Pallangyo, C.K.; Ziegler, P.K.; Greten, F.R. IKKbeta acts as a tumor suppressor in cancer-associated fibroblasts during intestinal tumorigenesis. J. Exp. Med. 2015, 212, 2253–2266. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NK-kappaB-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Pasparakis, M.; Kollias, G. IKKbeta in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J. Exp. Med. 2015, 212, 2235–2251. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hawkins, O.E.; Barham, W.; Gilchuk, P.; Boothby, M.; Ayers, G.D.; Joyce, S.; Karin, M.; Yull, F.E.; Richmond, A. Myeloid IKKbeta promotes antitumor immunity by modulating CCL11 and the innate immune response. Cancer Res. 2014, 74, 7274–7284. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Finkin, S.; Yuan, D.; Stein, I.; Taniguchi, K.; Weber, A.; Unger, K.; Browning, J.L.; Goossens, N.; Nakagawa, S.; Gunasekaran, G.; et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol. 2015, 16, 1235–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor κb activity is required for survival of activated b cell–like diffuse large b cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef] [PubMed]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NK-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Staudt, L.M. Oncogenic activation of NK-kappaB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000109. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.W.; Emre, N.C.; Kohlhammer, H.; Dave, S.S.; Davis, R.E.; Carty, S.; Lam, L.T.; Shaffer, A.L.; Xiao, W.; et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 13520–13525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, X.; Wood, L.D.; Lindman, M.; Jones, S.; Buckhaults, P.; Polyak, K.; Sukumar, S.; Carter, H.; Kim, D.; Karchin, R.; et al. Somatic mutations in the notch, NF-KB, PIK3CA, and hedgehog pathways in human breast cancers. Genes Chromosomes Cancer 2012, 51, 480–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pflueger, D.; Terry, S.; Sboner, A.; Habegger, L.; Esgueva, R.; Lin, P.C.; Svensson, M.A.; Kitabayashi, N.; Moss, B.J.; MacDonald, T.Y.; et al. Discovery of non-ets gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res. 2011, 21, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Bredel, M.; Scholtens, D.M.; Yadav, A.K.; Alvarez, A.A.; Renfrow, J.J.; Chandler, J.P.; Yu, I.L.; Carro, M.S.; Dai, F.; Tagge, M.J.; et al. Nfkbia deletion in glioblastomas. N. Engl. J. Med. 2011, 364, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NK-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Basseres, D.S.; Ebbs, A.; Levantini, E.; Baldwin, A.S. Requirement of the NK-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010, 70, 3537–3546. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yeddula, N.; Leblanc, M.; Ke, E.; Zhang, Y.; Oldfield, E.; Shaw, R.J.; Verma, I.M. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat. Cell Biol. 2012, 14, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniluk, J.; Liu, Y.; Deng, D.; Chu, J.; Huang, H.; Gaiser, S.; Cruz-Monserrate, Z.; Wang, H.; Ji, B.; Logsdon, C.D. An NK-kappaB pathway-mediated positive feedback loop amplifies ras activity to pathological levels in mice. J. Clin. Investig. 2012, 122, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin beta(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108. [Google Scholar] [CrossRef] [PubMed]
- Marazioti, A.; Lilis, I.; Vreka, M.; Apostolopoulou, H.; Kalogeropoulou, A.; Giopanou, I.; Giotopoulou, G.A.; Krontira, A.C.; Iliopoulou, M.; Kanellakis, N.I.; et al. Myeloid-derived interleukin-1beta drives oncogenic kras-NK-kappaBeta addiction in malignant pleural effusion. Nat. Commun. 2018, 9, 672. [Google Scholar] [CrossRef] [PubMed]
- Vreka, M.; Lilis, I.; Papageorgopoulou, M.; Giotopoulou, G.A.; Lianou, M.; Giopanou, I.; Kanellakis, N.I.; Spella, M.; Agalioti, T.; Armenis, V.; et al. IkappaB kinase alpha is required for development and progression of kras-mutant lung adenocarcinoma. Cancer Res. 2018, 78, 2939–2951. [Google Scholar] [CrossRef] [PubMed]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Sethi, G. Targeting transcription factor NK-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta 2010, 1805, 167–180. [Google Scholar] [PubMed]
- Voboril, R.; Weberova-Voborilova, J. Constitutive NK-kappaB activity in colorectal cancer cells: Impact on radiation-induced NK-kappaB activity, radiosensitivity, and apoptosis. Neoplasma 2006, 53, 518–523. [Google Scholar] [PubMed]
- Ishida, K.; Nishizuka, S.S.; Chiba, T.; Ikeda, M.; Kume, K.; Endo, F.; Katagiri, H.; Matsuo, T.; Noda, H.; Iwaya, T.; et al. Molecular marker identification for relapse prediction in 5-fu-based adjuvant chemotherapy in gastric and colorectal cancers. PLoS ONE 2012, 7, e43236. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NK-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Middleton, J.; Kim, T.; Lagana, A.; Piovan, C.; Secchiero, P.; Nuovo, G.J.; Cui, R.; Joshi, P.; Romano, G.; et al. A set of NK-kappaB-regulated micrornas induces acquired trail resistance in lung cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E3355–E3364. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.; Lau, E.Y.; Ching, R.H.; Cheng, B.Y.; Ma, M.K.; Ng, I.O.; Lee, T.K. Nuclear factor kappa B-mediated CD47 up-regulation promotes sorafenib resistance and its blockade synergizes the effect of sorafenib in hepatocellular carcinoma in mice. Hepatology 2015, 62, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mani, A.M.; Wu, Z.H. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. J. Cancer Metastasis Treat. 2017, 3, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Al-Katib, A.; Arnold, A.A.; Aboukameel, A.; Sosin, A.; Smith, P.; Mohamed, A.N.; Beck, F.W.; Mohammad, R.M. I-kappa-kinase-2 (IKK-2) inhibition potentiates vincristine cytotoxicity in non-hodgkin’s lymphoma. Mol. Cancer 2010, 9, 228. [Google Scholar] [CrossRef] [PubMed]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. Fas and NK-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Chariot, A. EGFR and NK-kappaB: Partners in cancer. Trends Mol. Med. 2015, 21, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.T.; Wright, G.; Davis, R.E.; Lenz, G.; Farinha, P.; Dang, L.; Chan, J.W.; Rosenwald, A.; Gascoyne, R.D.; Staudt, L.M. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood 2008, 111, 3701–3713. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuser, C.; Gotot, J.; Piotrowski, E.C.; Philipp, M.S.; Courreges, C.J.F.; Otte, M.S.; Guo, L.; Schmid-Burgk, J.L.; Hornung, V.; Heine, A.; et al. Prolonged IKKbeta inhibition improves ongoing CTL antitumor responses by incapacitating regulatory T cells. Cell Rep. 2017, 21, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Barnes, S.E.; Wang, Y.; Chen, L.; Molinero, L.L.; Gajewski, T.F.; Evaristo, C.; Alegre, M.L. T cell-NK-kappaB activation is required for tumor control in vivo. J. Immunother. Cancer 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Pauken, K.E.; Eagar, T.N.; Obu, T.; Wu, J.; Tang, Q.; Azuma, M.; Krummel, M.F.; Bluestone, J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009, 10, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wu, D.; Li, L.; Chai, Y.; Huang, J. PD-L1 and survival in solid tumors: A meta-analysis. PLoS ONE 2015, 10, e0131403. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, K.; Gunatilake, D.; Gallagher, S.J.; Tiffen, J.; Rizos, H.; Hersey, P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NK-kappaB. PLoS ONE 2015, 10, e0123410. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-kappaB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. Tnfalpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017, 8, 2256. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Dey, M. Immune system, friend or foe of oncolytic virotherapy? Front. Oncol. 2017, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Ou, P.; Rousso, C.; Bergeron, A.; Krishnan, R.; Pikor, L.; Chen, A.; Keller, B.A.; Ilkow, C.; Bell, J.C.; et al. Dimethyl fumarate potentiates oncolytic virotherapy through NK-kappaB inhibition. Sci. Transl. Med. 2018, 10, eaao1613. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, M.; Shah, N.R.; Felt, S.A.; Grdzelishvili, V.Z. Breaking resistance of pancreatic cancer cells to an attenuated vesicular stomatitis virus through a novel activity of IKK inhibitor TPCA-1. Virology 2015, 485, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Wilson, J.; Burke, F.; Kulbe, H.; Li, N.F.; Pluddemann, A.; Charles, K.; Gordon, S.; Balkwill, F.R. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J. Immunol. 2006, 176, 5023–5032. [Google Scholar] [CrossRef] [PubMed]
- Sawa-Wejksza, K.; Kandefer-Szerszen, M. Tumor-associated macrophages as target for antitumor therapy. Arch. Immunol. Ther. Exp. 2018, 66, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NK-kappaB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. P50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [PubMed]
- Torroella-Kouri, M.; Ma, X.; Perry, G.; Ivanova, M.; Cejas, P.J.; Owen, J.L.; Iragavarapu-Charyulu, V.; Lopez, D.M. Diminished expression of transcription factors nuclear factor kappaB and CCAAT/enhancer binding protein underlies a novel tumor evasion mechanism affecting macrophages of mammary tumor-bearing mice. Cancer Res. 2005, 65, 10578–10584. [Google Scholar] [CrossRef] [PubMed]
- Connelly, L.; Barham, W.; Onishko, H.M.; Chen, L.; Sherrill, T.P.; Zabuawala, T.; Ostrowski, M.C.; Blackwell, T.S.; Yull, F.E. NF-kappaB activation within macrophages leads to an anti-tumor phenotype in a mammary tumor lung metastasis model. Breast Cancer Res. 2011, 13, R83. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.P.; Mantovani, A. Targeting myelomonocytic cells to revert inflammation-dependent cancer promotion. Cancer Res. 2005, 65, 9113–9116. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Vicari, A.P.; Sangaletti, S.; Trinchieri, G.; Colombo, M.P. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005, 65, 3437–3446. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Biswas, S.K.; Lawrence, T.; Sica, A.; Lewis, C.E. Regulation of macrophage function in tumors: The multifaceted role of NK-kappaB. Blood 2009, 113, 3139–3146. [Google Scholar] [CrossRef] [PubMed]
- Gaddipati, S.; Lu, Q.; Kasetti, R.B.; Miller, M.C.; Lu, Q.; Trent, J.O.; Kaplan, H.J.; Li, Q. IKK2 inhibition using TPCA-1-loaded PLGA microparticles attenuates laser-induced choroidal neovascularization and macrophage recruitment. PLoS ONE 2015, 10, e0121185. [Google Scholar] [CrossRef] [PubMed]
- Enzler, T.; Sano, Y.; Choo, M.K.; Cottam, H.B.; Karin, M.; Tsao, H.; Park, J.M. Cell-selective inhibition of NK-kappaB signaling improves therapeutic index in a melanoma chemotherapy model. Cancer Discov. 2011, 1, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tan, Y.; Meng, T.; Liu, X.; Zhu, Y.; Hong, Y.; Yang, X.; Yuan, H.; Huang, X.; Hu, F. Simultaneous targeting therapy for lung metastasis and breast tumor by blocking the NK-kappaB signaling pathway using celastrol-loaded micelles. Drug Deliv. 2018, 25, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NK-kappaB conundrum: Embracing complexity to achieve specificity. Biomedicines 2017, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Capece, D.; Begalli, F.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. NF-kappaB in the crosshairs: Rethinking an old riddle. Int. J. Biochem. Cell Biol. 2018, 95, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Noy, A.; de Vos, S.; Thieblemont, C.; Martin, P.; Flowers, C.R.; Morschhauser, F.; Collins, G.P.; Ma, S.; Coleman, M.; Peles, S.; et al. Targeting bruton tyrosine kinase with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood 2017, 129, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Ghia, P.; Polliack, A.; Tam, C.; Suri, D.; Clow, F.; Kraljevic, S.; James, D.F.; Kipps, T.J. Randomized, multicenter, open-label, phase III study of the btk inhibitor ibrutinib versus chlorambucil in patients 65 years or older with treatment-naive CLL/SLL (RESONATE-2, PCYC-1115-CA). J. Clin. Oncol. 2013, 31, TPS7130. [Google Scholar]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in previously treated waldenstrom’s macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Miklos, D.; Cutler, C.S.; Arora, M.; Waller, E.K.; Jagasia, M.; Pusic, I.; Flowers, M.E.; Logan, A.C.; Nakamura, R.; Blazar, B.R.; et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood 2017, 130, 2243–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demchenko, Y.N.; Glebov, O.K.; Zingone, A.; Keats, J.J.; Bergsagel, P.L.; Kuehl, W.M. Classical and/or alternative NK-kappaB pathway activation in multiple myeloma. Blood 2010, 115, 3541–3552. [Google Scholar] [CrossRef] [PubMed]
- Rothwarf, D.M.; Zandi, E.; Natoli, G.; Karin, M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 1998, 395, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Strickland, I.; Ghosh, S. Use of cell permeable NBD peptides for suppression of inflammation. Ann. Rheum. Dis. 2006, 65 (Suppl. 3), iii75–iii82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habineza Ndikuyeze, G.; Gaurnier-Hausser, A.; Patel, R.; Baldwin, A.S.; May, M.J.; Flood, P.; Krick, E.; Propert, K.J.; Mason, N.J. A phase i clinical trial of systemically delivered nemo binding domain peptide in dogs with spontaneous activated B-cell like diffuse large B-cell lymphoma. PLoS ONE 2014, 9, e95404. [Google Scholar] [CrossRef] [PubMed]
- Vincendeau, M.; Hadian, K.; Messias, A.C.; Brenke, J.K.; Halander, J.; Griesbach, R.; Greczmiel, U.; Bertossi, A.; Stehle, R.; Nagel, D.; et al. Inhibition of canonical NF-κB signaling by a small molecule targeting nemo-ubiquitin interaction. Sci. Rep. 2016, 6, 18934. [Google Scholar] [CrossRef] [PubMed]
- Colomer, C.; Marruecos, L.; Vert, A.; Bigas, A.; Espinosa, L. NF-kappaB members left home: NF-kappaB-independent roles in cancer. Biomedicines 2017, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Ebbs, A.; Pasparakis, M.; Van Dyke, T.; Basseres, D.S.; Baldwin, A.S. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IkappaB kinase alpha (IKKalpha). J. Biol. Chem. 2014, 289, 25227–25240. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Antonia, R.J.; Baldwin, A.S. PI3K/Akt promotes feedforward mTORC2 activation through IKKalpha. Oncotarget 2016, 7, 21064–21075. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.I.; Bernstein, S.H.; Kahl, B.S.; Djulbegovic, B.; Robertson, M.J.; de Vos, S.; Epner, E.; Krishnan, A.; Leonard, J.P.; Lonial, S.; et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2006, 24, 4867–4874. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boothby, M. Specificity of SN50 for NK-kappa B? Nat. Immunol. 2001, 2, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Grinberg-Bleyer, Y.; Liao, W.; Maloney, D.; Wang, P.; Wu, Z.; Wang, J.; Bhatt, D.M.; Heise, N.; Schmid, R.M.; et al. An NK-kappaB transcription-factor-dependent lineage-specific transcriptional program promotes regulatory T cell identity and function. Immunity 2017, 47, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tam, W.F.; Hughes, C.C.W.; Rath, S.; Sen, R. c-Rel is a target of pentoxifylline-mediated inhibition of T lymphocyte activation. Immunity 1997, 6, 165–174. [Google Scholar] [CrossRef]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-kappaB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell 2017, 170, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Gerondakis, S. The c-Rel transcription factor in development and disease. Genes Cancer 2011, 2, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, L.; Baldwin, A.S., Jr.; Friedman, A.D.; Paz-Priel, I. Loss of IKKbeta but not NK-kappaB p65 skews differentiation towards myeloid over erythroid commitment and increases myeloid progenitor self-renewal and functional long-term hematopoietic stem cells. PLoS ONE 2015, 10, e0130441. [Google Scholar]
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.; Kung, A.; et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 2014, 56, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell 2018, 33, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Catrysse, L.; van Loo, G. Inflammation and the metabolic syndrome: The tissue-specific functions of NK-kappaB. Trends Cell Biol. 2017, 27, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Zheng, X.L.; Tang, C.K. Nuclear factor-kappaB activation as a pathological mechanism of lipid metabolism and atherosclerosis. Adv. Clin. Chem. 2015, 70, 1–30. [Google Scholar] [PubMed]
- Yan, J.; Greer, J.M. NK-kappa B, a potential therapeutic target for the treatment of multiple sclerosis. CNS Neurol. Disord. Drug Targets 2008, 7, 536–557. [Google Scholar] [CrossRef] [PubMed]
- Schuliga, M. NF-kappaB signaling in chronic inflammatory airway disease. Biomolecules 2015, 5, 1266–1283. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.; Carathers, M.; Li, Z.W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of IKK/NK-kappaB signaling in macrophages and myofibers promotes muscle degeneration in duchenne muscular dystrophy. J. Clin. Investig. 2007, 117, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Flood, P.M.; Qian, L.; Peterson, L.J.; Zhang, F.; Shi, J.S.; Gao, H.M.; Hong, J.S. Transcriptional factor NK-kappaB as a target for therapy in parkinson’s disease. Parkinsons Dis. 2011, 2011, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-kappaB in inflammatory bowel disease. J. Intern. Med. 2008, 263, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Armaka, M.; Ospelt, C.; Pasparakis, M.; Kollias, G. The p55TNFR-IKK2-Ripk3 axis orchestrates arthritis by regulating death and inflammatory pathways in synovial fibroblasts. Nat. Commun. 2018, 9, 618. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Andreakos, E.; Kiriakidis, S.; Mauri, C.; Bicknell, C.; Foxwell, B.; Cheshire, N.; Paleolog, E.; Feldmann, M. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 5634–5639. [Google Scholar] [CrossRef] [PubMed]
- Kempe, S.; Kestler, H.; Lasar, A.; Wirth, T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: Evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005, 33, 5308–5319. [Google Scholar] [CrossRef] [PubMed]
- Gareus, R.; Kotsaki, E.; Xanthoulea, S.; van der Made, I.; Gijbels, M.J.; Kardakaris, R.; Polykratis, A.; Kollias, G.; de Winther, M.P.; Pasparakis, M. Endothelial cell-specific NK-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008, 8, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Sui, Y.; Gizard, F.; Xu, J.; Rios-Pilier, J.; Helsley, R.N.; Han, S.S.; Zhou, C. Myeloid-specific IkappaB kinase beta deficiency decreases atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2869–2876. [Google Scholar] [CrossRef] [PubMed]
- Kanters, E.; Pasparakis, M.; Gijbels, M.J.; Vergouwe, M.N.; Partouns-Hendriks, I.; Fijneman, R.J.; Clausen, B.E.; Forster, I.; Kockx, M.M.; Rajewsky, K.; et al. Inhibition of NK-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J. Clin. Investig. 2003, 112, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Meylan, E.; Oliver, T.G.; Feldser, D.M.; Winslow, M.M.; Bronson, R.; Jacks, T. Response and resistance to NK-kappaB inhibitors in mouse models of lung adenocarcinoma. Cancer Discov. 2011, 1, 236–247. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
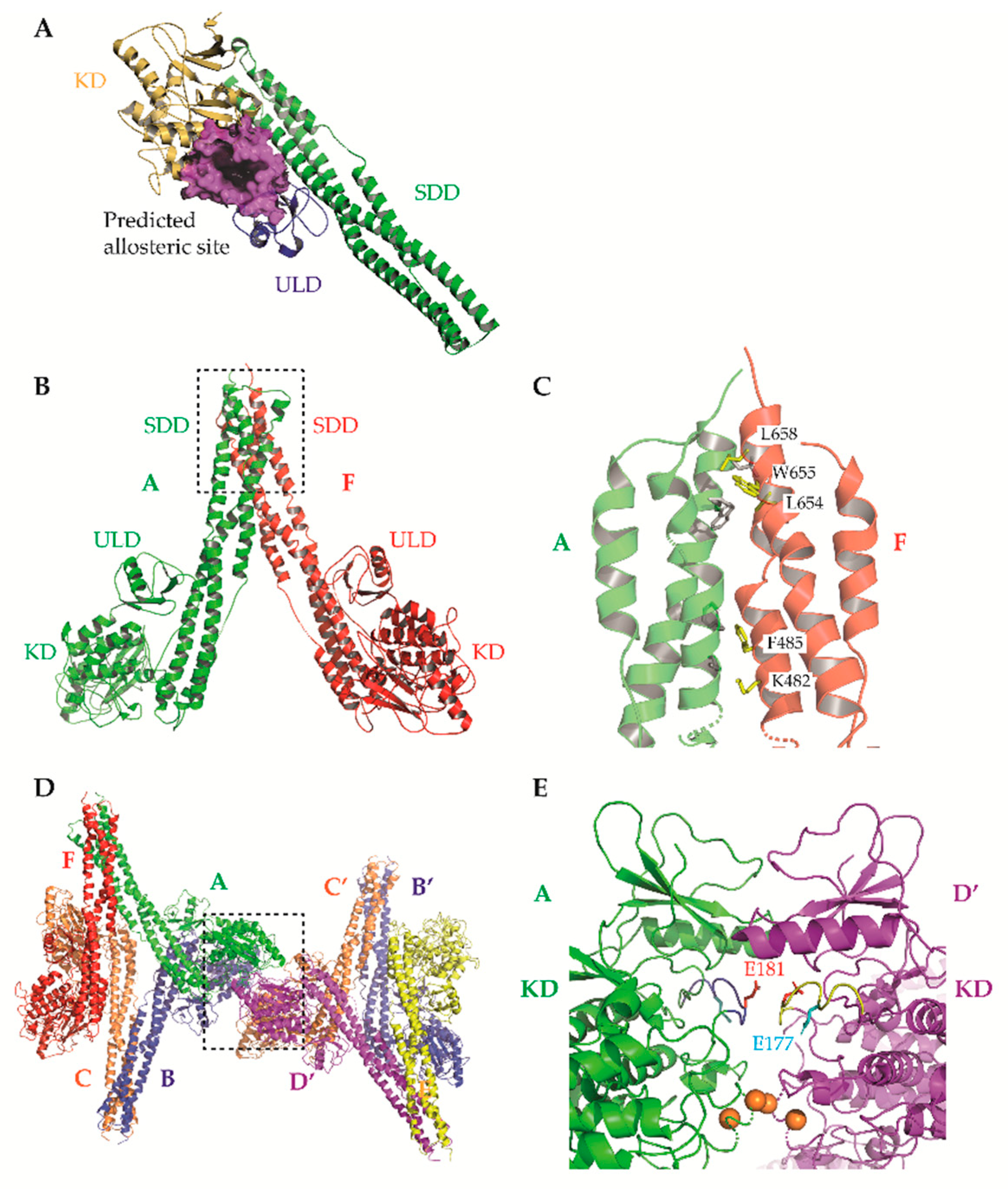{kind=link}
| Inhibitor | Mechanism | Ki/IC50 for IKKβ (nM) * [Ref] | Selectivity Over IKKα | Known Off-Targets | Bio-Availability | Pre-Clinical Therapeutic Efficacy |
|---|---|---|---|---|---|---|
| BI605906 (BIX02514) | ATP-competitive | 380 [40] | >300 fold (>100 µM) | >300-fold selectivity over 100 representative tyr/ser-thr kinases IGF1 (7.6 µM) | N/A | N/A |
| MLN120B | ATP-competitive | 60 [41] | >1000 fold (>100 µM) | >1000-fold selectivity over 30 representative tyr/ser-thr kinases | Good oral bio-availability | Multiple myeloma [42] Arthritis [43] |
| PHA-408 | ATP-competitive | 10–40 [44,45] | >350 fold (14 µM) | >100-fold selectivity over 30 representative tyr/ser-thr kinases PIM-1 (0.6 µM) | Good oral bio-availability | Arthritis [44] COPD [46,47] |
| TPCA-1 (IKK inhibitor IV) | ATP-competitive | 18 [48] | ~22-fold (400 nM) | STAT3 | Poor oral bio-availabilityAdministered intra-peritoneally | Arthritis [48] Nasal epithelium inflammation [49] Glioma [50] NSCLC [51] COPD [52] Wet AMD [53] |
| SC-514 | ATP-competitive | 3000–12,000 [54] | >15-fold (>200 µM) | CDK2/CycA (61 µM) Aurora A (71 µM) PRAK (75 µM) MSK (123 µM) | Poor oral bio-availabilityAdministered intra-peritoneally | Rat model of inflammation [54] Oral squamous cell carcinoma [55] Osteoclast-related disorders [56] Diabetic neuropathy [57] |
| LY2409881 | ATP-competitive | 30 [58] | > 10-fold | >10-fold selectivity over panel of representative tyr/ser-thr kinases | Administered intra-peritoneally | DLBCL [58] |
| PS-1145 | ATP-competitive | 100 [59,60] | N/A | [61] | Administered intra-peritoneally | Multiple myeloma [61] DLBCL [62] Graft-versus-host disease [60] Tobacco smoke-induced pulmonary inflammation [63] |
| Compound A (Bay 65-1942) | ATP-competitive | Ki for GST-IκBα = 4 nM [64] | >30 fold (135 nM) | IKKε, MKK4, MKK7, ERK-1, Syk, Lck, Fyn, PI3Kγ, PKA and PKC (IC50 > 10 µM) | Good oral bio-availability | KRAS-induced lung cancer [65] Chronic pulmonary inflammation [64] Ischemia–reperfusion injury [66] LPS-induced neurotoxicity [67] |
| IKK-16 (IKK Inhibitor VII) | ATP-competitive | 40–70 [68,69] | 5-fold (200 nM) | LRKK2 (50 nM) | Good oral bio-availability | Multiple organ failure associated with hemorrhagic shock [70] Sepsis-associated multiple organ dysfunction [71] Ventilation-induced lung injury [72] Acute kidney injury [73] |
| IMD-0354 (and pro-drug IMD-1041) | ATP-competitive | ~1µM [74,75] | N/A | N/A | Administered intra-peritoneally | CLL [76] Pancreatic cancer [77] Adult T-cell leukemia [78] Breast cancer [75] |
| ACHP (IKK inhibitor VIII) | ATP-competitive | 8.5 [79] | 30-fold (250 nM) | IKKε, Syk, MKK4 (IC50 > 20 µM) | Good oral bio-availability | Multiple myeloma [80] Adult T-cell leukemia [81] HIV-1 replication [82] |
| BMS-345541 | Allosteric | 300 [83] | ~13-fold (4000 nM) | >300-fold selectivity over a small panel of representative tyr/ser-thr kinases | Good oral bio-availability | Arthritis [84] Colitis [85] Cardiac graft rejection [86] T-ALL [87] Glioma [50] Prostate cancer [88] |
| Withaferin A | Cys179-binding | [89,90,91,92] | N/A | Broad spectrum inhibitor [93]Vimentin, HSP90, β-tubulin, Desmin, Annexin-A2, Notch-1, STAT1/3 | Poor oral bioavailability | N/A |
| BOT-64 | Ser-177/181 binding | 1000–3000 [94] | N/A | N/A | Administered intra-peritoneally | N/A |
| Ainsliadimer A | Cysteine-46 binding | 30 [95] | N/A | No significant activity against 340 human kinases at 200 nM | Administered intravenously | N/A |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prescott, J.A.; Cook, S.J. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 2018, 7, 115. https://doi.org/10.3390/cells7090115
Prescott JA, Cook SJ. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells. 2018; 7(9):115. https://doi.org/10.3390/cells7090115
Chicago/Turabian StylePrescott, Jack A., and Simon J. Cook. 2018. "Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors" Cells 7, no. 9: 115. https://doi.org/10.3390/cells7090115
APA StylePrescott, J. A., & Cook, S. J. (2018). Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells, 7(9), 115. https://doi.org/10.3390/cells7090115