Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease



Abstract
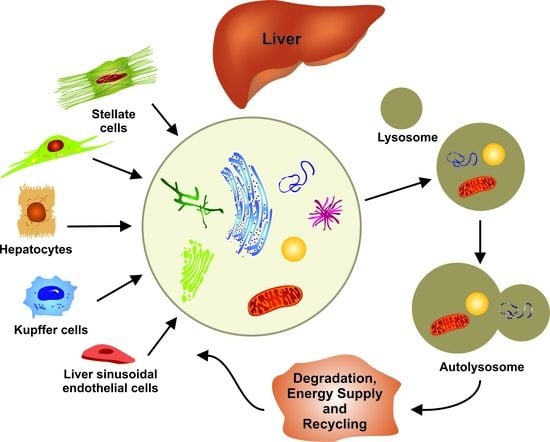
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Principal Functions and Molecular Mechanisms of Autophagy
3. Autophagy in Homeostasis of the Liver—Implications for Hereditary Liver Diseases
4. Autophagy in Liver Metabolism and Fatty Liver Disease
5. Autophagy in Liver Fibrosis and Cirrhosis
6. Autophagy in Liver Cancer
7. Therapeutic Implications and Outlook
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Sabatini, D.D.; Adesnik, M. Christian de Duve: Explorer of the cell who discovered new organelles by using a centrifuge. Proc. Natl. Acad. Sci. USA 2013, 110, 13234–13235. [Google Scholar] [CrossRef] [Green Version]
- Ktistakis, N.T. In praise of M. Anselmier who first used the term “autophagie” in 1859. Autophagy 2017, 13, 2015–2017. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of autophagosome biogenesis. Curr. Biol. 2012, 22, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Olsvik, H.L.; Svenning, S.; Abudu, Y.P.; Brech, A.; Stenmark, H.; Johansen, T.; Mejlvang, J. Endosomal microautophagy is an integrated part of the autophagic response to amino acid starvation. Autophagy 2018, 15, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Oku, M.; Sakai, Y. Three distinct types of microautophagy based on membrane dynamics and molecular machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Poillet-Perez, L.; Xie, X.; Zhan, L.; Yang, Y.; Sharp, D.W.; Hu, Z.S.; Su, X.; Maganti, A.; Jiang, C.; Lu, W.; et al. Autophagy maintains tumour growth through circulating arginine. Nature 2018, 563, 569–573. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of liver metabolism by autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef]
- Schneider, J.L.; Cuervo, A.M. Liver autophagy: Much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Moulis, M.; Vindis, C. Autophagy in metabolic age-related human diseases. Cells 2018, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M.; Batista-Gonzalez, A.; Merheb, E.; Aoun, M.L.; Tarabra, E.; Feng, D.; Sarparanta, J.; Merlo, P.; Botrè, F.; Schwartz, G.J.; et al. Autophagy regulates the liver clock and glucose metabolism by degrading CRY1. Cell Metab. 2018, 28, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, P.; Zhang, L.; Xiong, B.; Tao, J.; Guan, W.; Li, C.; Chen, C.; Gu, J.; Duanmu, J.; et al. Autophagy inhibition attenuates the induction of anti-inflammatory effect of catalpol in liver fibrosis. Biomed. Pharmacother. 2018, 103, 1262–1271. [Google Scholar] [CrossRef]
- Yang, J.; Pi, C.; Wang, G. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed. Pharmacother. 2018, 103, 699–707. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, M.; Wang, Q.; Hou, Y.; Li, L.; Weng, H.; Zhao, Y.; Chen, D.; Ding, H.; Guo, J.; et al. Alpha-fetoprotein inhibits autophagy to promote malignant behaviour in hepatocellular carcinoma cells by activating PI3K/AKT/mTOR signalling. Cell Death Dis. 2018, 9, 1027. [Google Scholar] [CrossRef]
- Liu, L.; Liao, J.Z.; He, X.X.; Li, P.Y. The role of autophagy in hepatocellular carcinoma: Friend or foe. Oncotarget 2017, 8, 57707–57722. [Google Scholar] [CrossRef]
- Teckman, J.H.; An, J.K.; Blomenkamp, K.; Schmidt, B.; Perlmutter, D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, 851–862. [Google Scholar] [CrossRef]
- Teckman, J.H.; Blomenkamp, K.S. Pathophysiology of alpha-1 antitrypsin deficiency liver disease. Methods Mol. Biol. 2017, 1639, 1–8. [Google Scholar] [CrossRef]
- Tang, Y.; Blomenkamp, K.S.; Fickert, P.; Trauner, M.; Teckman, J.H. NorUDCA promotes degradation of α1-antitrypsin mutant Z protein by inducing autophagy through AMPK/ULK1 pathway. PLoS ONE 2018, 13, e0200897. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Teckman, J.H. Alpha-1-antitrypsin deficiency liver disease. Clin. Liver Dis. 2018, 22, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Trautwein, C. Controlling autophagy: A new concept for clearing liver disease. Hepatology 2011, 53, 356–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, N.; Blomenkamp, K.; Annunziata, F.; Piccolo, P.; Mithbaokar, P.; Maria Sepe, R.; Vetrini, F.; Palmer, D.; Ng, P.; Polishchuk, E.; et al. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol. Med. 2013, 5, 397–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, S.; Annamali, M.; Blomenkamp, K.; Rudnick, D.; Halloran, D.; Brunt, E.M.; Teckman, J.H. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp. Biol. Med. 2010, 235, 700–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskirchen, S.; Kim, P.; Weiskirchen, R. Determination of copper poisoning in Wilson’s disease using laser ablation inductively coupled plasma spectrometry. Ann. Transl. Med. 2018. [Google Scholar] [CrossRef]
- Polishchuk, E.V.; Merolla, A.; Lichtmannegger, J.; Romano, A.; Indrieri, A.; Ilyechova, E.Y.; Concilli, M.; De Cegli, R.; Crispino, R.; Mariniello, M.; et al. Activation of autophagy, observed in liver tissues from patients with Wilson disease and from Atp7b-deficient animals, protects hepatocytes from copper-induced apoptosis. Gastroenterology 2018. [Google Scholar] [CrossRef]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic liver disease: Pathogenesis and current management. Alcohol Res. 2017, 38, 147–161. [Google Scholar]
- Chao, X.; Wang, S.; Zhao, K.; Li, Y.; Williams, J.A.; Li, T.; Chavan, H.; Krishnamurthy, P.; He, X.C.; Li, L.; et al. Impaired TFEB-mediated lysosome biogenesis and autophagy promote chronic ethanol-induced liver injury and steatosis in mice. Gastroenterology 2018, 155, 865–879. [Google Scholar] [CrossRef]
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global perspectives on non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Day, C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamming, D.W.; Bar-Peled, L. Lysosome: The metabolic signaling hub. Traffic 2019, 20, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Drižytė, K.; Casey, C.A.; McNiven, M.A. Hepatic lipophagy: New insights into autophagic catabolism of lipid droplets in the liver. Hepatol. Commun. 2017, 1, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaja, M.J. Function of autophagy in nonalcoholic fatty liver disease. Dig. Dis. Sci. 2016, 61, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Van Niekerk, G.; du Toit, A.; Loos, B.; Engelbrecht, A.M. Nutrient excess and autophagic deficiency: Explaining metabolic diseases in obesity. Metabolism 2018, 82, 14–21. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–555. [Google Scholar] [CrossRef]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Cho, C.S.; Park, H.W.; Ho, A.; Semple, I.A.; Kim, B.; Jang, I.; Park, H.; Reilly, S.; Saltiel, A.R.; Lee, J.H. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1-mediated p62/sequestosome 1 phosphorylation. Hepatology 2018, 68, 1331–1346. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ribeiro, M.; Iracheta-Vellve, A.; Lowe, P.; Ambade, A.; Satishchandran, A.; Bukong, T.; Catalano, D.; Kodys, K.; Szabo, G. Macrophage-specific HIF-1α contributes to impaired autophagic flux in non-alcoholic steatohepatitis. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Recent advances in understanding liver fibrosis: Bridging basic science and individualized treatment concepts. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Guixé-Muntet, S. The many-faced role of autophagy in liver diseases. J. Hepatol. 2018, 68, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Thoen, L.F.; Guimarães, E.L.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef]
- Kim, K.M.; Han, C.Y.; Kim, J.Y.; Cho, S.S.; Kim, Y.S.; Koo, J.H.; Lee, J.M.; Lim, S.C.; Kang, K.W.; Kim, J.S.; et al. Gα12 overexpression induced by miR-16 dysregulation contributes to liver fibrosis by promoting autophagy in hepatic stellate cells. J. Hepatol. 2018, 68, 493–504. [Google Scholar] [CrossRef]
- Duran, A.; Hernandez, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by binding to vitamin D receptor inhibits hepatic stellate cell activity, fibrosis, and liver cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef]
- Deng, J.; Huang, Q.; Wang, Y.; Shen, P.; Guan, F.; Li, J.; Huang, H.; Shi, C. Hypoxia-inducible factor-1alpha regulates autophagy to activate hepatic stellate cells. Biochem. Biophys. Res. Commun. 2014, 454, 328–334. [Google Scholar] [CrossRef]
- Fu, M.Y.; He, Y.J.; Lv, X.; Liu, Z.H.; Shen, Y.; Ye, G.R.; Deng, Y.M.; Shu, J.C. Transforming growth factor-β1 reduces apoptosis via autophagy activation in hepatic stellate cells. Mol. Med. Rep. 2014, 10, 1282–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zeng, C.; Zheng, B.; Liu, C.; Tang, M.; Jiang, Y.; Chang, Y.; Song, W.; Wang, Y.; Yang, C. HMGB1-induced autophagy facilitates hepatic stellate cells activation: A new pathway in liver fibrosis. Clin. Sci. 2018, 132, 1645–1667. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Lodder, J.; Denaës, T.; Chobert, M.N.; Wan, J.; El-Benna, J.; Pawlotsky, J.M.; Lotersztajn, S.; Teixeira-Clerc, F. Macrophage autophagy protects against liver fibrosis in mice. Autophagy 2015, 11, 1280–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Xu, L.; Jing, Y.; Han, Z.; Chen, X.; Cai, C.; Zhao, P.; Zhao, X.; Yang, L.; Wei, L. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-κB-IL1α/β-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett. 2017, 388, 198–207. [Google Scholar] [CrossRef]
- Habib, A.; Chokr, D.; Wan, J.; Hegde, P.; Mabire, M.; Siebert, M.; Ribeiro-Parenti, L.; Le Gall, M.; Lettéron, P.; Pilard, N.; et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut 2018. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, H.M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.Y.; Lan, S.H.; Wu, S.R.; Chiu, Y.C.; Lin, X.Z.; Su, I.J.; Tsai, T.F.; Yen, C.J.; Lu, T.H.; Liang, F.W.; et al. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Hepatology 2018, 68, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Lan, S.H.; Wu, S.Y.; Zuchini, R.; Lin, X.Z.; Su, I.J.; Tsai, T.F.; Lin, Y.J.; Wu, C.T.; Liu, H.S. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology 2014, 59, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Khambu, B.; Huda, N.; Chen, X.; Antoine, D.J.; Li, Y.; Dai, G.; Köhler, U.A.; Zong, W.X.; Waguri, S.; Werner, S.; et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J. Clin. Investig. 2018, 128, 2419–2435. [Google Scholar] [CrossRef]
- Di Fazio, P.; Matrood, S. Targeting autophagy in liver cancer. Transl. Gastroenterol. Hepatol. 2018, 3, 39. [Google Scholar] [CrossRef]
- Huang, F.; Wang, B.R.; Wang, Y.G. Role of autophagy in tumorigenesis, metastasis, targeted therapy and drug resistance of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 4643–4651. [Google Scholar] [CrossRef]
- Tai, W.T.; Shiau, C.W.; Chen, H.L.; Liu, C.Y.; Lin, C.S.; Cheng, A.L.; Chen, P.J.; Chen, K.F. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef]
- Lu, S.; Yao, Y.; Xu, G.; Zhou, C.; Zhang, Y.; Sun, J.; Jiang, R.; Shao, Q.; Chen, Y. CD24 regulates sorafenib resistance via activating autophagy in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 646. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, J.; Zhou, H.; Chen, Y.; Zhi, Y.; Zhang, B.; Chen, L.; Chu, X.; Wang, R.; Zhang, C. PU.1/microRNA-142-3p targets ATG5/ATG16L1 to inactivate autophagy and sensitize hepatocellular carcinoma cells to sorafenib. Cell Death Dis. 2018, 9, 312. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; Che, N.; Zhou, L.; Luk, S.T.; Kau, P.W.; Chai, S.; Ngan, E.S.; Lo, C.M.; Man, K.; Ding, J.; et al. Efficacy of annexin A3 blockade in sensitizing hepatocellular carcinoma to sorafenib and regorafenib. J. Hepatol. 2018, 69, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.T.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Liu, W.R.; Shi, G.M.; Gao, Q.; Wang, X.Y.; Song, K.; Fan, J.; et al. MicroRNA-30a suppresses autophagy-mediated anoikis resistance and metastasis in hepatocellular carcinoma. Cancer Lett. 2018, 412, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.F.; Shi, Y.H.; Ding, Z.B.; Ke, A.W.; Gu, C.Y.; Hui, B.; Zhou, J.; Qiu, S.J.; Dai, Z.; Fan, J. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and tumor metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Towers, C.G.; Thorburn, A. Therapeutic targeting of autophagy. EBioMedicine 2016, 14, 15–23. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F.; Weiskirchen, R. An update on the recent advances in antifibrotic therapy. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 1143–1152. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiskirchen, R.; Tacke, F. Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease. Cells 2019, 8, 16. https://doi.org/10.3390/cells8010016
Weiskirchen R, Tacke F. Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease. Cells. 2019; 8(1):16. https://doi.org/10.3390/cells8010016
Chicago/Turabian StyleWeiskirchen, Ralf, and Frank Tacke. 2019. "Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease" Cells 8, no. 1: 16. https://doi.org/10.3390/cells8010016
APA StyleWeiskirchen, R., & Tacke, F. (2019). Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease. Cells, 8(1), 16. https://doi.org/10.3390/cells8010016