Inflammasome-Mediated Inflammation in Liver Ischemia-Reperfusion Injury

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Ischemia-Reperfusion Injury, an Unresolved Problem in Clinical Practice
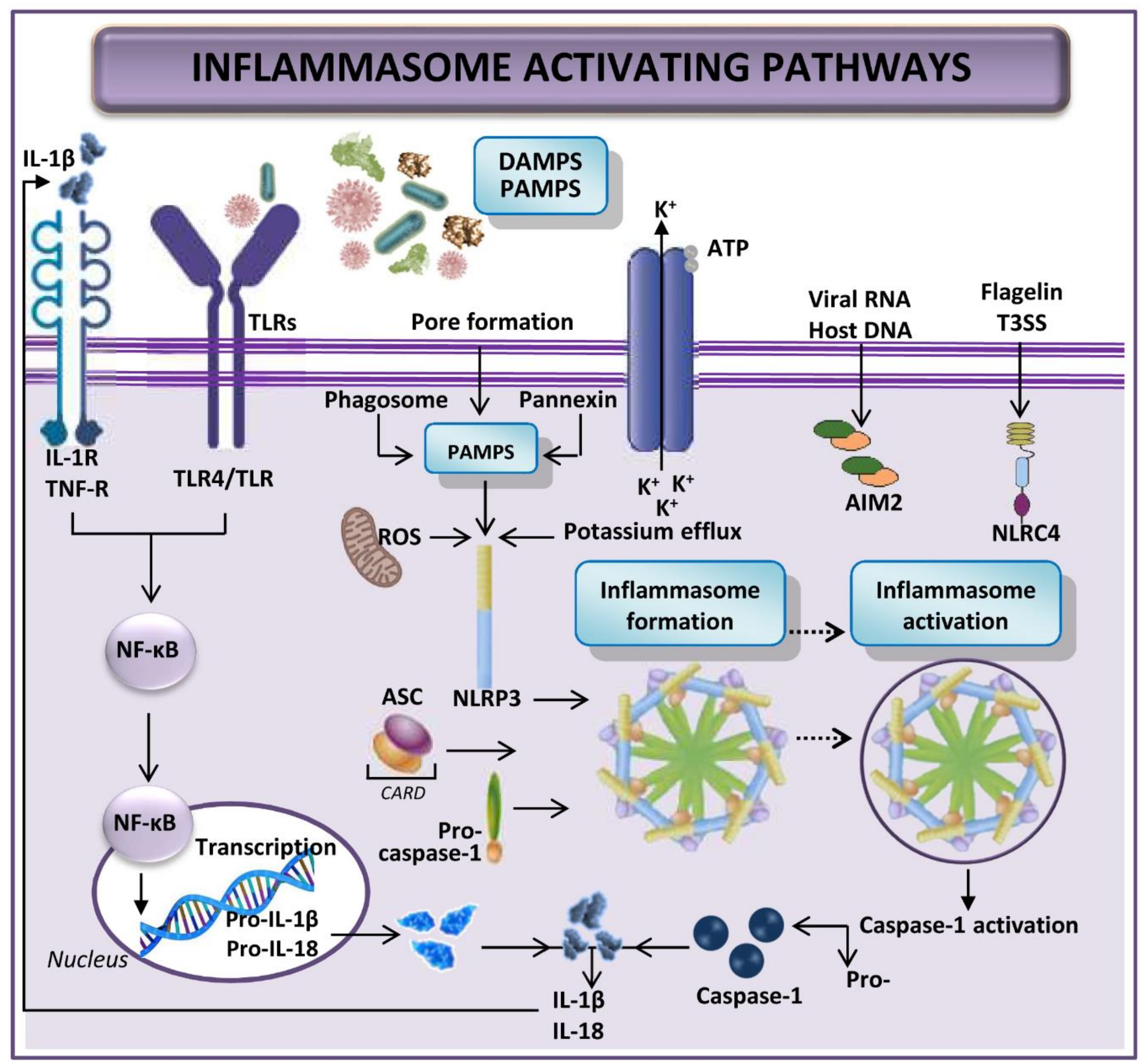
2. Inflammasome and Its Implications in Liver Disease
3. Relevance of Inflammasome in Hepatic Ischemia-Reperfusion Injury
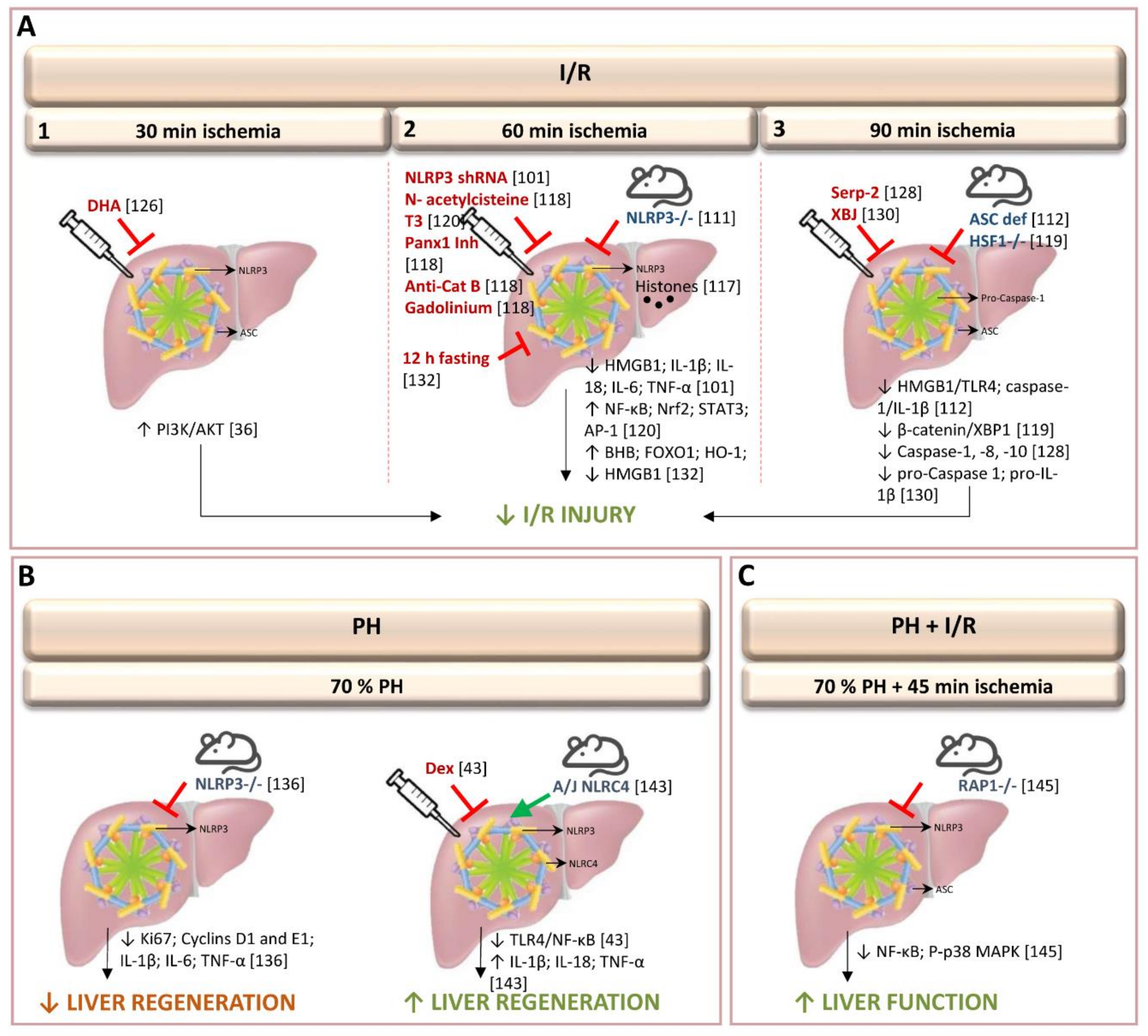
3.1. Inflammasome in Warm Ischemia-Reperfusion Associated with Liver Resection
3.1.1. Role of Inflammasome in Experimental Models of Warm Ischemia-Reperfusion Injury without Hepatic Resection
3.1.2. Role of Inflammasome in Experimental Models of Hepatectomy without Ischemia-Reperfusion
3.1.3. Role of Inflammasome in Experimental Models of Hepatic Resection under Vascular Occlusion
3.1.4. Clinical Results of Inflammasome Activation in Liver Resection
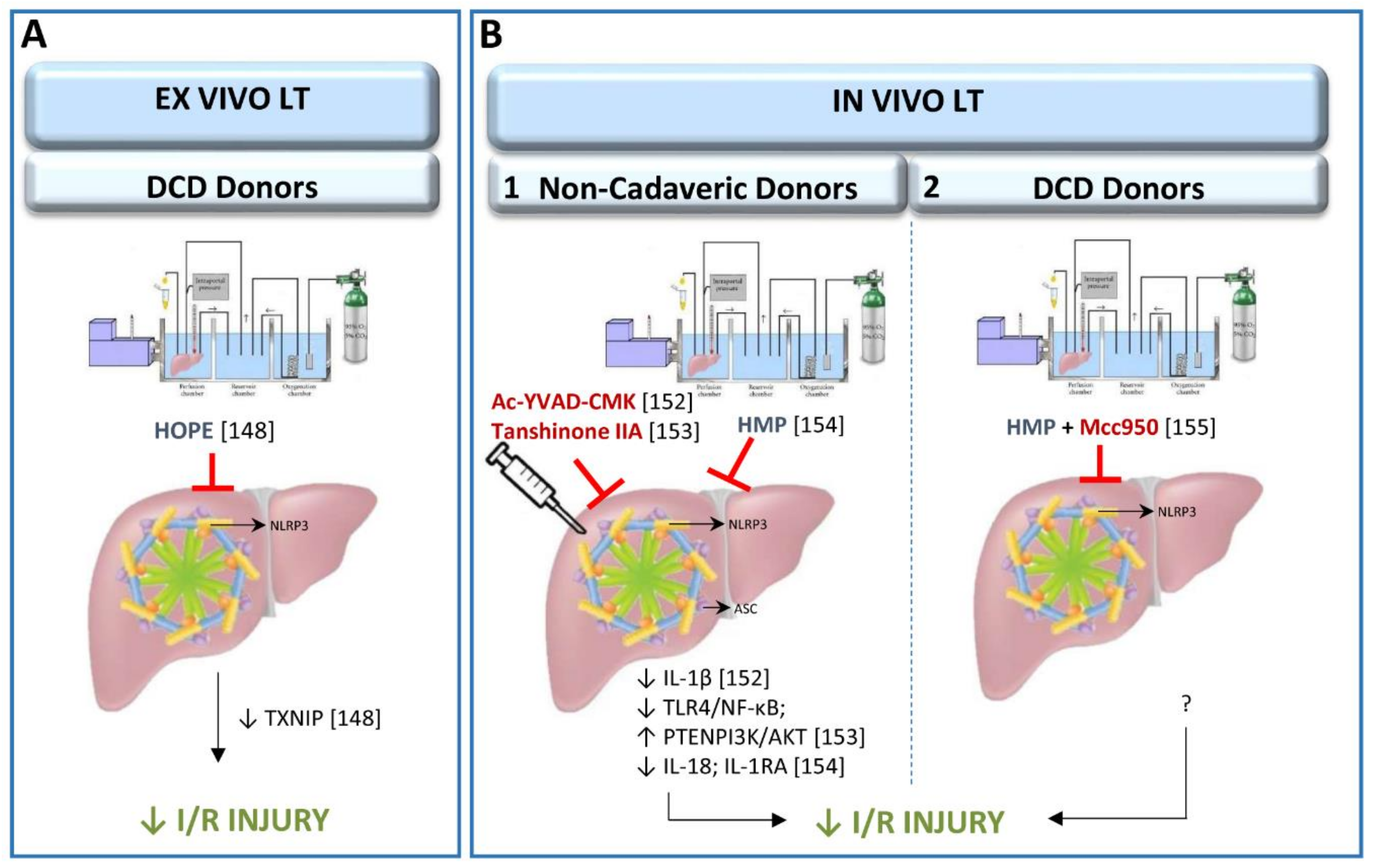
3.2. Inflammasome in Cold Ischemia-Reperfusion Associated with Liver Transplantation
3.2.1. Role of Inflammasome in Experimental Models of Ex Vivo Liver Transplantation
3.2.2. Role of Inflammasome in Experimental Models of In Vivo Liver Transplantation
3.2.3. Clinical Results of Inflammasome Activation in Liver Transplantation
4. Future Perspectives and Conclusion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AIM2 | Absent in melanoma 2 |
| AKT | Protein kinase B |
| AP-1 | Activator protein 1 |
| ASC | Apoptosis-associated speck-like protein containing a caspase recruitment domain |
| ATP | Adenosine triphosphate |
| BD | Brain death |
| BHB | β-hydroxybutyric acid |
| CARD | Carboxy-terminal caspase recruitment domain |
| Cat B | Cathepsin B |
| CCl4 | Carbon tetrachloride |
| DAMPs | Damage-associated molecular patterns |
| DCD | Cardiac-circulatory death |
| Dex | Dexmedetomidine |
| DHA | Docosahexaenoic acid |
| FOXO1 | Forkhead box protein O1 |
| HCC | Hepatocellular carcinoma |
| HMGB1 | High mobility group box 1 |
| HMP | Hypothermic machine perfusion |
| HO-1 | Heme oxygenase 1 |
| HOPE | Hypothermic oxygenated perfusion |
| HSF1 | Heat shock transcription factor 1 |
| I/R | Ischemia-reperfusion |
| IL | Interleukin |
| IL-1R | Interleukin-1 receptor |
| IL-1RA | IL-1R antagonist |
| LT | Liver transplantation |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLR | NOD-like receptors |
| NLRC4 | NLR family CARD domain-containing protein 4 |
| NLRP | NLR pyrin domain containing protein |
| NOD | Nucleotide-binding oligomerization domain |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| PAMPs | Pathogen-associated molecular patterns |
| Panx1 Inh | Pannexin-1 inhibitor |
| PH | Partial hepatectomy |
| PI3K | Phosphoinositide 3-kinase |
| PTEN | Phosphatase and tensin homolog |
| RAP1 | Repressor activator protein 1 |
| ROS | Reactive oxygen species |
| STAT3 | Signal transducer and activator of transcription 3 |
| T3 | 3,3’,5-triiodothyronine |
| TGF-β | Transforming growth factor-β |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| TXNIP | Thioredoxin-interacting protein |
| XBJ | Xuebijing |
| XBP1 | X-box-binding protein 1 |
References
- Peralta, C.; Jiménez-Castro, M.B.; Gracia-Sancho, J. Hepatic ischemia and reperfusion injury: Effects on the liver sinusoidal milieu. J. Hepatol. 2013, 59, 1094–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, P.; Li, W. Nitric oxide in liver ischemia-reperfusion injury. In Liver Pathophysiology; Muriel, P., Ed.; Elsevier: London, UK, 2017; Volume 8, pp. 125–127. [Google Scholar]
- Selzner, N.; Rudiger, H.; Graf, R.; Clavien, P. Protective strategies against ischemic injury of the liver. Gastroenterology 2003, 125, 917–936. [Google Scholar] [CrossRef]
- Jaeschke, H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am. J. Physiol. Gastrointest Liver Physiol. 2003, 284, G15–G26. [Google Scholar] [CrossRef] [PubMed]
- Montalvo-Jave, E.E.; Escalante-Tattersfield, T.; Ortega-Salgado, J.A.; Piña, E.; Geller, D.A. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J. Surg. Res. 2008, 147, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Villarreal Jr, G.; Zhang, Y.; Yu, J.X.; Liu, Y.; Tullius, S.G.; García-Cardeña, G. Flow cessation triggers endothelial dysfunction during organ cold storage conditions: Strategies for pharmacologic intervention. Transplantation 2010, 90, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Casillas-Ramírez, A.; Peralta, C. Molecular pathways in protecting the liver from ischaemia/reperfusion injury: A 2015 update. Clin. Sci. 2015, 129, 345–362. [Google Scholar] [CrossRef]
- Papadopoulos, D.; Siempis, T.; Theodorakou, E.; Tsoulfas, G. Hepatic ischemia and reperfusion injury and trauma: Current concepts. Arch. Trauma. Res. 2013, 2, 63–70. [Google Scholar] [CrossRef]
- Bilzer, M.; Gerbes, A.L. Preservation injury of the liver: Mechanisms and novel therapeutic strategies. J. Hepatol. 2000, 32, 508–515. [Google Scholar] [CrossRef]
- Mckeown, C.M.; Edwards, V.; Phillips, M.J.; Harvey, P.R.; Petrunka, C.N.; Strasberg, S.M. Sinusoidal lining cell damage: The critical injury in cold preservation ofliver allografts in the rat. Transplantation 1988, 46, 178–191. [Google Scholar] [CrossRef]
- Ikeda, T.; Yanaga, K.; Kishikawa, K.; Kakizoe, S.; Shimada, M.; Sugimachi, K. Ischemic injury in liver transplantation: Difference in injury sites between warm and cold ischemia in rats. Hepatology 1992, 16, 454–461. [Google Scholar] [CrossRef]
- Peralta, C.; Bartrons, R.; Riera, L.; Manzano, A.; Xaus, C.; Gelpí, E.; Rosello-Catafau, J. Hepatic preconditioning preserves energy metabolism during sustained ischemia. Am. J. Physiol. Gastrointest Liver Physiol. 2000, 279, G163–G171. [Google Scholar] [CrossRef] [PubMed]
- Gasbarrini, A.; Borle, A.B.; Farghali, H.; Bender, C.; Francavilla, A.; Van Thiel, D. Effect of anoxia on intracellular ATP, Na+i, Ca2+i, Mg2+i, and cytotoxicity in rat hepatocytes. J Biol. Chem. 1992, 267, 6654–6663. [Google Scholar] [PubMed]
- Caraceni, P.; Domenicali, M.; Vendemiale, G.; Grattagliano, I.; Pertosa, A.; Nardo, B.; Moselli-Labate, A.M.; Trevisani, F.; Palasciano, G.; Altomare, E.; et al. The reduced tolerance of rat fatty liver to ischemia reperfusion is associated with mitochondrial oxidative injury. J. Surg. Res. 2005, 124, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Caldwell-Kenkel, J.C.; Currin, R.T.; Tanaka, Y.; Thurman, R.G.; Lemasters, J.J. Reperfusion injury to endothelial cells following cold ischemic storage of rat livers. Hepatology 1989, 10, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Huet, P.M.; Nagaoka, M.R.; Desbiens, G.; Tarrab, E.; Brault, A.; Bralet, M.P.; Bilodeau, M. Sinusoidal endothelial cell and hepatocyte death following cold ischemia-warm reperfusion of the rat liver. Hepatology 2004, 39, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Marzi, I.; Zhong, Z.; Lemasters, J.J.; Thurman, R.G. Evidence that graft survival is not related to parenchymal cell viability in rat liver transplantation. The importance of nonparenchymal cells. Transplantation 1989, 48, 463–468. [Google Scholar] [CrossRef]
- Kukan, M.; Haddad, P.S. Role of hepatocytes and bile duct cells in preservation-reperfusion injury of liver grafts. Liver Transpl. 2001, 7, 381–400. [Google Scholar] [CrossRef]
- Ramalho, F.; Alfany-Fernandez, I.; Casillas-Ramírez, A.; Massip-Salcedo, M.; Serafín, A.; Rimola, A.; Arroyo, V.; Rodes, J.; Rosello-Catafau, J.; Peralta, C. Are angiotensin II receptor antagonists useful strategies in steatotic and nonsteatotic livers in conditions of partial hepatectomy under ischemia-reperfusion? J. Pharmacol. Exp. Ther. 2009, 329, 130–140. [Google Scholar] [CrossRef]
- Hayashi, H.; Chaudry, I.; Clemens, M.; Baue, A. Hepatic ischemia models for determining the effects of ATP-MgCl2 treatment. J. Surg. Res. 1986, 40, 167–175. [Google Scholar] [CrossRef]
- Cannistrà, M.; Ruggiero, M.; Zullo, A.; Gallelli, G.; Serafini, S.; Maria, M.; Naso, A.; Grande, R.; Serra, R.; Nardo, B. Hepatic ischemia reperfusion injury: A systematic review of literature and the role of current drugs and biomarkers. Int. J. Surg. 2016, 33 (Suppl. 1), S57–S70. [Google Scholar] [CrossRef]
- Chaudry, I.; Clemens, M.; Ohkawa, M.; Schleck, S.; Baue, A.E. Restoration of hepatocellular function and blood flow following hepatic ischemia with ATP–MgCl2. Adv. Shock Res. 1982, 8, 177–186. [Google Scholar] [PubMed]
- Hasselgren, P.; Jennische, E.; Fornander, J.; Hellman, A. No beneficial affect of ATPMgCl2 on impaired transmembrane potential and protein synthesis in liver ischemia. Acta Chir. Scand. 1982, 148, 601–607. [Google Scholar]
- Gonzalez-Flecha, B.; Cutrin, J.; Boveris, A. Time course and mechanism of oxidative stress and tissue damage in rat liver subjected to in vivo ischemia-reperfusion. J. Clin. Invest. 1993, 91, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Kawachi, S.; Hines, I.N.; Laroux, F.S.; Hoffman, J.; Bharwani, S.; Gray, L.; Leffer, D.; Grisham, M.B. Nitric oxide synthase and postischemic liver injury. Biochem. Biophys. Res. Commun. 2000, 276, 851–854. [Google Scholar] [CrossRef] [PubMed]
- Kuboki, S.; Shin, T.; Huber, N.; Eismann, T.; Galloway, E.; Schuster, R.; Blanchard, J.; Edwards, M.J.; Lentsch, A.B. Hepatocyte signaling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology 2008, 48, 1213–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Riel, W.G.; van Golen, R.F.; Reiniers, M.J.; Heger, M.; van Gulik, T.M. How much ischemia can the liver tolerate during resection? Hepatobiliary Surg. Nutr. 2016, 5, 58–71. [Google Scholar] [PubMed]
- Olthof, P.B.; van Golen, R.F.; Meijer, B.; van Beek, A.A.; Bennink, R.J.; Verheij, J.; van Gulik, T.M.; Heger, M. Warm ischemia time-dependent variation in liver damage, inflammation, and function in hepatic ischemia/reperfusion injury. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 375–385. [Google Scholar] [CrossRef]
- Fernández, L.; Heredia, N.; Grande, L.; Gómez, G.; Rimola, A.; Marco, A.; Gelpí, E.; Roselló-Catafau, J.; Peralta, C. Preconditioning protects liver and lung damage in rat liver transplantation: Role of xanthine/xanthine oxidase. Hepatology 2002, 36, 562–572. [Google Scholar] [CrossRef]
- D’Alessandro, A.M.; Kalayoglu, M.; Sollinger, H.W.; Hoffmann, R.M.; Reed, A.; Knechtle, S.J.; Pirsch, J.D.; Hafez, G.R.; Lorentzen, D.; Belzer, F.O. The predictive value of donor liver biopsies for the development of primary nonfunction after orthotopic liver transplantation. Transplantation 1991, 51, 157–163. [Google Scholar] [CrossRef]
- Loinaz, C.; Gonzalez, E.M. Marginal donors in liver transplantation. Hepatogastroenterology 2000, 47, 256–263. [Google Scholar]
- Rinella, M.E.; Alonso, E.; Rao, S.; Whitington, P.; Fryer, J.; Abecassis, M.; Superina, R.; Flamm, S.L.; Blei, A.T. Body mass index as a predictor of hepatic steatosis in living liver donors. Liver Transpl. 2001, 7, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Busuttil, R.W.; Tanaka, K. The utility of marginal donors in liver transplantation. Liver Transpl. 2003, 9, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, H.; Lewis, W.D.; Gordon, F.; Jenkins, R.; Khettry, U. Steatosis in donor and transplant liver biopsies. Hum. Pathol. 2000, 31, 1209–1213. [Google Scholar] [CrossRef] [PubMed]
- McCormack, L.; Dutkowski, P.; El-Badry, A.M.; Clavien, P.A. Liver transplantation using fatty livers: Always feasible? J. Hepatol. 2011, 54, 1055–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.C.; Fung, J.Y.; Chok, K.S.; Cheung, T.T.; Chan, A.C.; Sharr, W.W.; Dai, W.C.; Chan, S.C.; Lo, C.M. Excellent outcomes of liver transplantation using severely steatotic grafts from brain-dead donors. Liver Transpl. 2016, 22, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.A.; Berg, C.L.; Moylan, C.A. Nonalcoholic fatty liver disease: Key considerations before and after liver transplantation. Dig. Dis. Sci. 2016, 61, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Canelo, R.; Braun, F.; Sattler, B.; Klinge, B.; Lorf, T.; Ramadori, G.; Ringe, B. Is a fatty liver dangerous for transplantation? Transplant Proc. 1999, 31, 414–415. [Google Scholar] [CrossRef]
- Tashiro, H.; Kuroda, S.; Mikuriya, Y.; Ohdan, H. Ischemia-reperfusion injury in patients with fatty liver and the clinical impact of steatotic liver on hepatic surgery. Surg. Today 2014, 44, 1611–1625. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, A.L.; Lao, O.B.; Dick, A.A.; Bakthavatsalam, R.; Halldorson, J.B.; Yeh, M.M.; Upton, M.P.; Reyes, J.D.; Perkins, J.D. The biopsied donor liver: Incorporating macrosteatosis into high-risk donor assessment. Liver Transpl. 2010, 16, 874–884. [Google Scholar] [CrossRef] [PubMed]
- McCormack, L.; Petrowsky, H.; Jochum, W.; Furrer, K.; Clavien, P.A. Hepatic steatosis is a risk factor for postoperative complications after major hepatectomy: A matched case-control study. Ann. Surg. 2007, 245, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Veteläinen, R.; van Vliet, A.; Gouma, D.J.; van Gulik, T.M. Steatosis as a risk factor in liver surgery. Ann. Surg. 2007, 245, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Clavien, P.A.; Yadav, S.; Sindram, D.; Bentley, R.C. Protective effects of ischemic preconditioning for liver resection performed under inflow occlusion in humans. Ann. Surg. 2000, 232, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Behrns, K.E.; Tsiotos, G.G.; DeSouza, N.F.; Krishna, M.K.; Ludwig, J.; Nagorney, D.M. Hepatic steatosis as a potential risk factor for major hepatic resection. J. Gastrointest Surg. 1998, 2, 292–298. [Google Scholar] [CrossRef]
- de Meijer, V.E.; Kalish, B.T.; Puder, M.; Ijzermans, J.N. Systematic review and meta-analysis of steatosis as a risk factor in major hepatic resection. Br. J. Surg. 2010, 97, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Bachellier, P.; Rosso, E.; Pessaux, P.; Oussoultzoglou, E.; Nobili, C.; Panaro, F.; Jaeck, D. Risk factors for liver failure and mortality after hepatectomy associated with portal vein resection. Ann. Surg. 2011, 253, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Hamady, Z.Z.; Rees, M.; Welsh, F.K.; Toogood, G.J.; Prasad, K.R.; John, T.K.; Lodge, J.P. Fatty liver disease as a predictor of local recurrence following resection of colorectal liver metastases. Br. J. Surg. 2013, 100, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Truant, S.; Bouras, A.F.; Petrovai, G.; Buob, D.; Ernst, O.; Boleslawski, E.; Hebbar, M.; Pruvot, F.R. Volumetric gain of the liver after major hepatectomy in obese patients: A case-matched study in 84 patients. Ann. Surg. 2013, 258, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.; Torras, J.; Lladó, L.; Rafecas, A.; Serrano, T.; Lopez-Gordo, S.; Busquets, J.; Fabregat, J. The influence of steatosis on the short- and long-term results of resection of liver metastases from colorectal carcinoma. HPB (Oxford) 2016, 18, 389–896. [Google Scholar] [CrossRef] [PubMed]
- Jarnagin, W.R.; Gonen, M.; Fong, Y.; DeMatteo, R.P.; Ben-Porat, L.; Little, S.; Corvera, C.; Weber, S.; Blumgart, L.H. Improvement in perioperative outcome after hepatic resection: Analysis of 1,803 consecutive cases over the past decade. Ann. Surg. 2002, 236, 397–406. [Google Scholar] [CrossRef]
- Ijaz, S.; Yang, W.; Winslet, M.C.; Seifalian, A.M. Impairment of hepatic microcirculation in fatty liver. Microcirculation 2003, 10, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Peralta, C.; Brenner, C. Endoplasmic reticulum stress inhibition enhances liver tolerance to ischemia/reperfusion. Curr. Med. Chem. 2011, 18, 2016–2024. [Google Scholar] [CrossRef] [PubMed]
- Alfany-Fernandez, I.; Casillas-Ramirez, A.; Bintanel-Morcillo, M.; Brosnihan, K.B.; Ferrario, C.M.; Serafin, A.; Rimola, A.; Rodés, J.; Roselló-Catafau, J.; Peralta, C. Therapeutic targets in liver transplantation: Angiotensin II in nonsteatotic grafts and angiotensin-(1-7) in steatotic grafts. Am. J. Transplant 2009, 9, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Casillas-Ramírez, A.; Alfany-Fernández, I.; Massip-Salcedo, M.; Juan, M.E.; Planas, J.M.; Serafín, A.; Pallàs, M.; Rimola, A.; Rodés, J.; Peralta, C. Retinol-binding protein 4 and peroxisome proliferator-activated receptor-γ in steatotic liver transplantation. J. Pharmacol. Exp. Ther. 2011, 338, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Castro, M.B.; Elias-Miro, M.; Mendes-Braz, M.; Lemoine, A.; Rimola, A.; Rodés, J.; Casillas-Ramírez, A.; Peralta, C. Tauroursodeoxycholic acid affects PPARγ and TLR4 in Steatotic liver transplantation. Am. J. Transplant 2012, 12, 3257–3271. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Castro, M.B.; Casillas-Ramírez, A.; Mendes-Braz, M.; Massip-Salcedo, M.; Gracia-Sancho, J.; Elias-Miró, M.; Rodés, J.; Peralta, C. Adiponectin and resistin protect steatotic livers undergoing transplantation. J. Hepatol. 2013, 59, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Carrasco-Chaumel, E.; Serafín, A.; Xaus, C.; Grande, L.; Rimola, A.; Roselló-Catafau, J.; Peralta, C. Is ischemic preconditioning a useful strategy in steatotic liver transplantation? Am. J. Transplant 2004, 4, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xu, B.; Hock, C.E. Inhibition of nitric oxide synthesis by L-name exacerbates acute lung injury induced by hepatic ischemia-reperfusion. Shock 2001, 16, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Hato, S.; Urakami, A.; Yamano, T.; Uemura, T.; Ota, T.; Hirai, R.; Shimizu, N. Attenuation of liver and lung injury after hepatic ischemia and reperfusion by a cytokine-suppressive agent, FR167653. Eur. Surg. Res. 2001, 33, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Wanner, G.A.; Ertel, W.; Muller, P.; Höfer, Y.; Leiderer, R.; Menger, M.D.; Messmer, K. Liver ischemia and reperfusion induces a systemic inflammatory response through Kupffer cell activation. Shock 1996, 5, 34–40. [Google Scholar] [CrossRef]
- Watanabe, M.; Yamaguchi, K.; Chijiiwa, K.; Tanaka, M. FR167653 improves survival and pulmonary injury after partial hepatectomy under ischemia/reperfusion in rats. J. Surg. Res. 2001, 101, 146–151. [Google Scholar] [CrossRef]
- Franco-Gou, R.; Roselló-Catafau, J.; Peralta, C. Protection against lung damage in reduced-size liver transplantation. Crit. Care Med. 2006, 34, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Cornide-Petronio, M.E.; Jiménez-Castro, M.B.; Gracia-Sancho, J.; Peralta, C. Ischemic preconditioning directly or remotely applied on the liver to reduce ischemia-reperfusion injury in resections and transplantation. In Liver Disease and Surgery; Tsoulfas, G., Ed.; IntechOpen: London, UK, 2019; in press. [Google Scholar]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Núñez, G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Yao, X.; Li, H.; Xue, G.; Guo, Q.; Yang, G.; An, L.; Zhang, Y.; Meng, G. Cutting Edge: TRAF6 Mediates TLR/IL-1R Signaling-Induced Nontranscriptional Priming of the NLRP3 Inflammasome. J. Immunol. 2017, 199, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–842. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Marra, F. The inflammasome in liver disease. J. Hepatol. 2016, 65, 1055–1056. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Mehal, W.Z. Sterile inflammation in the liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef] [PubMed]
- Mohamadi, Y.; Mousavi, M.; Khanbabaei, H.; Salarinia, R.; Javankiani, S.; Hassanzadeh, G.; Momeni, F. The role of inflammasome complex in ischemia-reperfusion injury. J. Cell Biochem. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M. Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 2011, 11, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell. Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aachoui, Y.; Sagulenko, V.; Miao, E.A.; Stacey, K.J. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr. Opin. Microbiol. 2013, 16, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, G.; Csak, T. Inflammasomes in liver diseases. J. Hepatol. 2012, 57, 642–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muruve, D.A.; Pétrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poeck, H.; Bscheider, M.; Gross, O.; Finger, K.; Roth, S.; Rebsamen, M.; Hannesschläger, N.; Schlee, M.; Rothenfusser, S.; Barchet, W.; et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 2010, 11, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Mao, D.P.; Yudkovsky, N.; Bonneau, R.; Lorang, C.G.; Warren, S.E.; Leaf, I.A.; Aderem, A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2010, 107, 3076–3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Vinzing, M.; Eitel, J.; Lippmann, J.; Hocke, A.C.; Zahlten, J.; Slevogt, H.; N’guessan, P.D.; Günther, S.; Schmeck, B.; Hippenstiel, S.; et al. NAIP and Ipaf control Legionella pneumophila replication in human cells. J. Immunol. 2008, 180, 6808–6815. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Invest. 2012, 122, 3476–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Voican, C.S.; Njiké-Nakseu, M.; Boujedidi, H.; Barri-Ova, N.; Bouchet-Delbos, L.; Agostini, H.; Maitre, S.; Prévot, S.; Cassard-Doulcier, A.M.; Naveau, S.; et al. Alcohol withdrawal alleviates adipose tissue inflammation in patients with alcoholic liver disease. Liver Int. 2015, 35, 967–978. [Google Scholar] [CrossRef]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Gieling, R.G.; Wallace, K.; Han, Y.P. Interleukin-1 participates in the progression from liver injury to fibrosis. Am. J. Physiol. Gastrointest Liver Physiol. 2009, 296, G1324–G1331. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, Z.; Hou, R.; Yan, D.; Liu, C.; Chen, S.; Li, X.; Du, W. Expression of AIM2 is correlated with increased inflammation in chronic hepatitis B patients. Virol. J. 2015, 12, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imaeda, A.B.; Watanabe, A.; Sohail, M.A.; Mahmood, S.; Mohamadnejad, M.; Sutterwala, F.S.; Flavell, R.A.; Mehal, W.Z. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Invest. 2009, 119, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest. 2014, 94, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.H.; Wang, Y.Y.; Lu, J.; Zheng, Y.L.; Wu, D.M.; Li, M.Q.; Hu, B.; Zhang, Z.F.; Cheng, W.; Shan, Q. Luteoloside suppresses proliferation and metastasis of hepatocellular carcinoma cells by inhibition of NLRP3 inflammasome. PLoS ONE 2014, 9, e89961. [Google Scholar] [CrossRef] [PubMed]
- Shiffman, M.L.; Pockros, P.; McHutchison, J.G.; Schiff, E.R.; Morris, M.; Burgess, G. Clinical trial: The efficacy and safety of oral PF-03491390, a pancaspase inhibitor - a randomized placebo-controlled study in patients with chronic hepatitis C. Aliment. Pharmacol. Ther. 2010, 31, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Pockros, P.J.; Schiff, E.R.; Shiffman, M.L.; McHutchison, J.G.; Gish, R.G.; Afdhal, N.H.; Makhviladze, M.; Huyghe, M.; Hecht, D.; Oltersdorf, T.; et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology 2007, 46, 324–329. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.H.; Schipper, J.L.; Clark, A.C. The potential for caspases in drug discovery. Curr. Opin. Drug Discov. Devel. 2010, 13, 568–576. [Google Scholar]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Zhu, P.; Duan, L.; Chen, J.; Xiong, A.; Xu, Q.; Zhang, H.; Zheng, F.; Tan, Z.; Gong, F.; Fang, M. Gene silencing of NALP3 protects against liver ischemia-reperfusion injury in mice. Hum. Gene. Ther. 2011, 22, 853–864. [Google Scholar] [CrossRef]
- Jiménez-Castro, M.B.; Elias-Miró, M.; Casillas-Ramírez, A.; Peralta, C. Experimental Models in Liver Surgery. In Hepatic Surgery; Abdeldayem, H., Ed.; IntechOpen: London, UK, 2012; Volume 6, pp. 121–166. [Google Scholar]
- Fan, C.; Zwacka, R.M.; Engelhardt, J.F. Therapeutic approaches for ischemia/reperfusion injury in the liver. J. Mol. Med. 1999, 77, 577–592. [Google Scholar] [CrossRef]
- Okaya, T.; Lentsch, A.B. Hepatic expression of S32A/S36A IkappaBalpha does not reduce postischemic liver injury. J. Surg. Res. 2005, 124, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Sonnenday, C.J.; Warren, D.S.; Cooke, S.K.; Dietz, H.C.; Montgomery, R.A. A novel chimeric ribozyme vector produces potent inhibition of ICAM-1 expression on ischemic vascular endothelium. J. Gene. Med. 2004, 6, 1394–1402. [Google Scholar] [CrossRef]
- Coito, A.J.; Buelow, R.; Shen, X.D.; Amersi, F.; Moore, C.; Volk, H.D.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Heme oxygenase-1 gene transfer inhibits inducible nitric oxide synthase expression and protects genetically fat Zucker rat livers from ischemia-reperfusion injury. Transplantation 2002, 74, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Wakabayashi, G.; Takayanagi, A.; Shimazu, M.; Matsumoto, K.; Obara, H.; Shimizu, N.; Kitajima, M. Transfer of the interleukin-1 receptor antagonist gene into rat liver abrogates hepatic ischemia-reperfusion injury. Transplantation 2002, 74, 1434–1441. [Google Scholar] [CrossRef]
- Ke, B.; Lipshutz, G.S.; Kupiec-Weglinski, J.W. Gene therapy in liver ischemia and reperfusion injury. Curr. Pharm. Des. 2006, 12, 2969–2975. [Google Scholar] [CrossRef] [PubMed]
- Pachori, A.S.; Melo, L.G.; Hart, M.L.; Noiseux, N.; Zhang, L.; Morello, F.; Solomon, S.D.; Stahl, G.L.; Pratt, R.E.; Dzau, V.J. Hypoxia-regulated therapeutic gene as a preemptive treatment strategy against ischemia/reperfusion tissue injury. Proc. Natl. Acad. Sci. USA 2004, 101, 12282–12287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somia, N.; Verma, I.M. Gene therapy: Trials and tribulations. Nat. Rev. Genet. 2000, 1, 91–99. [Google Scholar] [CrossRef]
- Inoue, Y.; Shirasuna, K.; Kimura, H.; Usui, F.; Kawashima, A.; Karasawa, T.; Tago, K.; Dezaki, K.; Nishimura, S.; Sagara, J.; et al. NLRP3 regulates neutrophil functions and contributes to hepatic ischemia-reperfusion injury independently of inflammasomes. J. Immunol. 2014, 192, 4342–4351. [Google Scholar] [CrossRef]
- Kamo, N.; Ke, B.; Ghaffari, A.A.; Shen, X.D.; Busuttil, R.W.; Cheng, G.; Kupiec-Weglinski, J.W. ASC/caspase-1/IL-1β signaling triggers inflammatory responses by promoting HMGB1 induction in liver ischemia/reperfusion injury. Hepatology 2013, 58, 351–362. [Google Scholar] [CrossRef]
- Kato, A.; Gabay, C.; Okaya, T.; Lentsch, A.B. Specific role of interleukin-1 in hepatic neutrophil recruitment after ischemia/reperfusion. Am. J. Pathol. 2002, 161, 1797–1803. [Google Scholar] [CrossRef]
- Tan, Z.; Jiang, R.; Wang, X.; Wang, Y.; Lu, L.; Liu, Q.; Zheng, S.G.; Sun, B.; Ryffel, B. RORγt+IL-17+ neutrophils play a critical role in hepatic ischemia-reperfusion injury. J. Mol. Cell Biol. 2013, 5, 143–146. [Google Scholar] [CrossRef]
- Shito, M.; Wakabayashi, G.; Ueda, M.; Shimazu, M.; Shirasugi, N.; Endo, M.; Mukai, M.; Kitajima, M. Interleukin 1 receptor blockade reduces tumor necrosis factor production, tissue injury, and mortality after hepatic ischemia-reperfusion in the rat. Transplantation 1997, 63, 143–148. [Google Scholar] [CrossRef]
- Takeuchi, D.; Yoshidome, H.; Kato, A.; Ito, H.; Kimura, F.; Shimizu, H.; Ohtsuka, M.; Morita, Y.; Miyazaki, M. Interleukin 18 causes hepatic ischemia/reperfusion injury by suppressing anti-inflammatory cytokine expression in mice. Hepatology 2004, 39, 699–710. [Google Scholar] [CrossRef]
- Huang, H.; Chen, H.W.; Evankovich, J.; Yan, W.; Rosborough, B.R.; Nace, G.W.; Ding, Q.; Loughran, P.; Beer-Stolz, D.; Billiar, T.R.; et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J. Immunol. 2013, 191, 2665–2679. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, S.J.; Lee, S.M. Activation of NLRP3 and AIM2 inflammasomes in Kupffer cells in hepatic ischemia/reperfusion. FEBS J. 2015, 282, 259–270. [Google Scholar] [CrossRef]
- Yue, S.; Zhu, J.; Zhang, M.; Li, C.; Zhou, X.; Zhou, M.; Ke, M.; Busuttil, R.W.; Ying, Q.L.; Kupiec-Weglinski, J.W.; et al. The myeloid heat shock transcription factor 1/β-catenin axis regulates NLR family, pyrin domain-containing 3 inflammasome activation in mouse liver ischemia/reperfusion injury. Hepatology 2016, 64, 1683–1698. [Google Scholar] [CrossRef]
- Vargas, R.; Videla, L.A. Thyroid hormone suppresses ischemia-reperfusion-induced liver NLRP3 inflammasome activation: Role of AMP-activated protein kinase. Immunol. Lett. 2017, 184, 92–97. [Google Scholar] [CrossRef]
- Fernández, V.; Tapia, G.; Varela, P.; Castillo, I.; Mora, C.; Moya, F.; Orellana, M.; Videla, L.A. Redox up-regulated expression of rat liver manganese superoxide dismutase and Bcl-2 by thyroid hormone is associated with inhibitor of kappaB-alpha phosphorylation and nuclear factor-kappaB activation. J. Endocrinol. 2005, 186, 539–547. [Google Scholar] [CrossRef]
- Videla, L.A.; Cornejo, P.; Castillo, I.; Romanque, P. Thyroid hormone-induced regulatory interrelations in rat liver Nrf2-Keap1 signaling related toantioxidant enzyme expression. In Advances in Medicine and Biology; Berhardt, L.A., Ed.; Nova Science Publishers Inc.: New York, NY, USA, 2012; Volume 55, pp. 179–191. [Google Scholar]
- Tapia, G.; Fernández, V.; Pino, C.; Ardiles, L.; Videla, L.A. The acute-phaseresponse of the liver in relation to thyroid hormone-induced redox signalling. Free Radic. Biol. Med. 2006, 40, 1628–1635. [Google Scholar] [CrossRef]
- Fernández, V.; Reyes, S.; Bravo, S.; Sepúlveda, R.; Romanque, P.; Santander, G.; Castillo, I.; Varela, P.; Tapia, G.; Videla, L.A. Involvement of Kupffer cell-dependent signaling in T3-inducedhepatocyte proliferation in vivo. Biol. Chem. 2007, 388, 831–837. [Google Scholar] [CrossRef]
- Hwang, J.K.; Yu, H.N.; Noh, E.M.; Kim, J.M.; Hong, O.Y.; Youn, H.J.; Jung, S.H.; Kwon, K.B.; Kim, J.S.; Lee, Y.R. DHA blocks TPA-induced cell invasion by inhibiting MMP-9 expression via suppression of the PPAR-gamma/NF-kappaB pathway in MCF-7 cells. Oncol. Lett. 2017, 13, 243–249. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, F.; Cao, Y.; Zhang, J.; Shi, P.; Sun, X.; Zhang, F.; Tong, L. DHA attenuates hepatic ischemia reperfusion injury by inhibiting pyroptosis and activating PI3K/Akt pathway. Eur. J. Pharmacol. 2018, 835, 1–10. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Hornung, V. Pore formation by GSDMD is the effector mechanism of pyroptosis. EMBO J. 2016, 35, 2167–2169. [Google Scholar] [CrossRef] [Green Version]
- Yaron, J.R.; Chen, H.; Ambadapadi, S.; Zhang, L.; Tafoya, A.M.; Munk, B.H.; Wakefield, D.N.; Fuentes, J.; Marques, B.J.; Harripersaud, K.; et al. Serp-2, a virus-derived apoptosis and inflammasome inhibitor, attenuates liver ischemia-reperfusion injury in mice. J. Inflamm. 2019, 16, 12. [Google Scholar] [CrossRef]
- Yang, M.; Antoine, D.J.; Weemhoff, J.L.; Jenkins, R.E.; Farhood, A.; Park, B.K.; Jaeschke, H. Biomarkers distinguish apoptotic and necrotic cell death during hepatic ischemia/reperfusion injury in mice. Liver Transpl. 2014, 20, 1372–1382. [Google Scholar] [CrossRef]
- Liu, X.; Hu, Z.; Zhou, B.; Li, X.; Tao, R. Chinese Herbal Preparation Xuebijing Potently Inhibits Inflammasome Activation in Hepatocytes and Ameliorates Mouse Liver Ischemia-Reperfusion Injury. PLoS ONE 2015, 10, e0131436. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Verweij, M.; Brand, K.; van de Ven, M.; Goemaere, N.; van den Engel, S.; Chu, T.; Forrer, F.; Müller, C.; de Jong, M.; et al. Short-term dietary restriction and fasting precondition against ischemia reperfusion injury in mice. Aging Cell 2010, 9, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, T.; Uchida, Y.; Kadono, K.; Hirao, H.; Kawasoe, J.; Watanabe, T.; Ueda, S.; Okajima, H.; Terajima, H.; Uemoto, S. Up-regulation of FOXO1 and reduced inflammation by β-hydroxybutyric acid are essential diet restriction benefits against liver injury. Proc. Natl. Acad. Sci. USA 2019, 116, 13533–13542. [Google Scholar] [CrossRef]
- Verweij, M.; van Ginhoven, T.M.; Mitchell, J.R.; Sluiter, W.; van den Engel, S.; Roest, H.P.; Torabi, E.; Ijzermans, J.N.; Hoeijmakers, J.H.; de Bruin, R.W. Preoperative fasting protects mice against hepatic ischemia/reperfusion injury: Mechanisms and effects on liver regeneration. Liver Transpl. 2011, 17, 695–704. [Google Scholar] [CrossRef]
- Qin, J.; Zhou, J.; Dai, X.; Zhou, H.; Pan, X.; Wang, X.; Zhang, F.; Rao, J.; Lu, L. Short-term starvation attenuates liver ischemia-reperfusion injury (IRI) by Sirt1-autophagy signaling in mice. Am. J. Transl. Res. 2016, 8, 3364–3375. [Google Scholar]
- Rickenbacher, A.; Jang, J.H.; Limani, P.; Ungethüm, U.; Lehmann, K.; Oberkofler, C.E.; Weber, A.; Graf, R.; Humar, B.; Clavien, P.A. Fasting protects liver from ischemic injury through Sirt1-mediated downregulation of circulating HMGB1 in mice. J. Hepatol. 2014, 61, 301–308. [Google Scholar] [CrossRef]
- Ando, T.; Ito, H.; Kanbe, A.; Hara, A.; Seishima, M. Deficiency of NALP3 Signaling Impairs Liver Regeneration After Partial Hepatectomy. Inflammation 2017, 40, 1717–1725. [Google Scholar] [CrossRef]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef]
- Yamada, Y.; Kirillova, I.; Peschon, J.J.; Fausto, N. Initiation of liver growth by tumor necrosis factor: Deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 1441–1446. [Google Scholar] [CrossRef] [Green Version]
- Böhm, F.; Köhler, U.A.; Speicher, T.; Werner, S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol. Med. 2010, 2, 294–305. [Google Scholar] [CrossRef]
- Lv, M.; Zeng, H.; He, Y.; Zhang, J.; Tan, G. Dexmedetomidine promotes liver regeneration in mice after 70% partial hepatectomy by suppressing NLRP3 inflammasome not TLR4/NFκB. Int. Immunopharmacol. 2018, 54, 46–51. [Google Scholar] [CrossRef]
- Yu, S.X.; Chen, W.; Hu, X.Z.; Feng, S.Y.; Li, K.Y.; Qi, S.; Lei, Q.Q.; Hu, G.Q.; Li, N.; Zhou, F.H.; et al. Liver X receptors agonists suppress NLRP3 inflammasome activation. Cytokine 2017, 91, 30–37. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- DeSantis, D.A.; Ko, C.W.; Wang, L.; Lee, P.; Croniger, C.M. Constitutive Activation of the Nlrc4 Inflammasome Prevents Hepatic Fibrosis and Promotes Hepatic Regeneration after Partial Hepatectomy. Mediators Inflamm. 2015, 2015, 909827. [Google Scholar] [CrossRef]
- Casillas-Ramírez, A.; Escobedo-Medina, S.G.; Cordero-Pérez, P.; Jiménez-Castro, M.B.; Peralta, C. Ischemia-reperfusion injury and oxidative stress. In Gastrointestinal Tissue: Oxidative Stress and Dietary Antioxidants; Gracia-Gracia, J., Salvadó, J., Eds.; Elsevier: London, UK, 2016; pp. 141–154. [Google Scholar]
- Liu, H.; Lo, C.M.; Yeung, O.W.H.; Li, C.X.; Liu, X.B.; Qi, X.; Ng, K.T.P.; Liu, J.; Ma, Y.Y.; Lam, Y.F.; et al. NLRP3 inflammasome induced liver graft injury through activation of telomere-independent RAP1/KC axis. J. Pathol. 2017, 242, 284–296. [Google Scholar] [CrossRef]
- Ling, C.C.; Ng, K.T.; Shao, Y.; Geng, W.; Xiao, J.W.; Liu, H.; Li, C.X.; Liu, X.B.; Ma, Y.Y.; Yeung, W.H.; et al. Post-transplant endothelial progenitor cell mobilization via CXCL10/CXCR3 signaling promotes liver tumor growth. J. Hepatol. 2014, 60, 103–109. [Google Scholar] [CrossRef]
- Sonohara, F.; Inokawa, Y.; Kanda, M.; Nishikawa, Y.; Yamada, S.; Fujii, T.; Sugimoto, H.; Kodera, Y.; Nomoto, S. Association of Inflammasome Components in Background Liver with Poor Prognosis After Curatively-resected Hepatocellular Carcinoma. Anticancer Res. 2017, 37, 293–300. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Ye, S.; Zeng, C.; Xue, S.; Hu, X.; Zhang, X.; Gao, S.; Xiong, Y.; He, X.; Vivalda, S.; et al. Hypothermic oxygenated perfusion (HOPE) attenuates ischemia/reperfusion injury in the liver through inhibition of the TXNIP/NLRP3 inflammasome pathway in a rat model of donation after cardiac death. FASEB J. 2018, in press. [Google Scholar] [CrossRef]
- Schlegel, A.; Kron, P.; Dutkowski, P. Hypothermic machine perfusion in liver transplantation. Curr. Opin. Organ. Transplant 2016, 21, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Liu, Y.F.; Yang, L. Advantages of dual hypothermic oxygenated machine perfusion over simple cold storage in the preservation of liver from porcine donors after cardiac death. Clin Transplant 2015, 29, 820–828. [Google Scholar] [CrossRef]
- Westerkamp, A.C.; Karimian, N.; Matton, A.P.; Mahboub, P.; van Rijn, R.; Wiersema-Buist, J.; de Boer, M.T.; Leuvenink, H.G.; Gouw, A.S.; Lisman, T.; et al. Oxygenated hypothermic machine perfusion after static cold storage improves hepatobiliary functionof extendedcriteriadonorlivers. Transplantation 2016, 100, 825–835. [Google Scholar] [CrossRef]
- Hong, B.J.; Liu, H.; Wang, Z.H.; Zhu, Y.X.; Su, L.Y.; Zhang, M.X.; Xu, K.; Chen, J.Z. Inflammasome activation involved in early inflammation reaction after liver transplantation. Immunol. Lett. 2017, 190, 265–271. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Zhang, W.; Gong, J.; Cheng, Y. Pre-conditioning with tanshinone IIA attenuates the ischemia/reperfusion injury caused by liver grafts via regulation of HMGB1 in rat Kupffer cells. Biomed. Pharmacother. 2017, 89, 1392–1400. [Google Scholar] [CrossRef]
- Sadowsky, D.; Zamora, R.; Barclay, D.; Yin, J.; Fontes, P.; Vodovotz, Y. Machine Perfusion of Porcine Livers with Oxygen-Carrying Solution Results in Reprogramming of Dynamic Inflammation Networks. Front Pharmacol. 2016, 7, 413. [Google Scholar] [CrossRef]
- Yu, Y.; Cheng, Y.; Pan, Q.; Zhang, Y.J.; Jia, D.G.; Liu, Y.F. Effect of the Selective NLRP3 Inflammasome Inhibitor mcc950 on Transplantation Outcome in a Pig Liver Transplantation Model with Organs From Donors After Circulatory Death Preserved by Hypothermic Machine Perfusion. Transplantation 2019, 103, 353–362. [Google Scholar] [CrossRef]
- Karp, S.J.; Johnson, S.; Evenson, A.; Curry, M.P.; Manning, D.; Malik, R.; Lake-Bakaar, G.; Lai, M.; Hanto, D. Minimising cold ischaemic time is essential in cardiac death donor-associated liver transplantation. HPB Oxford 2011, 13, 411–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.; Dominguez-Gil, B.; Greer, D.M.; Manara, A.R.; Souter, M.J. Organ donation after circulatory death: Current status and future potential. Intensive Care Med. 2019, 45, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.X.; Na, N.; Li, J.J.; Fan, L.; Weng, R.H.; Jiang, N. Outcomes of Controlled Donation After Cardiac Death Compared with Donation After Brain Death in Liver Transplantation: A Systematic Review and Meta-analysis. Transplant Proc. 2018, 50, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Selzner, M.; Clavien, P.A. Fatty liver in liver transplantation and surgery. Semin. Liver Dis. 2001, 21, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, H.; Li, Z.; Shi, B.; Wang, P.; Wang, C.; Fan, J.; Sun, H.; Wang, P.; Qin, X.; et al. C7 genotype of the donor may predict early bacterial infection after liver transplantation. Sci. Rep. 2016, 6, 24121. [Google Scholar] [CrossRef]
- Baccarani, U.; Isola, M.; Adani, G.L.; Avellini, C.; Lorenzin, D.; Rossetto, A.; Currò, G.; Comuzzi, C.; Toniutto, P.; Risaliti, A.; et al. Steatosis of the hepatic graft as a risk factor for post-transplant biliary complications. Clin. Transplant 2010, 24, 631–635. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Castro, M.B.; Cornide-Petronio, M.E.; Gracia-Sancho, J.; Peralta, C. Inflammasome-Mediated Inflammation in Liver Ischemia-Reperfusion Injury. Cells 2019, 8, 1131. https://doi.org/10.3390/cells8101131
Jiménez-Castro MB, Cornide-Petronio ME, Gracia-Sancho J, Peralta C. Inflammasome-Mediated Inflammation in Liver Ischemia-Reperfusion Injury. Cells. 2019; 8(10):1131. https://doi.org/10.3390/cells8101131
Chicago/Turabian StyleJiménez-Castro, Mónica B., María Eugenia Cornide-Petronio, Jordi Gracia-Sancho, and Carmen Peralta. 2019. "Inflammasome-Mediated Inflammation in Liver Ischemia-Reperfusion Injury" Cells 8, no. 10: 1131. https://doi.org/10.3390/cells8101131
APA StyleJiménez-Castro, M. B., Cornide-Petronio, M. E., Gracia-Sancho, J., & Peralta, C. (2019). Inflammasome-Mediated Inflammation in Liver Ischemia-Reperfusion Injury. Cells, 8(10), 1131. https://doi.org/10.3390/cells8101131