Altered VEGF Splicing Isoform Balance in Tumor Endothelium Involves Activation of Splicing Factors Srpk1 and Srsf1 by the Wilms’ Tumor Suppressor Wt1

 ,
,  ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Culture
2.3. Endothelial Cell Isolation
2.4. RT-PCR and Quantitative RT-PCR
2.5. Tissue Samples and Immunohistology
2.6. Cloning and Transient Transfection Experiments
2.7. Chromatin Immunoprecipitation Assay
2.8. Statistical Analysis
3. Results and Discussion
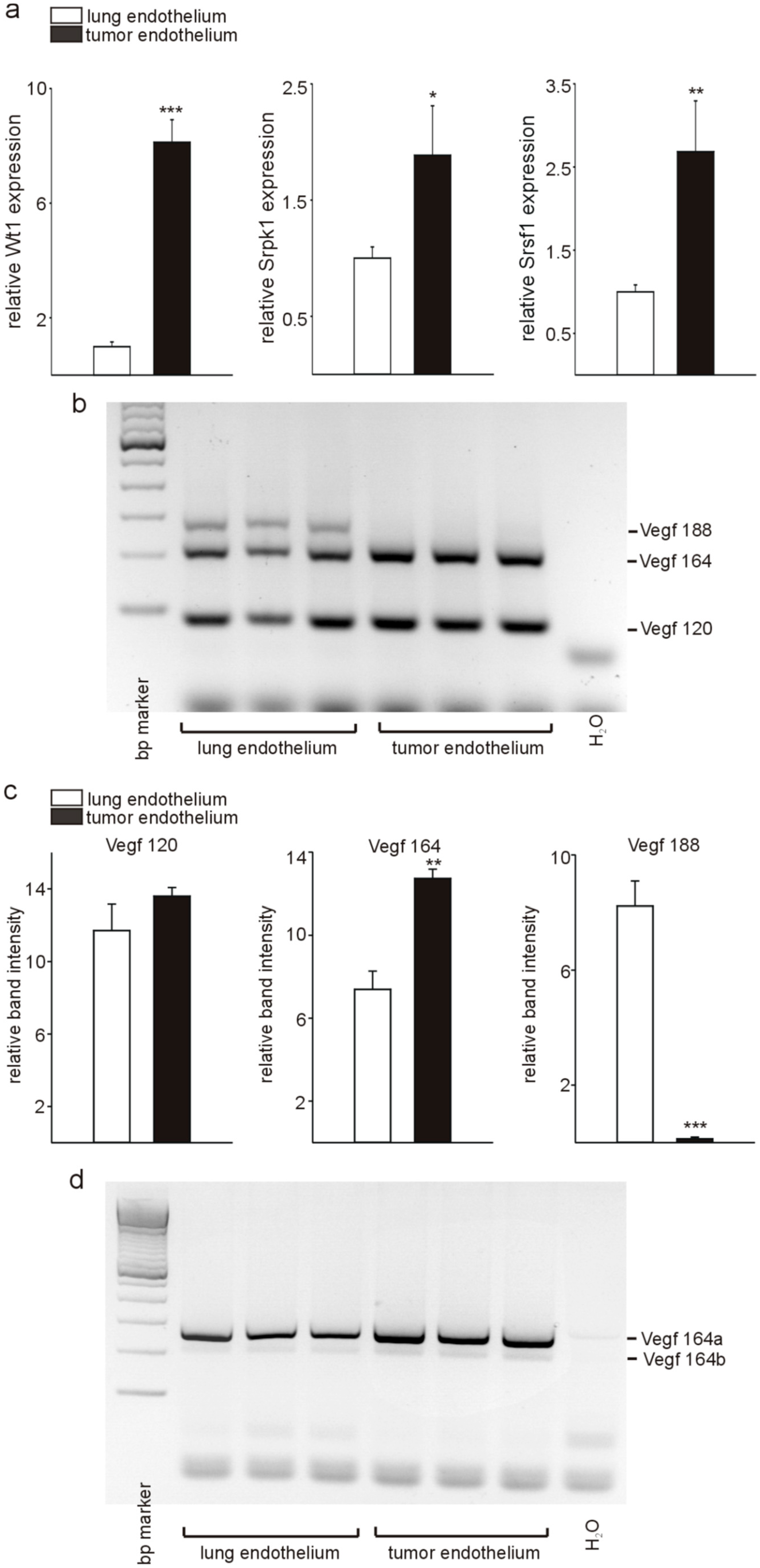
3.1. Wt1, Srpk1, and Srsf1 Are highly Expressed in Tumor versus Lung Endothelium
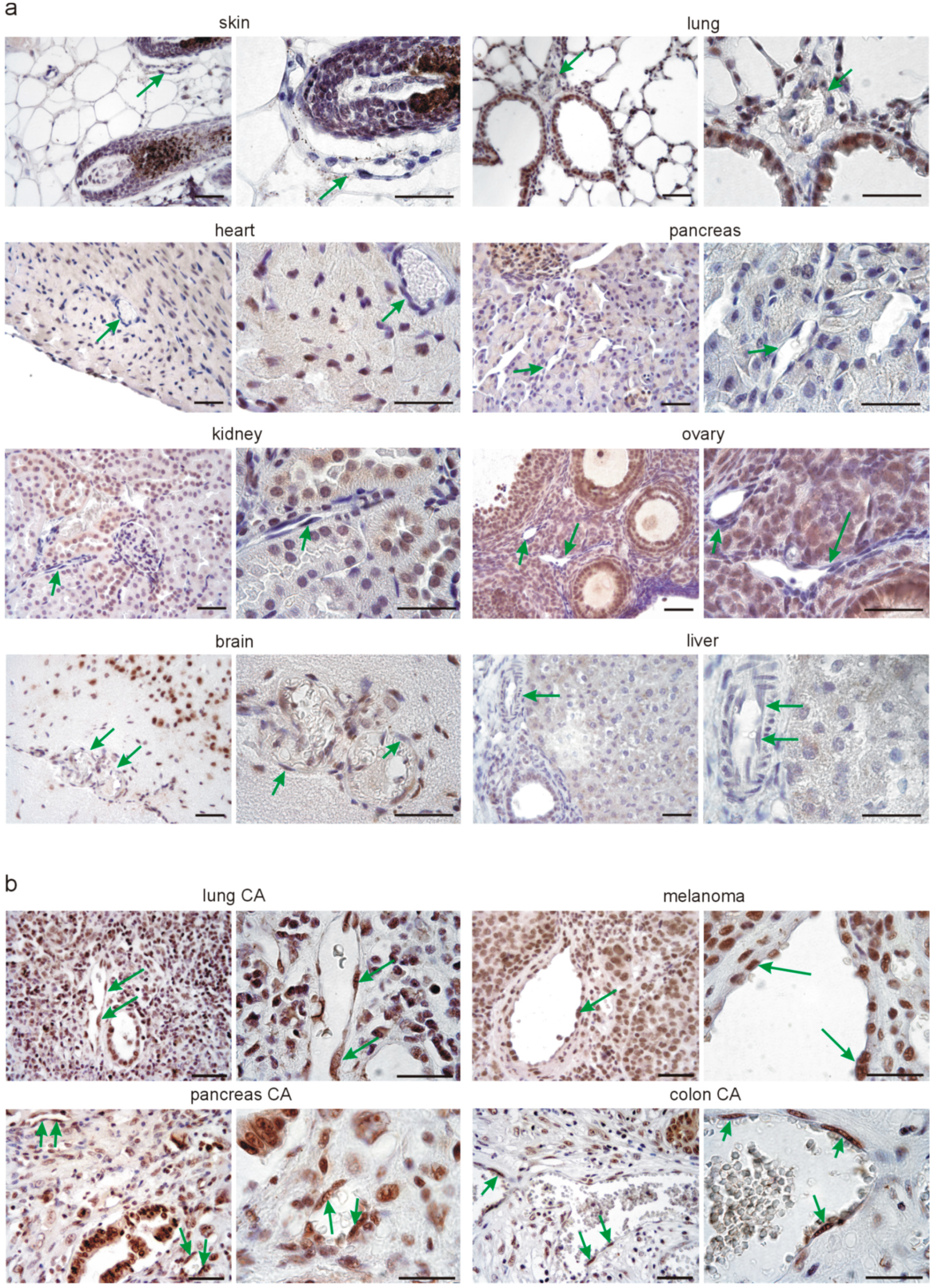
3.2. Srsf1 Protein Is Differentially Expressed in Normal Tissue Endothelium Compared to Tumor Endothelium
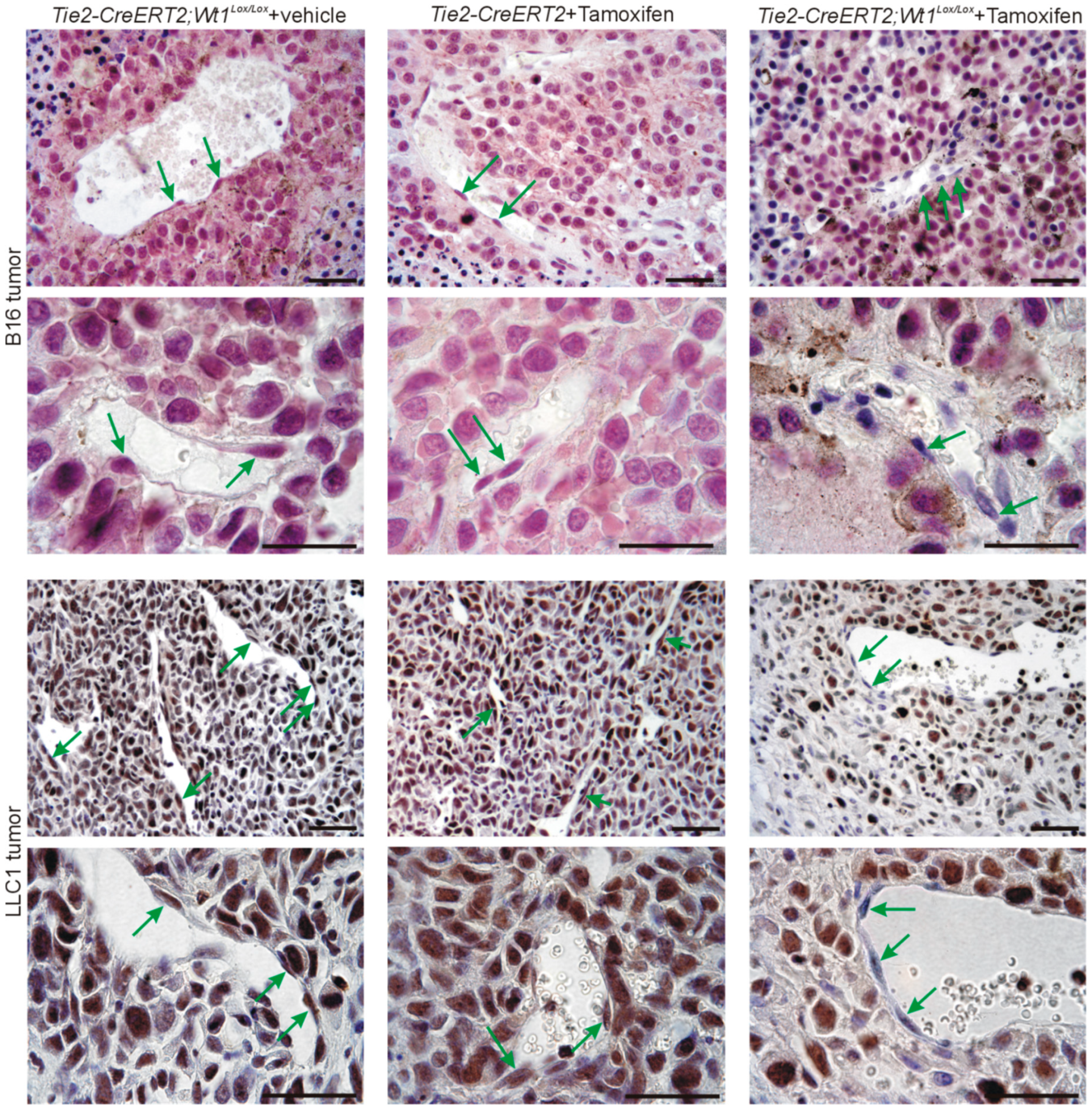
3.3. Inducible Vascular-Specific Knockout of Wt1 Abolishes Nuclear Endothelial Srsf1 Expression
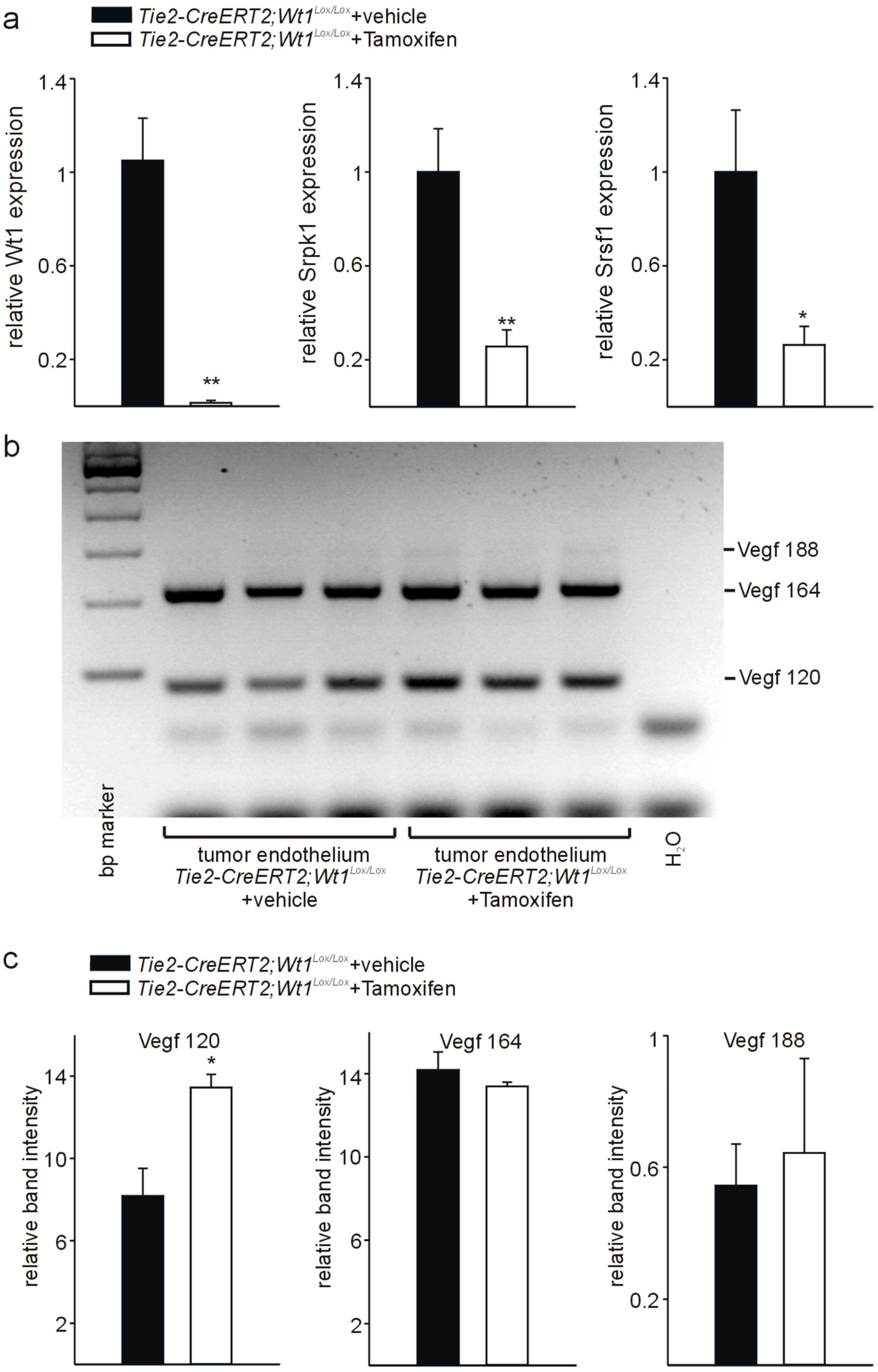
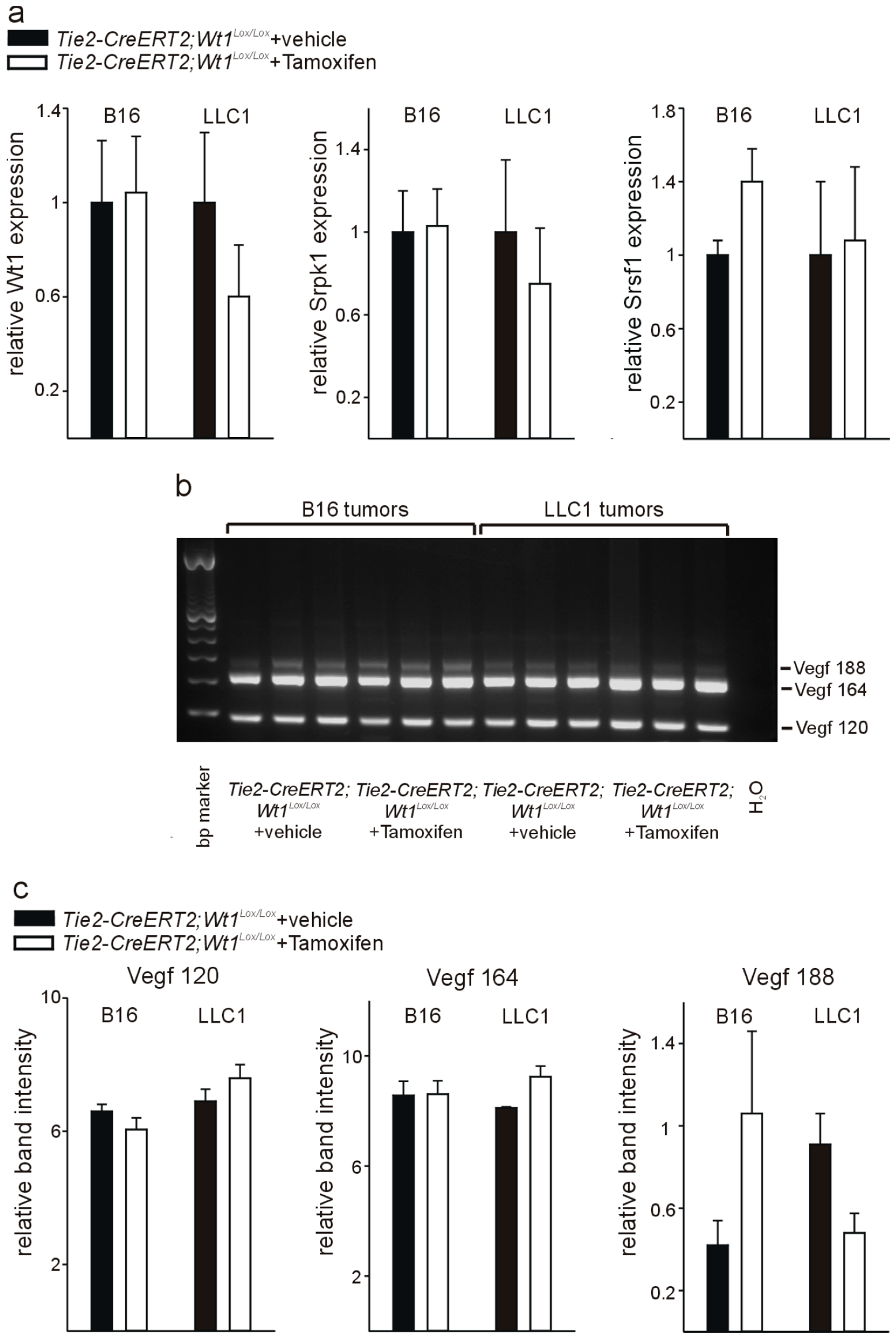
3.4. Knockout of Wt1 in Tumor Endothelium Affects Srpk1, and Srsf1 Expression and Vegf Splicing
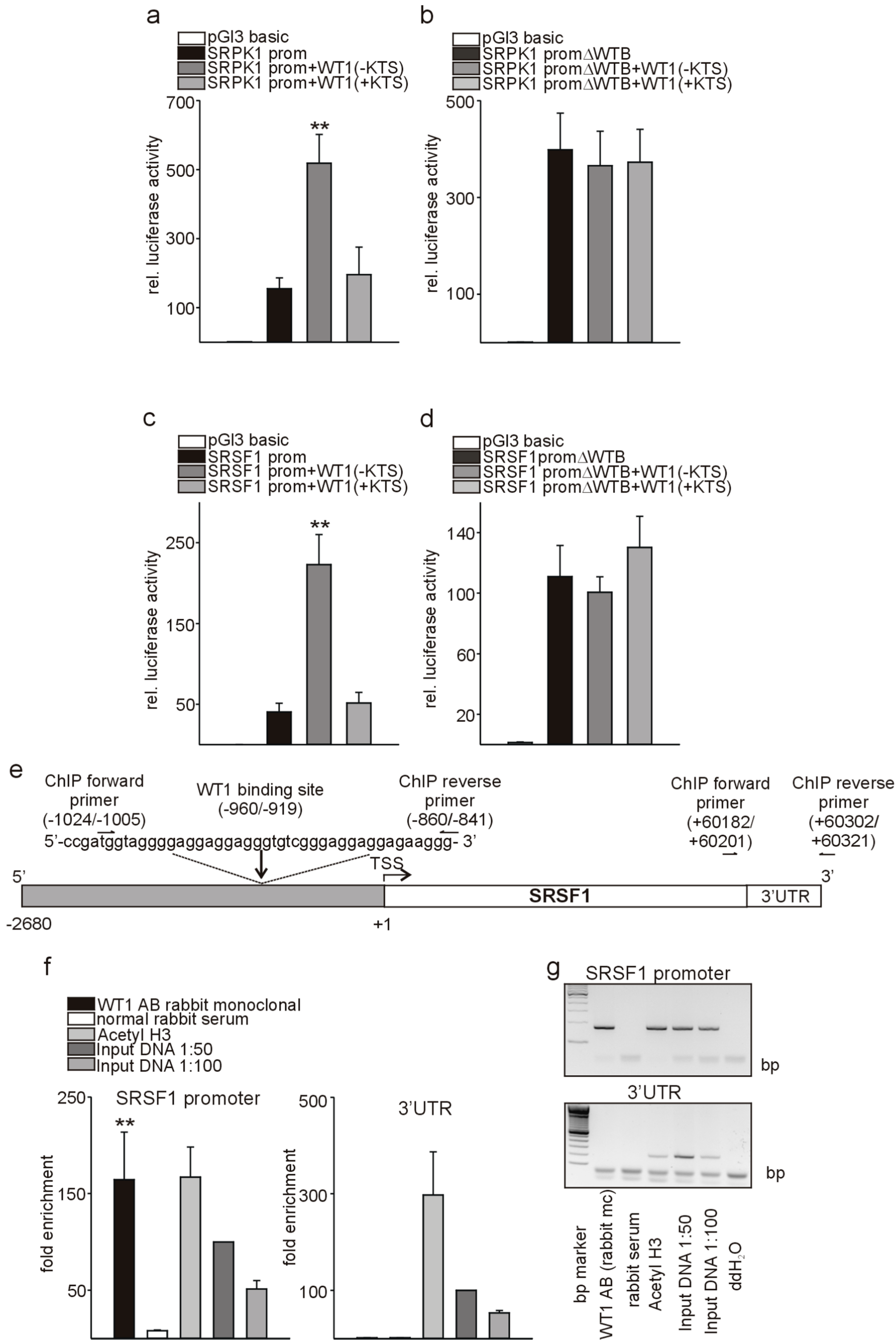
3.5. Wt1 Activates Srpk1 and Srsf1 in Endothelial Cells
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef]
- Augustin, H.G.; Koh, G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017, 357, eaal2379. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Perruzzi, C.A.; Feder, J.; Dvorak, H.F. A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res. 1986, 46, 5629–5632. [Google Scholar]
- Keck, P.J.; Hauser, S.D.; Krivi, G.; Sanzo, K.; Warren, T.; Feder, J.; Connolly, D.T. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 1989, 246, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380, 435–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damert, A.; Miquerol, L.; Gertsenstein, M.; Risau, W.; Nagy, A. Insufficient VEGFA activity in yolk sac endoderm compromises haematopoietic and endothelial differentiation. Development 2002, 129, 1881–1892. [Google Scholar] [PubMed]
- Miquerol, L.; Gertsenstein, M.; Harpal, K.; Rossant, J.; Nagy, A. Multiple developmental roles of VEGF suggested by a LacZ-tagged allele. Dev. Biol. 1999, 212, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Miquerol, L.; Langille, B.L.; Nagy, A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development 2000, 127, 3941–3946. [Google Scholar] [PubMed]
- El Alaoui-Lasmaili, K.; Faivre, B. Antiangiogenic therapy: Markers of response, “normalization” and resistance. Crit. Rev. Oncol. Hematol. 2018, 128, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.P.; Levy, N.S.; Wegner, S.; Goldberg, M.A. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J. Biol. Chem. 1995, 270, 13333–13340. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- McCarty, G.; Awad, O.; Loeb, D.M. WT1 protein directly regulates expression of vascular endothelial growth factor and is a mediator of tumor response to hypoxia. J. Biol. Chem. 2011, 286, 43634–43643. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.D.; Wagner, N.; Wellmann, S.; Schley, G.; Bondke, A.; Theres, H.; Scholz, H. Oxygen-regulated expression of the Wilms’ tumor suppressor Wt1 involves hypoxia-inducible factor-1 (HIF-1). FASEB J. 2003, 17, 1364–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, P.; Wagner, K.D.; Hofman, P.; Van Obberghen, E. RNA Activation of the Vascular Endothelial Growth Factor Gene (VEGF) Promoter by Double-Stranded RNA and Hypoxia: Role of Noncoding VEGF Promoter Transcripts. Mol. Cell Biol. 2016, 36, 1480–1493. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Palumbo, I.; Grosso, M.; Slater, N.; Miles, C.G. WT1 regulates murine hematopoiesis via maintenance of VEGF isoform ratio. Blood 2013, 122, 188–192. [Google Scholar] [CrossRef] [Green Version]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Ladomery, M.R.; Harper, S.J.; Bates, D.O. Alternative splicing in angiogenesis: The vascular endothelial growth factor paradigm. Cancer Lett. 2007, 249, 133–142. [Google Scholar] [CrossRef] [PubMed]
- E, G.; Cao, Y.; Bhattacharya, S.; Dutta, S.; Wang, E.; Mukhopadhyay, D. Endogenous vascular endothelial growth factor-A (VEGF-A) maintains endothelial cell homeostasis by regulating VEGF receptor-2 transcription. J. Biol. Chem. 2012, 287, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.D.; Cherfils-Vicini, J.; Hosen, N.; Hohenstein, P.; Gilson, E.; Hastie, N.D.; Michiels, J.F.; Wagner, N. The Wilms’ tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat. Commun. 2014, 5, 5852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, K.D.; Wagner, N.; Guo, J.K.; Elger, M.; Dallman, M.J.; Bugeon, L.; Schedl, A. An inducible mouse model for PAX2-dependent glomerular disease: Insights into a complex pathogenesis. Curr. Biol. 2006, 16, 793–800. [Google Scholar] [CrossRef] [PubMed]
- El Maï, M.; Wagner, K.D.; Michiels, J.F.; Ambrosetti, D.; Borderie, A.; Destree, S.; Renault, V.; Djerbi, N.; Giraud-Panis, M.J.; Gilson, E.; et al. The Telomeric Protein TRF2 Regulates Angiogenesis by Binding and Activating the PDGFRβ Promoter. Cell Rep. 2014, 9, 1047–1060. [Google Scholar] [CrossRef] [Green Version]
- Sobczak, M.; Dargatz, J.; Chrzanowska-Wodnicka, M. Isolation and culture of pulmonary endothelial cells from neonatal mice. J. Vis. Exp. 2010, 46, e2316. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; Nakamura, K.; MacLauchlan, S.; Ngo, D.T.; Shimizu, I.; Fuster, J.J.; Katanasaka, Y.; Yoshida, S.; Qiu, Y.; Yamaguchi, T.P.; et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat. Med. 2014, 20, 1464–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keber, R.; Motaln, H.; Wagner, K.D.; Debeljak, N.; Rassoulzadegan, M.; Ačimovič, J.; Rozman, D.; Horvat, S. Mouse knockout of the cholesterogenic cytochrome P450 lanosterol 14alpha-demethylase (Cyp51) resembles Antley-Bixler syndrome. J. Biol. Chem. 2011, 286, 29086–29097. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.; Milligan, S.G.; Graham, S.V. Human papillomavirus type 16 E2 protein transcriptionally activates the promoter of a key cellular splicing factor, SF2/ASF. J. Virol. 2009, 83, 357–367. [Google Scholar] [CrossRef]
- Wagner, N.; Michiels, J.F.; Schedl, A.; Wagner, K.D. The Wilms’ tumour suppressor WT1 is involved in endothelial cell proliferation and migration: Expression in tumour vessels in vivo. Oncogene 2008, 27, 3662–3672. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.C.; Giang, K.; Chakrabarti, S.; Ma, C.T.; Huynh, N.; Hagopian, J.C.; Dorrestein, P.C.; Fu, X.D.; Adams, J.A.; Ghosh, G. A sliding docking interaction is essential for sequential and processive phosphorylation of an SR protein by SRPK1. Mol. Cell 2008, 29, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Lin, R.I.; Tarn, W.Y. Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 10154–10159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacic, M.; Edwards, N.A.; Merrill, M.J. Differential expression of vascular endothelial growth factor (vascular permeability factor) forms in rat tissues. Growth Factors 1995, 12, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.; Wong, M.P.; Yuen, S.T.; Leung, S.Y.; Chung, L.P. Tissue-specific expression pattern of vascular endothelial growth factor isoforms in the malignant transformation of lung and colon. Hum. Pathol. 1998, 29, 910–914. [Google Scholar] [CrossRef]
- Medford, A.R.; Douglas, S.K.; Godinho, S.I.; Uppington, K.M.; Armstrong, L.; Gillespie, K.M.; van Zyl, B.; Tetley, T.D.; Ibrahim, N.B.; Millar, A.B. Vascular Endothelial Growth Factor (VEGF) isoform expression and activity in human and murine lung injury. Respir. Res. 2009, 10, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, A.; Lin, C.Y.; Chou, C.H.; Shih, C.M.; Chen, C.Y.; Cheng, H.W.; Chen, Y.F.; Chen, J.J.; Chen, J.H.; Yang, P.C.; et al. Functional and structural characteristics of tumor angiogenesis in lung cancers overexpressing different VEGF isoforms assessed by DCE- and SSCE-MRI. PLoS ONE 2011, 6, e16062. [Google Scholar] [CrossRef] [PubMed]
- Nishi, M.; Abe, Y.; Tomii, Y.; Tsukamoto, H.; Kijima, H.; Yamazaki, H.; Ohnishi, Y.; Iwasaki, M.; Inoue, H.; Ueyama, Y.; et al. Cell binding isoforms of vascular endothelial growth factor-A (VEGF189) contribute to blood flow-distant metastasis of pulmonary adenocarcinoma. Int. J. Oncol. 2005, 26, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, M.; Toullec, A.; Buteau-Lozano, H.; Abdelkarim, M.; Vacher, S.; Velasco, G.; Christofari, M.; Pocard, M.; Bieche, I.; Perrot-Applanat, M. MDA-MB-231 breast cancer cells overexpressing single VEGF isoforms display distinct colonisation characteristics. Br. J. Cancer 2015, 113, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Nowak, D.G.; Amin, E.M.; Rennel, E.S.; Hoareau-Aveilla, C.; Gammons, M.; Damodoran, G.; Hagiwara, M.; Harper, S.J.; Woolard, J.; Ladomery, M.R.; et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: A novel therapeutic strategy for angiogenesis. J. Biol. Chem. 2010, 285, 5532–5540. [Google Scholar] [CrossRef]
- Nowak, D.G.; Woolard, J.; Amin, E.M.; Konopatskaya, O.; Saleem, M.A.; Churchill, A.J.; Ladomery, M.R.; Harper, S.J.; Bates, D.O. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 2008, 121 Pt 20, 3487–3495. [Google Scholar] [CrossRef] [Green Version]
- Cui, T.G.; Foster, R.R.; Saleem, M.; Mathieson, P.W.; Gillatt, D.A.; Bates, D.O.; Harper, S.J. Differentiated human podocytes endogenously express an inhibitory isoform of vascular endothelial growth factor (VEGF165b) mRNA and protein. Am. J. Physiol. Renal Physiol. 2004, 286, F767–F773. [Google Scholar] [CrossRef] [Green Version]
- Varey, A.H.; Rennel, E.S.; Qiu, Y.; Bevan, H.S.; Perrin, R.M.; Raffy, S.; Dixon, A.R.; Paraskeva, C.; Zaccheo, O.; Hassan, A.B.; et al. VEGF 165 b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: Balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br. J. Cancer 2008, 98, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Li, Y.; Ning, J.; Sun, D.; Lin, L.; Liu, X. HnRNP A1/A2 and SF2/ASF regulate alternative splicing of interferon regulatory factor-3 and affect immunomodulatory functions in human non-small cell lung cancer cells. PLoS ONE 2013, 8, e62729. [Google Scholar] [CrossRef] [PubMed]
- Crosby, J.R.; Kaminski, W.E.; Schatteman, G.; Martin, P.J.; Raines, E.W.; Seifert, R.A.; Bowen-Pope, D.F. Endothelial cells of hematopoietic origin make a significant contribution to adult blood vessel formation. Circ. Res. 2000, 87, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Mallinjoud, P.; Villemin, J.P.; Mortada, H.; Polay Espinoza, M.; Desmet, F.O.; Samaan, S.; Chautard, E.; Tranchevent, L.C.; Auboeuf, D. Endothelial, epithelial, and fibroblast cells exhibit specific splicing programs independently of their tissue of origin. Genome Res. 2014, 24, 511–521. [Google Scholar] [CrossRef]
- Davies, R.C.; Calvio, C.; Bratt, E.; Larsson, S.H.; Lamond, A.I.; Hastie, N.D. WT1 interacts with the splicing factor U2AF65 in an isoform-dependent manner and can be incorporated into spliceosomes. Genes Dev. 1998, 12, 3217–3225. [Google Scholar] [CrossRef] [Green Version]
- Larsson, S.H.; Charlieu, J.P.; Miyagawa, K.; Engelkamp, D.; Rassoulzadegan, M.; Ross, A.; Cuzin, F.; van Heyningen, V.; Hastie, N.D. Subnuclear localization of WT1 in splicing or transcription factor domains is regulated by alternative splicing. Cell 1995, 81, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.D.; Wagner, N.; Schedl, A. The complex life of WT1. J. Cell Sci. 2003, 116 Pt 9, 1653–1658. [Google Scholar] [CrossRef] [Green Version]
- Roberts, S.G. Transcriptional regulation by WT1 in development. Curr. Opin. Genet. Dev. 2005, 15, 542–547. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| Wt1 forward | CCA GCT CAG TGA AAT GGA CA |
| Wt1 reverse | CTG TAC TGG GCA CCA CAG AG |
| Vegfa Exon 4 forward | CAC AGC AGA TGT GAA TGC AG [18] |
| Vegfa Exon 8 reverse | CCT TCC TGC AGC CTG GCT C [18] |
| Vegf164a/b forward | CAG AAA ATC ACT GTG AGC CTT GTT [26] |
| Vegf164a/b reverse | ATT AAG GAC TGT TCT GTC AA [26] |
| Srpk1 forward | CCA AGT GAA GAT CGC AGA CC |
| Srpk1 reverse | TCT TCA GTG AAA TGC TTG TGC |
| Srsf1 forward | TCC GAG AAC AGA GTG GTT GTC |
| Srsf1 reverse | CAT ACA TCA CCT GCC TCA CG |
| Rplp0 forward | CAC TGG TCT AGG ACC CGA GAA G [27] |
| Rplp0 reverse | GGT GCC TCT GGA GAT TTT CG [27] |
| Gapdh forward | CCA ATG TGT CCG TCG TGG ATC T [27] |
| Gapdh reverse | GTT GAA GTC GCA GGA GAC AAC C [27] |
| Actb forward | CTT CCT CCC TGG AGA AGA GC [27] |
| Actb reverse | ATG CCA CAG GAT TCC ATA CC [27] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, K.-D.; El Maï, M.; Ladomery, M.; Belali, T.; Leccia, N.; Michiels, J.-F.; Wagner, N. Altered VEGF Splicing Isoform Balance in Tumor Endothelium Involves Activation of Splicing Factors Srpk1 and Srsf1 by the Wilms’ Tumor Suppressor Wt1. Cells 2019, 8, 41. https://doi.org/10.3390/cells8010041
Wagner K-D, El Maï M, Ladomery M, Belali T, Leccia N, Michiels J-F, Wagner N. Altered VEGF Splicing Isoform Balance in Tumor Endothelium Involves Activation of Splicing Factors Srpk1 and Srsf1 by the Wilms’ Tumor Suppressor Wt1. Cells. 2019; 8(1):41. https://doi.org/10.3390/cells8010041
Chicago/Turabian StyleWagner, Kay-Dietrich, Mounir El Maï, Michael Ladomery, Tareg Belali, Nathalie Leccia, Jean-François Michiels, and Nicole Wagner. 2019. "Altered VEGF Splicing Isoform Balance in Tumor Endothelium Involves Activation of Splicing Factors Srpk1 and Srsf1 by the Wilms’ Tumor Suppressor Wt1" Cells 8, no. 1: 41. https://doi.org/10.3390/cells8010041
APA StyleWagner, K. -D., El Maï, M., Ladomery, M., Belali, T., Leccia, N., Michiels, J. -F., & Wagner, N. (2019). Altered VEGF Splicing Isoform Balance in Tumor Endothelium Involves Activation of Splicing Factors Srpk1 and Srsf1 by the Wilms’ Tumor Suppressor Wt1. Cells, 8(1), 41. https://doi.org/10.3390/cells8010041