The Molecular Links between Cell Death and Inflammasome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
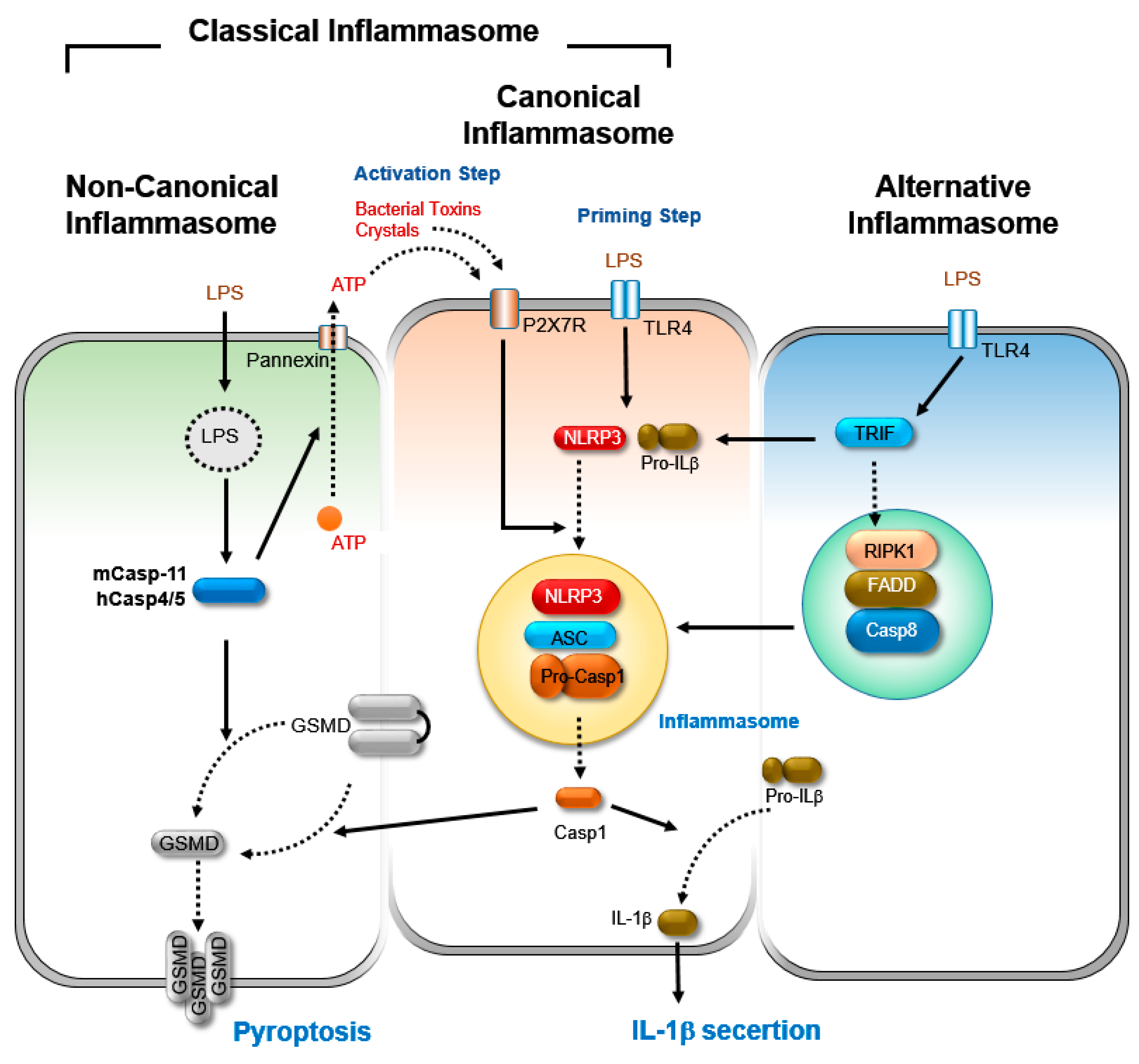
2. Inflammasome
2.1. Canonical Inflammasome Activation
2.2. Non-Canonical Inflammasome Activation
2.3. Alternative Inflammasome Activation
3. Programmed Cell Death
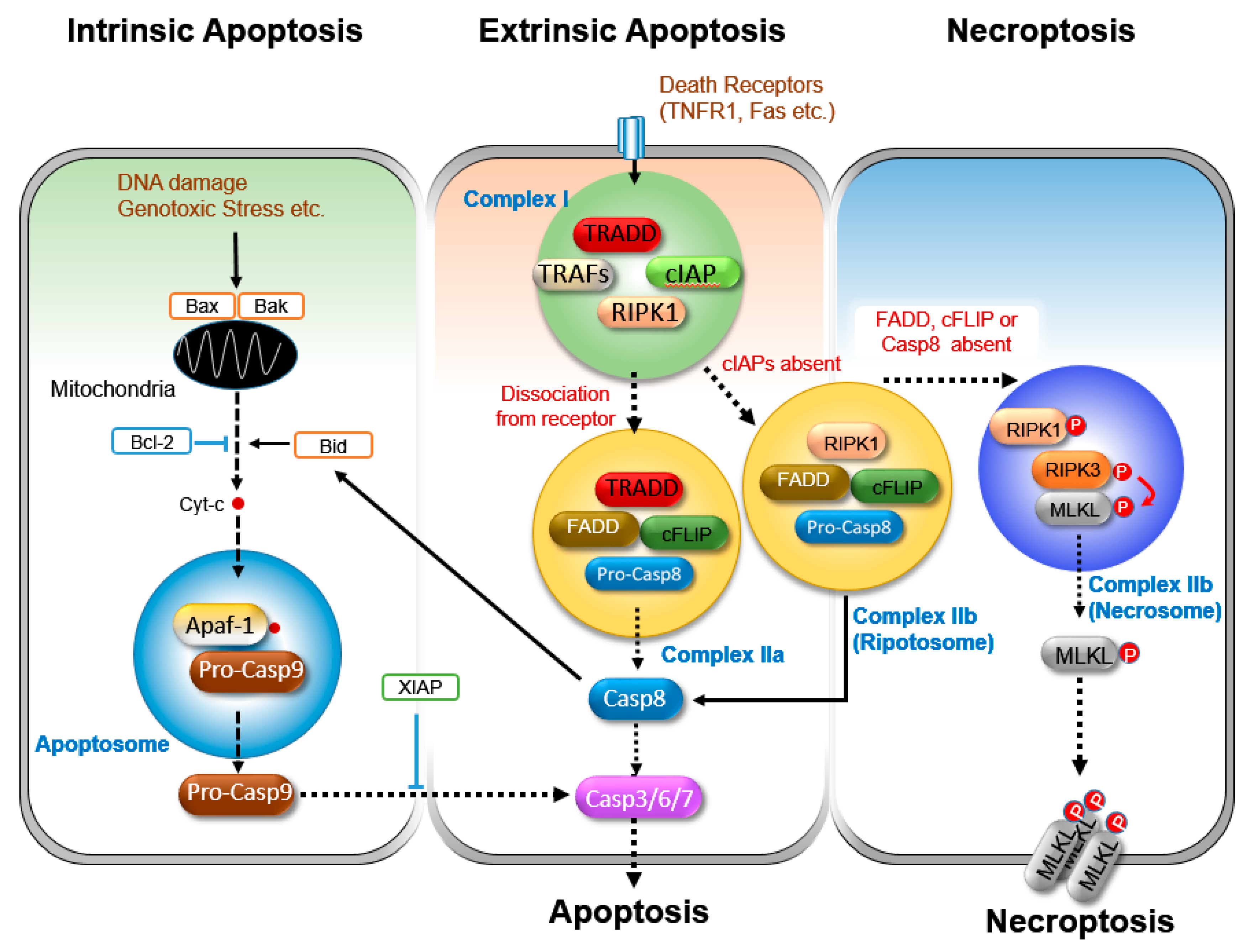
3.1. Apoptosis
3.1.1. Intrinsic Apoptosis
3.1.2. Extrinsic Apoptosis
3.1.3. Regulation of Extrinsic Apoptosis
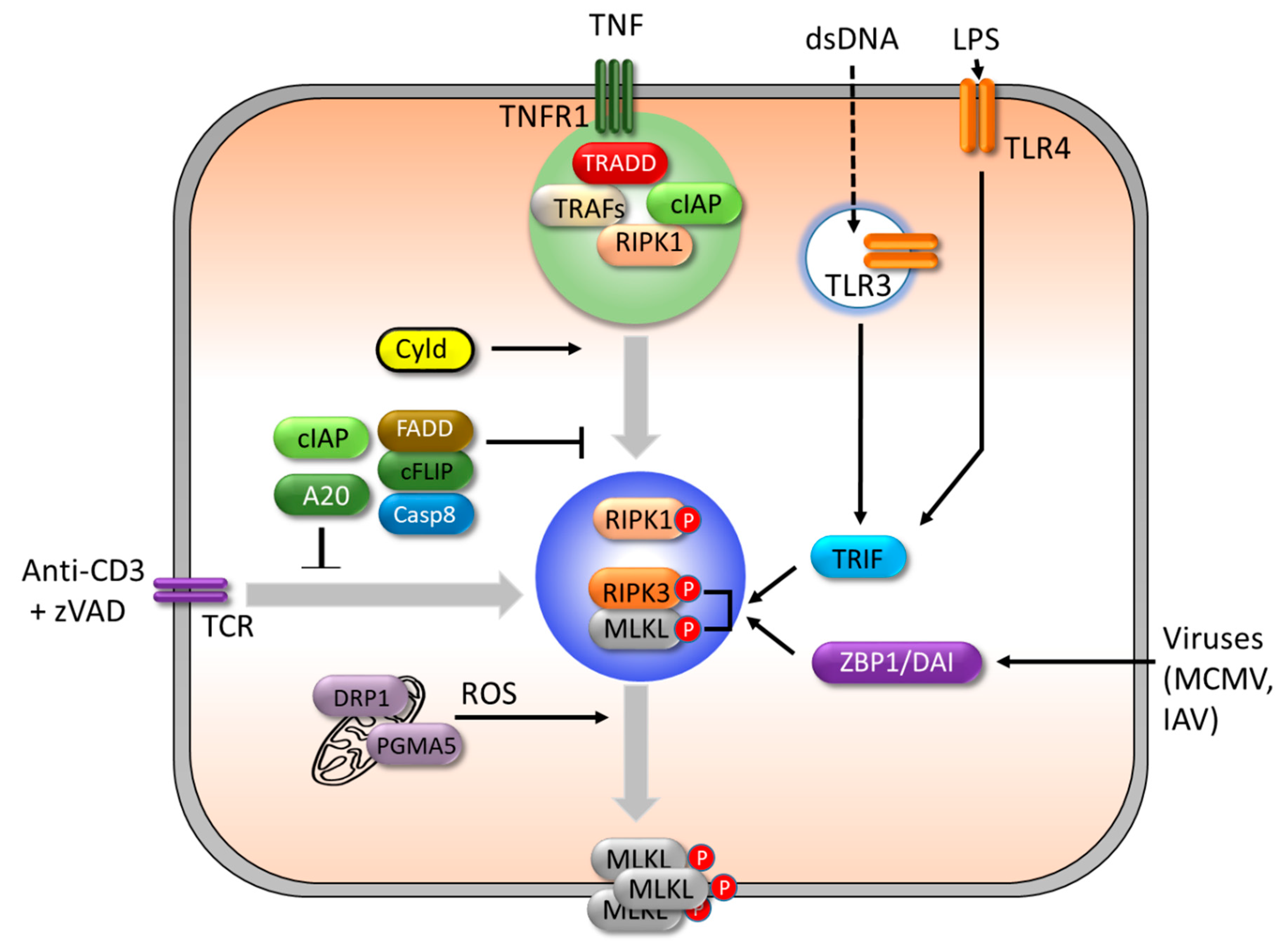
3.2. Necroptosis
3.2.1. Initiation of Necroptosis
3.2.2. Execution of Necroptosis
3.2.3. Regulation of Necroptosis.
4. Cell Death Regulators Associated with the Inflammasome.
4.1. Caspase-8 and cFLIP
4.2. A20
4.3. RIPK1/3
4.4. MLKL
4.5. PGAM5/DRP1
5. Conclusions
Funding
Conflicts of Interest
References
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Cordoba-Rodriguez, R.; Fang, H.; Lankford, C.S.; Frucht, D.M. Anthrax lethal toxin rapidly activates caspase-1/ICE and induces extracellular release of interleukin (IL)-1beta and IL-18. J. Biol. Chem. 2004, 279, 20563–20566. [Google Scholar] [CrossRef]
- Cirelli, K.M.; Gorfu, G.; Hassan, M.A.; Printz, M.; Crown, D.; Leppla, S.H.; Grigg, M.E.; Saeij, J.P.; Moayeri, M. Inflammasome sensor NLRP1 controls rat macrophage susceptibility to Toxoplasma gondii. PLoS Pathog. 2014, 10, e1003927. [Google Scholar] [CrossRef] [PubMed]
- Ewald, S.E.; Chavarria-Smith, J.; Boothroyd, J.C. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 2014, 82, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.C.; Mogridge, J. Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect. Immun. 2013, 81, 570–579. [Google Scholar] [CrossRef]
- Masters, S.L.; Gerlic, M.; Metcalf, D.; Preston, S.; Pellegrini, M.; O’Donnell, J.A.; McArthur, K.; Baldwin, T.M.; Chevrier, S.; Nowell, C.J.; et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 2012, 37, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Bergstralh, D.T.; O’Connor, W.; Morrison, A.C.; Taxman, D.J.; Duncan, J.A.; Barnoy, S.; Venkatesan, M.M.; Flavell, R.A.; Deshmukh, M.; et al. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2007, 2, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.; Sugimoto, N.; Tobe, T. Enteropathogenic Escherichia coli Uses NleA to Inhibit NLRP3 Inflammasome Activation. PLoS Pathog. 2015, 11, e1005121. [Google Scholar] [CrossRef]
- Liu, Z.; Zaki, M.H.; Vogel, P.; Gurung, P.; Finlay, B.B.; Deng, W.; Lamkanfi, M.; Kanneganti, T.D. Role of inflammasomes in host defense against Citrobacter rodentium infection. J. Biol. Chem. 2012, 287, 16955–16964. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef] [Green Version]
- Kanneganti, T.D.; Body-Malapel, M.; Amer, A.; Park, J.H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.W.; Alnemri, E.S. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef]
- Chen, I.Y.; Ichinohe, T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015, 23, 55–63. [Google Scholar] [CrossRef]
- da Costa, L.S.; Outlioua, A.; Anginot, A.; Akarid, K.; Arnoult, D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis. 2019, 10, 346. [Google Scholar] [CrossRef]
- Lightfield, K.L.; Persson, J.; Brubaker, S.W.; Witte, C.E.; von Moltke, J.; Dunipace, E.A.; Henry, T.; Sun, Y.H.; Cado, D.; Dietrich, W.F.; et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 2008, 9, 1171–1178. [Google Scholar] [CrossRef]
- Miao, E.A.; Alpuche-Aranda, C.M.; Dors, M.; Clark, A.E.; Bader, M.W.; Miller, S.I.; Aderem, A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 2006, 7, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Amer, A.; Body-Malapel, M.; Kanneganti, T.D.; Ozoren, N.; Jagirdar, R.; Inohara, N.; Vandenabeele, P.; Bertin, J.; Coyle, A.; et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 2006, 7, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Mao, D.P.; Yudkovsky, N.; Bonneau, R.; Lorang, C.G.; Warren, S.E.; Leaf, I.A.; Aderem, A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2010, 107, 3076–3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Rayamajhi, M.; Zak, D.E.; Chavarria-Smith, J.; Vance, R.E.; Miao, E.A. Cutting edge: Mouse NAIP1 detects the type III secretion system needle protein. J. Immunol. 2013, 191, 3986–3989. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Burgk, J.L.; Chauhan, D.; Schmidt, T.; Ebert, T.S.; Reinhardt, J.; Endl, E.; Hornung, V. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J. Biol. Chem. 2016, 291, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef]
- Jones, J.W.; Kayagaki, N.; Broz, P.; Henry, T.; Newton, K.; O’Rourke, K.; Chan, S.; Dong, J.; Qu, Y.; Roose-Girma, M.; et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. USA 2010, 107, 9771–9776. [Google Scholar] [CrossRef]
- Warren, S.E.; Armstrong, A.; Hamilton, M.K.; Mao, D.P.; Leaf, I.A.; Miao, E.A.; Aderem, A. Cutting edge: Cytosolic bacterial DNA activates the inflammasome via Aim2. J. Immunol. 2010, 185, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taxman, D.J.; Huang, M.T.; Ting, J.P. Inflammasome inhibition as a pathogenic stealth mechanism. Cell Host Microbe 2010, 8, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Modulation of inflammasome pathways by bacterial and viral pathogens. J. Immunol. 2011, 187, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, G.; Leon-Juarez, M.; Garcia-Cordero, J.; Meza-Sanchez, D.E.; Cedillo-Barron, L. Inflammasomes and its importance in viral infections. Immunol. Res. 2016, 64, 1101–1117. [Google Scholar] [CrossRef] [Green Version]
- Komada, T.; Muruve, D.A. The role of inflammasomes in kidney disease. Nat. Rev. Nephrol. 2019, 15, 501–520. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Vince, J.E.; Silke, J. The intersection of cell death and inflammasome activation. Cell Mol. Life Sci. 2016, 73, 2349–2367. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.N.; Wang, C.; Willette-Brown, J.; Herjan, T.; Gulen, M.F.; Zhou, H.; Bulek, K.; Franchi, L.; Sato, T.; Alnemri, E.S.; et al. IKKalpha negatively regulates ASC-dependent inflammasome activation. Nat. Commun. 2014, 5, 4977. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Lamkanfi, M. K(+) drops tilt the NLRP3 inflammasome. Immunity 2013, 38, 1085–1088. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Nunez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Vanaja, S.K.; Waggoner, L.; Sokolovska, A.; Becker, C.; Stuart, L.M.; Leong, J.M.; Fitzgerald, K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 2012, 150, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef]
- Gavrilin, M.A.; Bouakl, I.J.; Knatz, N.L.; Duncan, M.D.; Hall, M.W.; Gunn, J.S.; Wewers, M.D. Internalization and phagosome escape required for Francisella to induce human monocyte IL-1beta processing and release. Proc. Natl. Acad. Sci. USA 2006, 103, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Gavrilin, M.A.; Mitra, S.; Seshadri, S.; Nateri, J.; Berhe, F.; Hall, M.W.; Wewers, M.D. Pyrin critical to macrophage IL-1beta response to Francisella challenge. J. Immunol. 2009, 182, 7982–7989. [Google Scholar] [CrossRef] [PubMed]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Gaidt, M.M.; Hornung, V. Alternative inflammasome activation enables IL-1beta release from living cells. Curr. Opin. Immunol. 2017, 44, 7–13. [Google Scholar] [CrossRef]
- Renehan, A.G.; Booth, C.; Potten, C.S. What is apoptosis, and why is it important? BMJ 2001, 322, 1536–1538. [Google Scholar] [CrossRef] [Green Version]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, A.M. The apoptosome: Heart and soul of the cell death machine. Neoplasia 1999, 1, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.M.; Adrain, C.; Duriez, P.J.; Creagh, E.M.; Martin, S.J. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J. 2004, 23, 2134–2145. [Google Scholar] [CrossRef] [Green Version]
- Esposti, M.D. The roles of Bid. Apoptosis 2002, 7, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.E.; Krammer, P.H. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr. Opin. Immunol. 1998, 10, 545–551. [Google Scholar] [CrossRef]
- Suliman, A.; Lam, A.; Datta, R.; Srivastava, R.K. Intracellular mechanisms of TRAIL: Apoptosis through mitochondrial-dependent and -independent pathways. Oncogene 2001, 20, 2122–2133. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Shu, H.B.; Pan, M.G.; Goeddel, D.V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 1996, 84, 299–308. [Google Scholar] [CrossRef]
- Zheng, L.; Bidere, N.; Staudt, D.; Cubre, A.; Orenstein, J.; Chan, F.K.; Lenardo, M. Competitive control of independent programs of tumor necrosis factor receptor-induced cell death by TRADD and RIP1. Mol. Cell Biol. 2006, 26, 3505–3513. [Google Scholar] [CrossRef] [PubMed]
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the immune system. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Silke, J. The regulation of TNF signalling: What a tangled web we weave. Curr. Opin. Immunol. 2011, 23, 620–626. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- McQuade, T.; Cho, Y.; Chan, F.K. Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem. J. 2013, 456, 409–415. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.X.; He, Y.W. A role for c-FLIP(L) in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death Differ. 2013, 20, 188–197. [Google Scholar] [CrossRef]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.T.; Onizawa, M.; Hammer, G.E.; Turer, E.E.; Yin, Q.; Damko, E.; Agelidis, A.; Shifrin, N.; Advincula, R.; Barrera, J.; et al. Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity 2013, 38, 896–905. [Google Scholar] [CrossRef]
- He, K.L.; Ting, A.T. A20 inhibits tumor necrosis factor (TNF) alpha-induced apoptosis by disrupting recruitment of TRADD and RIP to the TNF receptor 1 complex in Jurkat T cells. Mol. Cell Biol. 2002, 22, 6034–6045. [Google Scholar] [CrossRef]
- Hitomi, J.; Christofferson, D.E.; Ng, A.; Yao, J.; Degterev, A.; Xavier, R.J.; Yuan, J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008, 135, 1311–1323. [Google Scholar] [CrossRef]
- Moquin, D.M.; McQuade, T.; Chan, F.K. CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE 2013, 8, e76841. [Google Scholar] [CrossRef]
- O’Donnell, M.A.; Legarda-Addison, D.; Skountzos, P.; Yeh, W.C.; Ting, A.T. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr. Biol. 2007, 17, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israel, A.; Wallach, D.; Courtois, G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Brummelkamp, T.R.; Nijman, S.M.; Dirac, A.M.; Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 2003, 424, 797–801. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, pdb–prot087379. [Google Scholar] [CrossRef]
- Quan, L.T.; Caputo, A.; Bleackley, R.C.; Pickup, D.J.; Salvesen, G.S. Granzyme B is inhibited by the cowpox virus serpin cytokine response modifier A. J. Biol. Chem. 1995, 270, 10377–10379. [Google Scholar] [CrossRef]
- Zhou, Q.; Snipas, S.; Orth, K.; Muzio, M.; Dixit, V.M.; Salvesen, G.S. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J. Biol. Chem. 1997, 272, 7797–7800. [Google Scholar] [CrossRef]
- Chan, F.K.; Shisler, J.; Bixby, J.G.; Felices, M.; Zheng, L.; Appel, M.; Orenstein, J.; Moss, B.; Lenardo, M.J. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem. 2003, 278, 51613–51621. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, S.Q.; Liang, Y.; Zhou, X.; Chen, W.; Li, L.; Wu, J.; Zhuang, Q.; Chen, C.; Li, J.; et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 2015, 17, 229–242. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Liu, S.; Yu, X.; Li, L.; Shi, C.; He, W.; Li, J.; Xu, L.; Hu, Z.; et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. USA 2014, 111, 15438–15443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Omoto, S.; Harris, P.A.; Finger, J.N.; Bertin, J.; Gough, P.J.; Kaiser, W.J.; Mocarski, E.S. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 2015, 17, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Schock, S.N.; Chandra, N.V.; Sun, Y.; Irie, T.; Kitagawa, Y.; Gotoh, B.; Coscoy, L.; Winoto, A. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ. 2017, 24, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue-Gervais, I.G.; Labbe, K.; Dagenais, M.; Dupaul-Chicoine, J.; Champagne, C.; Morizot, A.; Skeldon, A.; Brincks, E.L.; Vidal, S.M.; Griffith, T.S.; et al. Cellular inhibitor of apoptosis protein cIAP2 protects against pulmonary tissue necrosis during influenza virus infection to promote host survival. Cell Host Microbe 2014, 15, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Polykratis, A.; Hermance, N.; Zelic, M.; Roderick, J.; Kim, C.; Van, T.M.; Lee, T.H.; Chan, F.K.M.; Pasparakis, M.; Kelliher, M.A. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 2014, 193, 1539–1543. [Google Scholar] [CrossRef]
- Koehler, H.; Cotsmire, S.; Langland, J.; Kibler, K.V.; Kalman, D.; Upton, J.W.; Mocarski, E.S.; Jacobs, B.L. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc. Natl. Acad. Sci. USA 2017, 114, 11506–11511. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef]
- Mack, C.; Sickmann, A.; Lembo, D.; Brune, W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc. Natl. Acad. Sci. USA 2008, 105, 3094–3099. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J. Biol. Chem. 2008, 283, 16966–16970. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010, 7, 302–313. [Google Scholar] [CrossRef]
- Daley-Bauer, L.P.; Roback, L.; Crosby, L.N.; McCormick, A.L.; Feng, Y.; Kaiser, W.J.; Mocarski, E.S. Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc. Natl. Acad. Sci. USA 2017, 114, E2786–E2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, Y.; Peng, S.; Yu, X.; Li, W.; Shi, F.; Luo, X.; Tang, M.; Tan, Z.; Bode, A.M.; et al. Epstein-Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination. Cell Death Dis. 2018, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Deng, B.; Liao, Y.; Shan, L.; Yin, F.; Wang, Z.; Zeng, H.; Zuo, D.; Hua, Y.; Cai, Z. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 2013, 13, 580. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, E.A. Phospho-DeltaNp63alpha-responsive microRNAs contribute to the regulation of necroptosis in squamous cell carcinoma upon cisplatin exposure. FEBS Lett. 2015, 589, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Koo, G.B.; Morgan, M.J.; Lee, D.G.; Kim, W.J.; Yoon, J.H.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Hong, S.S.; et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184. [Google Scholar] [CrossRef] [PubMed]
- Kitur, K.; Parker, D.; Nieto, P.; Ahn, D.S.; Cohen, T.S.; Chung, S.; Wachtel, S.; Bueno, S.; Prince, A. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015, 11, e1004820. [Google Scholar] [CrossRef]
- Hsu, H.; Huang, J.; Shu, H.B.; Baichwal, V.; Goeddel, D.V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 1996, 4, 387–396. [Google Scholar] [CrossRef]
- Kelliher, M.A.; Grimm, S.; Ishida, Y.; Kuo, F.; Stanger, B.Z.; Leder, P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 1998, 8, 297–303. [Google Scholar] [CrossRef]
- Dannappel, M.; Vlantis, K.; Kumari, S.; Polykratis, A.; Kim, C.; Wachsmuth, L.; Eftychi, C.; Lin, J.; Corona, T.; Hermance, N.; et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 2014, 513, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Newton, K.; Dugger, D.L.; Wickliffe, K.E.; Kapoor, N.; de Almagro, M.C.; Vucic, D.; Komuves, L.; Ferrando, R.E.; French, D.M.; Webster, J.; et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 2014, 343, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Orozco, S.; Yatim, N.; Werner, M.R.; Tran, H.; Gunja, S.Y.; Tait, S.W.; Albert, M.L.; Green, D.R.; Oberst, A. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014, 21, 1511–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, C.P.; Weinlich, R.; Rodriguez, D.A.; Cripps, J.G.; Quarato, G.; Gurung, P.; Verbist, K.C.; Brewer, T.L.; Llambi, F.; Gong, Y.N.; et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 2014, 157, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, X.; McQuade, T.; Li, J.; Chan, F.K.; Zhang, J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 2011, 471, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [PubMed]
- Maelfait, J.; Liverpool, L.; Bridgeman, A.; Ragan, K.B.; Upton, J.W.; Rehwinkel, J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 2017, 36, 2529–2543. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, H.; Ragan, K.B.; Guo, H.; Gilley, R.P.; Landsteiner, V.J.; Kaiser, W.J.; Upton, J.W. Murine cytomegalovirus IE3-dependent transcription is required for DAI/ZBP1-mediated necroptosis. EMBO Rep. 2017, 18, 1429–1441. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. MLKL regulates necrotic plasma membrane permeabilization. Cell Res. 2014, 24, 139–140. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Goossens, V.; Grootjans, S.; Van den Haute, C.; Vanlangenakker, N.; Dondelinger, Y.; Roelandt, R.; Bruggeman, I.; Goncalves, A.; Bertrand, M.J.; et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 2014, 5, e1004. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, W.; Yan, Y.; Gong, T.; Han, J.; Tian, Z.; Zhou, R. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol. 2014, 15, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Zheng, X.; Wang, Z.A.; Chen, X.; He, W.T.; Zhang, Y.; Xu, J.G.; Zhao, H.; Shi, W.; Wang, X.; et al. The MLKL Channel in Necroptosis Is an Octamer Formed by Tetramers in a Dyadic Process. Mol. Cell Biol. 2017, 37, e00497-16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Y.; He, W.; Sun, L. Necrosome core machinery: MLKL. Cell Mol. Life Sci. 2016, 73, 2153–2163. [Google Scholar] [CrossRef]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL forms cation channels. Cell Res. 2016, 26, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef]
- Varfolomeev, E.E.; Schuchmann, M.; Luria, V.; Chiannilkulchai, N.; Beckmann, J.S.; Mett, I.L.; Rebrikov, D.; Brodianski, V.M.; Kemper, O.C.; Kollet, O.; et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998, 9, 267–276. [Google Scholar] [CrossRef]
- Yeh, W.C.; Itie, A.; Elia, A.J.; Ng, M.; Shu, H.B.; Wakeham, A.; Mirtsos, C.; Suzuki, N.; Bonnard, M.; Goeddel, D.V.; et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 2000, 12, 633–642. [Google Scholar] [CrossRef]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.P.; Oberst, A.; Weinlich, R.; Janke, L.J.; Kang, T.B.; Ben-Moshe, T.; Mak, T.W.; Wallach, D.; Green, D.R. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012, 1, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes. Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.A.; Perez-Jimenez, E.; Oberst, A.; Ng, A.; Massoumi, R.; Xavier, R.; Green, D.R.; Ting, A.T. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 2011, 13, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Imre, G.; Larisch, S.; Rajalingam, K. Ripoptosome: A novel IAP-regulated cell death-signalling platform. J. Mol. Cell Biol. 2011, 3, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Reiley, W.W.; Chang, M.; Jin, W.; Lee, A.J.; Zhang, M.; Sun, S.C. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev. Cell 2007, 13, 705–716. [Google Scholar] [CrossRef]
- Onizawa, M.; Oshima, S.; Schulze-Topphoff, U.; Oses-Prieto, J.A.; Lu, T.; Tavares, R.; Prodhomme, T.; Duong, B.; Whang, M.I.; Advincula, R.; et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 2015, 16, 618–627. [Google Scholar] [CrossRef] [Green Version]
- Maelfait, J.; Vercammen, E.; Janssens, S.; Schotte, P.; Haegman, M.; Magez, S.; Beyaert, R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J. Exp. Med. 2008, 205, 1967–1973. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; Wong, W.W.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Stammler, D.; Eigenbrod, T.; Menz, S.; Frick, J.S.; Sweet, M.J.; Shakespear, M.R.; Jantsch, J.; Siegert, I.; Wolfle, S.; Langer, J.D.; et al. Inhibition of Histone Deacetylases Permits Lipopolysaccharide-Mediated Secretion of Bioactive IL-1beta via a Caspase-1-Independent Mechanism. J. Immunol. 2015, 195, 5421–5431. [Google Scholar] [CrossRef] [PubMed]
- Shenderov, K.; Riteau, N.; Yip, R.; Mayer-Barber, K.D.; Oland, S.; Hieny, S.; Fitzgerald, P.; Oberst, A.; Dillon, C.P.; Green, D.R.; et al. Cutting edge: Endoplasmic reticulum stress licenses macrophages to produce mature IL-1beta in response to TLR4 stimulation through a caspase-8- and TRIF-dependent pathway. J. Immunol. 2014, 192, 2029–2033. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; Kaptein, T.M.; Wevers, B.A.; Theelen, B.; van der Vlist, M.; Boekhout, T.; Geijtenbeek, T.B. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012, 13, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.B.; Yang, S.H.; Toth, B.; Kovalenko, A.; Wallach, D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 2013, 38, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef] [PubMed]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Nunez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonopoulos, C.; El Sanadi, C.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Proapoptotic chemotherapeutic drugs induce noncanonical processing and release of IL-1beta via caspase-8 in dendritic cells. J. Immunol. 2013, 191, 4789–4803. [Google Scholar] [CrossRef]
- Bossaller, L.; Chiang, P.I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 2012, 189, 5508–5512. [Google Scholar] [CrossRef]
- Antonopoulos, C.; Russo, H.M.; El Sanadi, C.; Martin, B.N.; Li, X.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J. Biol. Chem. 2015, 290, 20167–20184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Tourlomousis, P.; Hopkins, L.; Monie, T.P.; Fitzgerald, K.A.; Bryant, C.E. Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1beta production. J. Immunol. 2013, 191, 5239–5246. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Wu, Y.H.; Kuo, W.C.; Wu, Y.J.; Yang, K.T.; Chen, S.T.; Jiang, S.T.; Gordy, C.; He, Y.W.; Lai, M.Z. Participation of c-FLIP in NLRP3 and AIM2 inflammasome activation. Cell Death Differ. 2014, 21, 451–461. [Google Scholar] [CrossRef]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef]
- Vande Walle, L.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.D.; van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Yabal, M.; Muller, N.; Adler, H.; Knies, N.; Gross, C.J.; Damgaard, R.B.; Kanegane, H.; Ringelhan, M.; Kaufmann, T.; Heikenwalder, M.; et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 2014, 7, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Weng, D.; Marty-Roix, R.; Ganesan, S.; Proulx, M.K.; Vladimer, G.I.; Kaiser, W.J.; Mocarski, E.S.; Pouliot, K.; Chan, F.K.; Kelliher, M.A.; et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 7391–7396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip, N.H.; Dillon, C.P.; Snyder, A.G.; Fitzgerald, P.; Wynosky-Dolfi, M.A.; Zwack, E.E.; Hu, B.; Fitzgerald, L.; Mauldin, E.A.; Copenhaver, A.M.; et al. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-kappaB and MAPK signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7385–7390. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Chan, F.K. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J. Immunol. 2015, 194, 1938–1944. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Lin, H.; Wen, J.; Sun, Q.; Gao, Y.; Xu, X.; Wang, J.; Zhang, J.; Weng, D. The kinase receptor-interacting protein 1 is required for inflammasome activation induced by endoplasmic reticulum stress. Cell Death Dis. 2018, 9, 641. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, K.D.; Davis, M.A.; Daniels, B.P.; Olsen, T.M.; Ralli-Jain, P.; Tait, S.W.; Gale, M., Jr.; Oberst, A. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1beta Independently of Gasdermin-D. J. Immunol. 2017, 198, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fan, C.; Zhang, H.; Zhao, Q.; Liu, Y.; Xu, C.; Xie, Q.; Wu, X.; Yu, X.; Zhang, J.; et al. MLKL and FADD Are Critical for Suppressing Progressive Lymphoproliferative Disease and Activating the NLRP3 Inflammasome. Cell Rep. 2016, 16, 3247–3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriwaki, K.; Farias Luz, N.; Balaji, S.; De Rosa, M.J.; O’Donnell, C.L.; Gough, P.J.; Bertin, J.; Welsh, R.M.; Chan, F.K. The Mitochondrial Phosphatase PGAM5 Is Dispensable for Necroptosis but Promotes Inflammasome Activation in Macrophages. J. Immunol. 2016, 196, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Liu, G.; Liu, Q.; Zhou, Y. Swine Influenza Virus Induces RIPK1/DRP1-Mediated Interleukin-1 Beta Production. Viruses 2018, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Shi, L.; Wang, Z.; Zhou, J.; Manaenko, A.; Reis, C.; Chen, S.; Zhang, J. RIP1-RIP3-DRP1 pathway regulates NLRP3 inflammasome activation following subarachnoid hemorrhage. Exp. Neurol. 2017, 295, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, Z.; Guan, Q.; Qiu, F.; Li, Y.; Liu, Z.; Zhang, H.; Dong, H.; Zhang, Z. PEDF Inhibits the Activation of NLRP3 Inflammasome in Hypoxia Cardiomyocytes through PEDF Receptor/Phospholipase A2. Int. J. Mol. Sci. 2016, 17, 2064. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-H.; Kang, T.-B. The Molecular Links between Cell Death and Inflammasome. Cells 2019, 8, 1057. https://doi.org/10.3390/cells8091057
Lee K-H, Kang T-B. The Molecular Links between Cell Death and Inflammasome. Cells. 2019; 8(9):1057. https://doi.org/10.3390/cells8091057
Chicago/Turabian StyleLee, Kwang-Ho, and Tae-Bong Kang. 2019. "The Molecular Links between Cell Death and Inflammasome" Cells 8, no. 9: 1057. https://doi.org/10.3390/cells8091057
APA StyleLee, K. -H., & Kang, T. -B. (2019). The Molecular Links between Cell Death and Inflammasome. Cells, 8(9), 1057. https://doi.org/10.3390/cells8091057