The Neglected Insulin: IGF-II, a Metabolic Regulator with Implications for Diabetes, Obesity, and Cancer



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Impediments to Progress
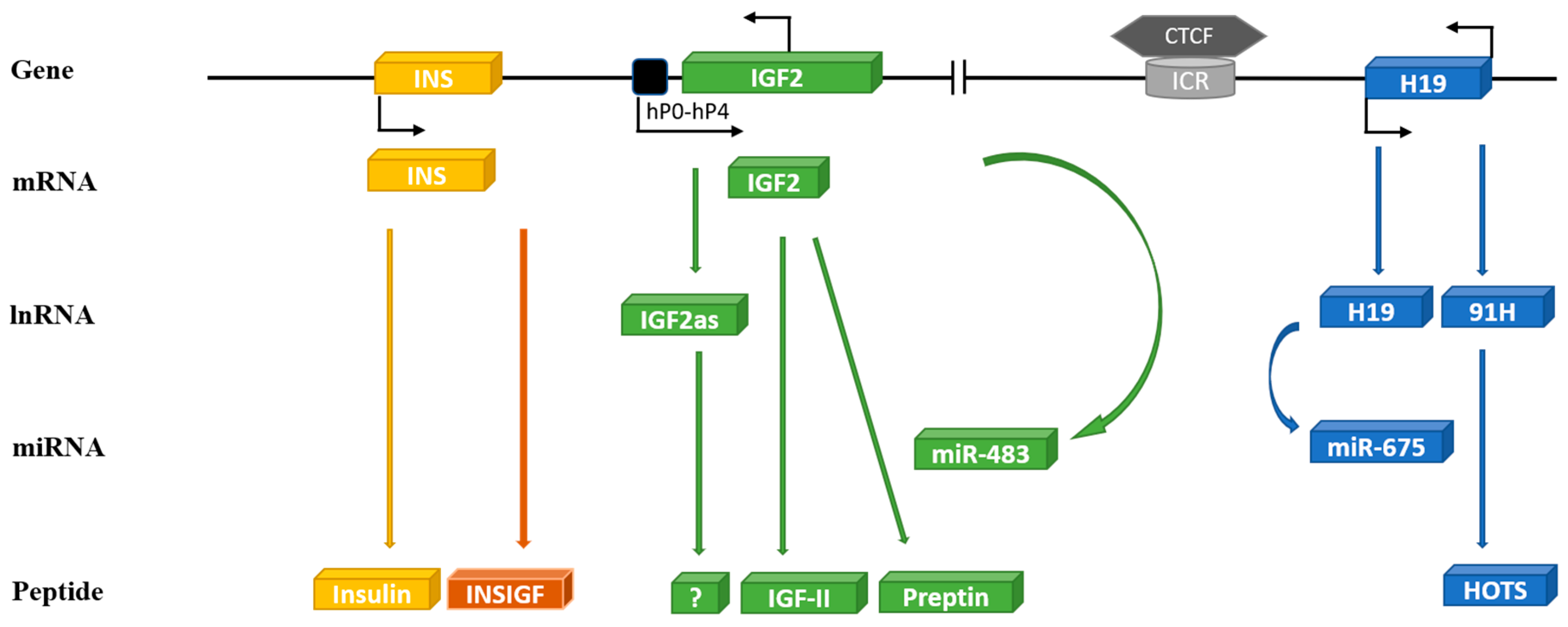
3. IGF-II: An Imprinted Multifunctional Metabolic Genetic Loci
3.1. Preptin
3.2. H19
3.3. miR-675
3.4. H19 as a miRNA Decoy
3.5. H19 Interactions with Proteins
3.6. 91H
3.7. miR-483
3.8. Other Transcripts
3.8.1. The INS/IGF-II Overlapping Region (INSIGF) Read-Through
3.8.2. IGF-II Antisense
3.9. IMPs
3.10. GRP94
3.11. Receptors
3.11.1. Insulin and IGF-I Receptors
3.11.2. IGF-II Receptor
3.12. IGF Binding Proteins
4. Indications for a Role in Diabetes and Obesity
4.1. Preptin
4.2. miR-483
4.3. H19
4.4. IMPs
4.5. IGFBPs
4.6. Receptors
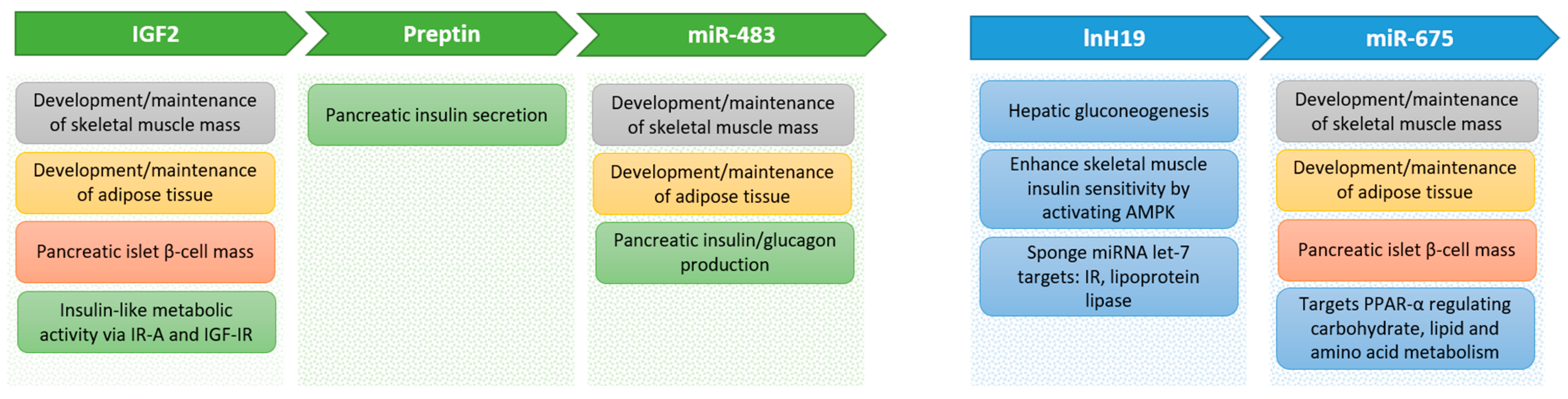
5. Metabolic Effects of Components of the IGF-II Locus
5.1. IGF-II
5.2. Preptin
5.3. miR-483
5.4. H19
5.5. miR-675
5.6. IMPs
5.7. Receptors
6. Role of IGF-II in Pancreatic Islet Function
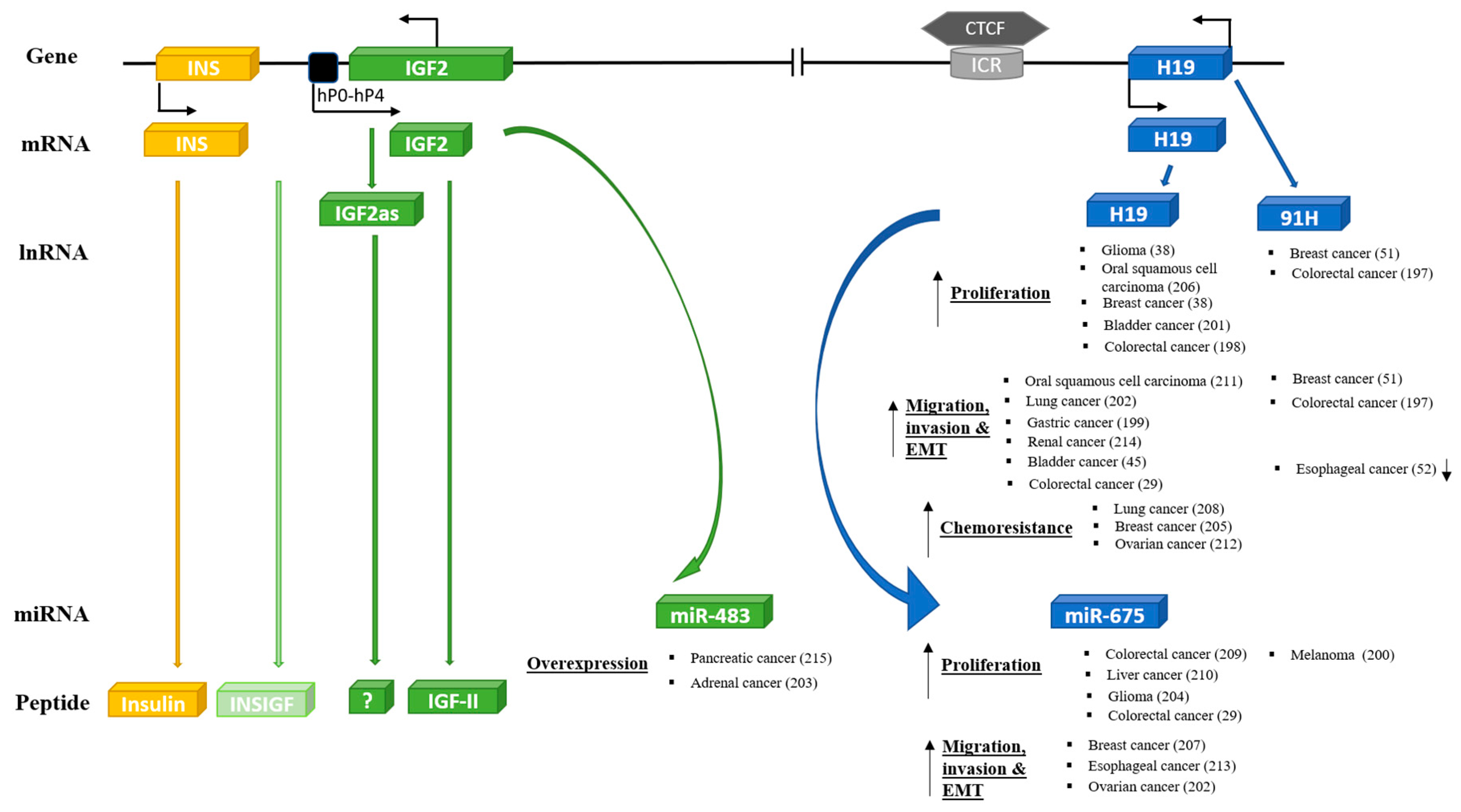
7. IGF-II Locus and Cancer
7.1. IGF-II
7.2. Other Components of the IGF-II/H19 Locus
8. Hypothesis: A Fundamental Metabolic Role for IGF-II
Author Contributions
Funding
Conflicts of Interest
References
- Holly, J.M. The IGF-II enigma. Growth Horm. Igf. Res. 1998, 8, 183–184. [Google Scholar] [CrossRef]
- De Meyts, P. Insulin and its receptor: Structure, function and evolution. Bioessays 2004, 26, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Salmon, W.D., Jr.; Daughaday, W.H. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J. Lab. Clin. Med. 1957, 49, 825–836. [Google Scholar] [PubMed]
- Cotterill, A.M.; Holly, J.M.; Davies, S.C.; Coulson, V.J.; Price, P.A.; Wass, J.A. The insulin-like growth factors and their binding proteins in a case of non-islet-cell tumour-associated hypoglycaemia. J. Endocrinol. 1991, 131, 303–311. [Google Scholar] [CrossRef]
- Holly, J.M.; Perks, C.M. Insulin-like growth factor physiology: What we have learned from human studies. Endocrinol. Metab. Clin. North. Am. 2012, 41, 249–263. [Google Scholar] [CrossRef]
- Rotwein, P. The complex genetics of human insulin-like growth factor 2 are not reflected in public databases. J. Biol. Chem. 2018, 293, 4324–4333. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, M.Z. Igf2-H19, an imprinted tandem gene, is an important regulator of embryonic development, a guardian of proliferation of adult pluripotent stem cells, a regulator of longevity, and a ‘passkey’ to cancerogenesis. Folia Histochem. Cytobiol. 2012, 50, 171–179. [Google Scholar] [CrossRef]
- Reik, W.; Constancia, M.; Dean, W.; Davies, K.; Bowden, L.; Murrell, A.; Feil, R.; Walter, J.; Kelsey, G. Igf2 imprinting in development and disease. Int. J. Dev. Biol. 2000, 44, 145–150. [Google Scholar]
- Monk, D.; Sanches, R.; Arnaud, P.; Apostolidou, S.; Hills, F.A.; Abu-Amero, S.; Murrell, A.; Friess, H.; Reik, W.; Stanier, P.; et al. Imprinting of IGF2 P0 transcript and novel alternatively spliced INS-IGF2 isoforms show differences between mouse and human. Hum. Mol Genet. 2006, 15, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Hu, J.F.; Qiu, X.; Ling, J.; Chen, H.; Wang, S.; Hou, A.; Vu, T.H.; Hoffman, A.R. CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive complex 2 intrachromosomal loop. Mol. Cell Biol. 2008, 28, 6473–6482. [Google Scholar] [CrossRef]
- Murrell, A.; Heeson, S.; Reik, W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat. Genet. 2004, 36, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Vu, T.H.; Lu, Q.; Ling, J.Q.; Li, T.; Hou, A.; Wang, S.K.; Chen, H.L.; Hu, J.F.; Hoffman, A.R. A complex deoxyribonucleic acid looping configuration associated with the silencing of the maternal Igf2 allele. Mol. Endocrinol. 2008, 22, 1476–1488. [Google Scholar] [CrossRef]
- Zhang, H.; Niu, B.; Hu, J.F.; Ge, S.; Wang, H.; Li, T.; Ling, J.; Steelman, B.N.; Qian, G.; Hoffman, A.R. Interruption of intrachromosomal looping by CCCTC binding factor decoy proteins abrogates genomic imprinting of human insulin-like growth factor II. J. Cell Biol. 2011, 193, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzi, S.; Abi Habib, W.; Netchine, I. Beckwith-Wiedemann and Russell-Silver Syndromes: From new molecular insights to the comprehension of imprinting regulation. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Ounap, K. Silver-Russell Syndrome and Beckwith-Wiedemann Syndrome: Opposite Phenotypes with Heterogeneous Molecular Etiology. Mol. Syndr. 2016, 7, 110–121. [Google Scholar] [CrossRef]
- Dai, Y.M.; Wang, Z.; Li, J.; Gu, X.; Zheng, M.; Zhou, J.; Ye, X.; Yao, J.; Cui, I.; Hu, Y.; et al. Imprinting status of IGF2 in cord blood cells of Han Chinese newborns. Int. J. Mol. Sci. 2007, 8, 273–283. [Google Scholar] [CrossRef]
- Rancourt, R.C.; Harris, H.R.; Barault, L.; Michels, K.B. The prevalence of loss of imprinting of H19 and IGF2 at birth. FASEB J. 2013, 27, 3335–3343. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, C.M.; Phillips, A.R.; Cooper, G.J. Preptin derived from proinsulin-like growth factor II (proIGF-II) is secreted from pancreatic islet beta-cells and enhances insulin secretion. Biochem. J. 2001, 360, 431–439. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Pachnis, V.; Belayew, A.; Tilghman, S.M. Locus unlinked to alpha-fetoprotein under the control of the murine raf and Rif genes. Proc. Natl. Acad. Sci. USA 1984, 81, 5523–5527. [Google Scholar] [CrossRef]
- Brannan, C.I.; Dees, E.C.; Ingram, R.S.; Tilghman, S.M. The product of the H19 gene may function as an RNA. Mol. Cell Biol. 1990, 10, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Matouk, I.; Ayesh, B.; Schneider, T.; Ayesh, S.; Ohana, P.; de-Groot, N.; Hochberg, A.; Galun, E. Oncofetal splice-pattern of the human H19 gene. Biochem. Biophys. Res. Commun. 2004, 318, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Cullen, B.R. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA 2007, 13, 313–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat. Cell Biol. 2012, 14, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.; Haig, D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. 1991, 7, 45–49. [Google Scholar] [CrossRef]
- Farzi-Molan, A.; Babashah, S.; Bakhshinejad, B.; Atashi, A.; Fakhr Taha, M. Down-regulation of the non-coding RNA H19 and its derived miR-675 is concomitant with up-regulation of insulin-like growth factor receptor type 1 during neural-like differentiation of human bone marrow mesenchymal stem cells. Cell Biol. Int. 2018, 42, 940–948. [Google Scholar] [CrossRef]
- Lv, J.; Wang, L.; Zhang, J.; Lin, R.; Wang, L.; Sun, W.; Wu, H.; Xin, S. Long noncoding RNA H19-derived miR-675 aggravates restenosis by targeting PTEN. Biochem. Biophys. Res. Commun. 2018, 497, 1154–1161. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. (Lausanne) 2018, 9, 338. [Google Scholar] [CrossRef] [Green Version]
- Tsang, W.P.; Ng, E.K.; Ng, S.S.; Jin, H.; Yu, J.; Sung, J.J.; Kwok, T.T. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 2010, 31, 350–358. [Google Scholar] [CrossRef]
- Huang, Y.; Zheng, Y.; Jin, C.; Li, X.; Jia, L.; Li, W. Long Non-coding RNA H19 Inhibits Adipocyte Differentiation of Bone Marrow Mesenchymal Stem Cells through Epigenetic Modulation of Histone Deacetylases. Sci. Rep. 2016, 6, 28897. [Google Scholar] [CrossRef] [Green Version]
- Su, J.L.; Chen, P.S.; Johansson, G.; Kuo, M.L. Function and regulation of let-7 family microRNAs. MicroRNA 2012, 1, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Kallen, A.N.; Zhou, X.B.; Xu, J.; Qiao, C.; Ma, J.; Yan, L.; Lu, L.; Liu, C.; Yi, J.S.; Zhang, H.; et al. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol. Cell 2013, 52, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wu, F.; Zhou, J.; Yan, L.; Jurczak, M.J.; Lee, H.Y.; Yang, L.; Mueller, M.; Zhou, X.B.; Dandolo, L.; et al. The H19/let-7 double-negative feedback loop contributes to glucose metabolism in muscle cells. Nucleic Acids Res. 2014, 42, 13799–13811. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichael, G.G.; Taylor, H.S.; et al. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene 2017, 36, 2345–2354. [Google Scholar] [CrossRef]
- Zhu, H.; Shyh-Chang, N.; Segre, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef]
- Zhou, X.; Ye, F.; Yin, C.; Zhuang, Y.; Yue, G.; Zhang, G. The Interaction Between MiR-141 and lncRNA-H19 in Regulating Cell Proliferation and Migration in Gastric Cancer. Cell Physiol. Biochem. 2015, 36, 1440–1452. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, L.; Zhong, T.; Mueller, M.; Men, Y.; Zhang, N.; Xie, J.; Giang, K.; Chung, H.; Sun, X.; et al. H19 lncRNA alters DNA methylation genome wide by regulating S-adenosylhomocysteine hydrolase. Nat. Commun. 2015, 6, 10221. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Li, Y.; Ren, K.; Li, X.; Han, X.; Wang, J. Long non-coding RNA H19 promotes the proliferation and invasion of breast cancer through upregulating DNMT1 expression by sponging miR-152. J. Biochem. Mol. Toxicol. 2017, 31. [Google Scholar] [CrossRef]
- Yang, F.; Bi, J.; Xue, X.; Zheng, L.; Zhi, K.; Hua, J.; Fang, G. Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J. 2012, 279, 3159–3165. [Google Scholar] [CrossRef]
- Goyal, N.; Tiwary, S.; Kesharwani, D.; Datta, M. Long non-coding RNA H19 inhibition promotes hyperglycemia in mice by upregulating hepatic FoxO1 levels and promoting gluconeogenesis. J. Mol. Med. (Berl.) 2019, 97, 115–126. [Google Scholar] [CrossRef]
- Zheng, Z.H.; Wu, D.M.; Fan, S.H.; Zhang, Z.F.; Chen, G.Q.; Lu, J. Upregulation of miR-675-5p induced by lncRNA H19 was associated with tumor progression and development by targeting tumor suppressor p53 in non-small cell lung cancer. J. Cell. Biochem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Runge, S.; Nielsen, F.C.; Nielsen, J.; Lykke-Andersen, J.; Wewer, U.M.; Christiansen, J. H19 RNA binds four molecules of insulin-like growth factor II mRNA-binding protein. J. Biol. Chem. 2000, 275, 29562–29569. [Google Scholar] [CrossRef] [PubMed]
- Giovarelli, M.; Bucci, G.; Ramos, A.; Bordo, D.; Wilusz, C.J.; Chen, C.Y.; Puppo, M.; Briata, P.; Gherzi, R. H19 long noncoding RNA controls the mRNA decay promoting function of KSRP. Proc. Natl. Acad. Sci. USA 2014, 111, E5023–E5028. [Google Scholar] [CrossRef] [PubMed]
- Gherzi, R.; Chen, C.Y.; Ramos, A.; Briata, P. KSRP controls pleiotropic cellular functions. Semin. Cell Dev. Biol. 2014, 34, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Li, Z.; Wang, W.; Zeng, Y.; Liu, Z.; Qiu, J. Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Lett. 2013, 333, 213–221. [Google Scholar] [CrossRef]
- Zhuo, C.; Jiang, R.; Lin, X.; Shao, M. LncRNA H19 inhibits autophagy by epigenetically silencing of DIRAS3 in diabetic cardiomyopathy. Oncotarget 2017, 8, 1429–1437. [Google Scholar] [CrossRef]
- Baric, I. Inherited disorders in the conversion of methionine to homocysteine. J. Inherit. Metab. Dis. 2009, 32, 459–471. [Google Scholar] [CrossRef]
- Monnier, P.; Martinet, C.; Pontis, J.; Stancheva, I.; Ait-Si-Ali, S.; Dandolo, L. H19 lncRNA controls gene expression of the Imprinted Gene Network by recruiting MBD1. Proc. Natl. Acad. Sci. USA 2013, 110, 20693–20698. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, Y.; Huang, T.; Cheng, A.S.; Yu, J.; Kang, W.; To, K.F. The Interplay of LncRNA-H19 and Its Binding Partners in Physiological Process and Gastric Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 450. [Google Scholar] [CrossRef]
- Berteaux, N.; Aptel, N.; Cathala, G.; Genton, C.; Coll, J.; Daccache, A.; Spruyt, N.; Hondermarck, H.; Dugimont, T.; Curgy, J.J.; et al. A novel H19 antisense RNA overexpressed in breast cancer contributes to paternal IGF2 expression. Mol. Cell Biol. 2008, 28, 6731–6745. [Google Scholar] [CrossRef]
- Vennin, C.; Spruyt, N.; Robin, Y.M.; Chassat, T.; Le Bourhis, X.; Adriaenssens, E. The long non-coding RNA 91H increases aggressive phenotype of breast cancer cells and up-regulates H19/IGF2 expression through epigenetic modifications. Cancer Lett. 2017, 385, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; He, B.; Pan, Y.; Xu, Y.; Li, R.; Deng, Q.; Sun, H.; Wang, S. Long non-coding RNA 91H contributes to the occurrence and progression of esophageal squamous cell carcinoma by inhibiting IGF2 expression. Mol. Carcinog. 2015, 54, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Onyango, P.; Feinberg, A.P. A nucleolar protein, H19 opposite tumor suppressor (HOTS), is a tumor growth inhibitor encoded by a human imprinted H19 antisense transcript. Proc. Natl. Acad. Sci. USA 2011, 108, 16759–16764. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Tie, Y.; Xu, C.; Zhang, Z.; Zhu, J.; Shi, Y.; Jiang, H.; Sun, Z.; Zheng, X. Identification of human fetal liver miRNAs by a novel method. FEBS Lett. 2005, 579, 3849–3854. [Google Scholar] [CrossRef] [Green Version]
- Ma, N.; Wang, X.; Qiao, Y.; Li, F.; Hui, Y.; Zou, C.; Jin, J.; Lv, G.; Peng, Y.; Wang, L.; et al. Coexpression of an intronic microRNA and its host gene reveals a potential role for miR-483-5p as an IGF2 partner. Mol. Cell Endocrinol. 2011, 333, 96–101. [Google Scholar] [CrossRef]
- Liu, M.; Roth, A.; Yu, M.; Morris, R.; Bersani, F.; Rivera, M.N.; Lu, J.; Shioda, T.; Vasudevan, S.; Ramaswamy, S.; et al. The IGF2 intronic miR-483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes Dev. 2013, 27, 2543–2548. [Google Scholar] [CrossRef]
- Kanatsuna, N.; Taneera, J.; Vaziri-Sani, F.; Wierup, N.; Larsson, H.E.; Delli, A.; Skarstrand, H.; Balhuizen, A.; Bennet, H.; Steiner, D.F.; et al. Autoimmunity against INS-IGF2 protein expressed in human pancreatic islets. J. Biol. Chem. 2013, 288, 29013–29023. [Google Scholar] [CrossRef]
- Gao, S.; Lin, Z.; Li, C.; Wang, Y.; Yang, L.; Zou, B.; Chen, J.; Li, J.; Feng, D.; Song, Z.; et al. lncINS-IGF2 Promotes Cell Proliferation and Migration by Promoting G1/S Transition in Lung Cancer. Technol. Cancer Res. Treat. 2019, 18, 1533033818823029. [Google Scholar] [CrossRef]
- Okutsu, T.; Kuroiwa, Y.; Kagitani, F.; Kai, M.; Aisaka, K.; Tsutsumi, O.; Kaneko, Y.; Yokomori, K.; Surani, M.A.; Kohda, T.; et al. Expression and imprinting status of human PEG8/IGF2AS, a paternally expressed antisense transcript from the IGF2 locus, in Wilms’ tumors. J. Biochem. 2000, 127, 475–483. [Google Scholar] [CrossRef]
- Duart-Garcia, C.; Braunschweig, M.H. The Igf2as transcript is exported into cytoplasm and associated with polysomes. Biochem. Genet. 2013, 51, 119–130. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, T.; Wang, F.; Gong, B.; Xie, W.; Ma, M.; Yang, X. Long Noncoding RNA IGF2AS is Acting as an Epigenetic Tumor Suppressor in Human Prostate Cancer. Urology 2019, 124, 310 e311–310 e318. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Luo, Y.; Chen, G.; Liu, H.; Tian, N.; Zen, X.; Liu, Q. Long noncoding RNA IGF2AS regulates high-glucose induced apoptosis in human retinal pigment epithelial cells. Iubmb. Life 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, B.; Li, B.; Song, C.; Diao, H.; Guo, Z.; Li, Z.; Zhang, J. Inhibition of long noncoding RNA IGF2AS promotes angiogenesis in type 2 diabetes. Biomed. Pharm. 2017, 92, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Degrauwe, N.; Suva, M.L.; Janiszewska, M.; Riggi, N.; Stamenkovic, I. IMPs: An RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016, 30, 2459–2474. [Google Scholar] [CrossRef] [PubMed]
- Dai, N.; Rapley, J.; Angel, M.; Yanik, M.F.; Blower, M.D.; Avruch, J. mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev. 2011, 25, 1159–1172. [Google Scholar] [CrossRef] [Green Version]
- Ni, M.; Lee, A.S. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007, 581, 3641–3651. [Google Scholar] [CrossRef] [Green Version]
- Ostrovsky, O.; Ahmed, N.T.; Argon, Y. The chaperone activity of GRP94 toward insulin-like growth factor II is necessary for the stress response to serum deprivation. Mol. Biol. Cell 2009, 20, 1855–1864. [Google Scholar] [CrossRef]
- Wanderling, S.; Simen, B.B.; Ostrovsky, O.; Ahmed, N.T.; Vogen, S.M.; Gidalevitz, T.; Argon, Y. GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion. Mol. Biol. Cell 2007, 18, 3764–3775. [Google Scholar] [CrossRef]
- Ostrovsky, O.; Eletto, D.; Makarewich, C.; Barton, E.R.; Argon, Y. Glucose regulated protein 94 is required for muscle differentiation through its control of the autocrine production of insulin-like growth factors. Biochim. Biophys. Acta 2010, 1803, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Marzec, M.; Hawkes, C.P.; Eletto, D.; Boyle, S.; Rosenfeld, R.; Hwa, V.; Wit, J.M.; van Duyvenvoorde, H.A.; Oostdijk, W.; Losekoot, M.; et al. A Human Variant of Glucose-Regulated Protein 94 That Inefficiently Supports IGF Production. Endocrinology 2016, 157, 1914–1928. [Google Scholar] [CrossRef]
- Siddle, K. Molecular basis of signaling specificity of insulin and IGF receptors: Neglected corners and recent advances. Front. Endocrinol. 2012, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [PubMed]
- Morcavallo, A.; Gaspari, M.; Pandini, G.; Palummo, A.; Cuda, G.; Larsen, M.R.; Vigneri, R.; Belfiore, A. Research resource: New and diverse substrates for the insulin receptor isoform A revealed by quantitative proteomics after stimulation with IGF-II or insulin. Mol. Endocrinol. 2011, 25, 1456–1468. [Google Scholar] [CrossRef] [PubMed]
- Pandini, G.; Conte, E.; Medico, E.; Sciacca, L.; Vigneri, R.; Belfiore, A. IGF-II binding to insulin receptor isoform A induces a partially different gene expression profile from insulin binding. Ann. N. Y. Acad. Sci. 2004, 1028, 450–456. [Google Scholar] [CrossRef]
- Sacco, A.; Morcavallo, A.; Pandini, G.; Vigneri, R.; Belfiore, A. Differential signaling activation by insulin and insulin-like growth factors I and II upon binding to insulin receptor isoform A. Endocrinology 2009, 150, 3594–3602. [Google Scholar] [CrossRef]
- Morrione, A.; Valentinis, B.; Xu, S.Q.; Yumet, G.; Louvi, A.; Efstratiadis, A.; Baserga, R. Insulin-like growth factor II stimulates cell proliferation through the insulin receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 3777–3782. [Google Scholar] [CrossRef] [Green Version]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef]
- Leibiger, B.; Leibiger, I.B.; Moede, T.; Kemper, S.; Kulkarni, R.N.; Kahn, C.R.; de Vargas, L.M.; Berggren, P.O. Selective insulin signaling through A and B insulin receptors regulates transcription of insulin and glucokinase genes in pancreatic beta cells. Mol. Cell 2001, 7, 559–570. [Google Scholar] [CrossRef]
- Uhles, S.; Moede, T.; Leibiger, B.; Berggren, P.O.; Leibiger, I.B. Isoform-specific insulin receptor signaling involves different plasma membrane domains. J. Cell Biol. 2003, 163, 1327–1337. [Google Scholar] [CrossRef] [Green Version]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef]
- Slaaby, R.; Schaffer, L.; Lautrup-Larsen, I.; Andersen, A.S.; Shaw, A.C.; Mathiasen, I.S.; Brandt, J. Hybrid receptors formed by insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) have low insulin and high IGF-1 affinity irrespective of the IR splice variant. J. Biol. Chem. 2006, 281, 25869–25874. [Google Scholar] [CrossRef] [PubMed]
- Martin-Kleiner, I.; Gall Troselj, K. Mannose-6-phosphate/insulin-like growth factor 2 receptor (M6P/IGF2R) in carcinogenesis. Cancer Lett. 2010, 289, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; MacDonald, R.G.; Thinakaran, G.; Kar, S. Insulin-Like Growth Factor-II/Cation-Independent Mannose 6-Phosphate Receptor in Neurodegenerative Diseases. Mol. Neurobiol. 2017, 54, 2636–2658. [Google Scholar] [CrossRef] [PubMed]
- Bergman, D.; Halje, M.; Nordin, M.; Engstrom, W. Insulin-like growth factor 2 in development and disease: A mini-review. Gerontology 2013, 59, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Jirtle, R.L.; Hoffman, A.R. Cross-species clues of an epigenetic imprinting regulatory code for the IGF2R gene. Cytogenet. Genome Res. 2006, 113, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Lau, M.M.; Stewart, C.E.; Liu, Z.; Bhatt, H.; Rotwein, P.; Stewart, C.L. Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev. 1994, 8, 2953–2963. [Google Scholar] [CrossRef] [PubMed]
- Appell, K.C.; Simpson, I.A.; Cushman, S.W. Characterization of the stimulatory action of insulin on insulin-like growth factor II binding to rat adipose cells. Differences in the mechanism of insulin action on insulin-like growth factor II receptors and glucose transporters. J. Biol. Chem. 1988, 263, 10824–10829. [Google Scholar]
- Lonnroth, P.; Appell, K.C.; Wesslau, C.; Cushman, S.W.; Simpson, I.A.; Smith, U. Insulin-induced subcellular redistribution of insulin-like growth factor II receptors in the rat adipose cell. Counterregulatory effects of isoproterenol, adenosine, and cAMP analogues. J. Biol. Chem. 1988, 263, 15386–15391. [Google Scholar]
- Wardzala, L.J.; Simpson, I.A.; Rechler, M.M.; Cushman, S.W. Potential mechanism of the stimulatory action of insulin on insulin-like growth factor II binding to the isolated rat adipose cell. Apparent redistribution of receptors cycling between a large intracellular pool and the plasma membrane. J. Biol. Chem. 1984, 259, 8378–8383. [Google Scholar]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–212. [Google Scholar] [CrossRef]
- Holly, J.; Perks, C. The role of insulin-like growth factor binding proteins. Neuroendocrinology 2006, 83, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Cwyfan-Hughes, S.C.; van der Stappen, J.W.; Sansom, J.; Burton, J.L.; Donnelly, M.; Holly, J.M. Insulin-like growth factors (IGFs) and IGF-binding proteins in human skin interstitial fluid. J. Clin. Endocrinol. Metab. 1995, 80, 2940–2945. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, D.R.; Underwood, L.E. Nutritional regulation of IGF-I and IGF binding proteins. Annu. Rev. Nutr. 1991, 11, 393–412. [Google Scholar] [CrossRef] [PubMed]
- Bar, R.S.; Clemmons, D.R.; Boes, M.; Busby, W.H.; Booth, B.A.; Dake, B.L.; Sandra, A. Transcapillary permeability and subendothelial distribution of endothelial and amniotic fluid insulin-like growth factor binding proteins in the rat heart. Endocrinology 1990, 127, 1078–1086. [Google Scholar] [CrossRef]
- Holly, J.M. The physiological role of IGFBP-1. Acta Endocrinol. (Copenh.) 1991, 124, 55–62. [Google Scholar]
- Frystyk, J.; Skjaerbaek, C.; Vestbo, E.; Fisker, S.; Orskov, H. Circulating levels of free insulin-like growth factors in obese subjects: The impact of type 2 diabetes. Diabetes Metab. Res. Rev. 1999, 15, 314–322. [Google Scholar] [CrossRef]
- Belobrajdic, D.P.; Frystyk, J.; Jeyaratnaganthan, N.; Espelund, U.; Flyvbjerg, A.; Clifton, P.M.; Noakes, M. Moderate energy restriction-induced weight loss affects circulating IGF levels independent of dietary composition. Eur. J. Endocrinol. 2010, 162, 1075–1082. [Google Scholar] [CrossRef] [Green Version]
- Bevan, S.J.; Parry-Billings, M.; Opara, E.; Liu, C.T.; Dunger, D.B.; Newsholme, E.A. The effect of insulin-like growth factor II on glucose uptake and metabolism in rat skeletal muscle in vitro. Biochem. J. 1992, 286, 561–565. [Google Scholar] [CrossRef] [Green Version]
- Faienza, M.F.; Santoro, N.; Lauciello, R.; Calabro, R.; Giordani, L.; Di Salvo, G.; Ventura, A.; Delvecchio, M.; Perrone, L.; Del Giudice, E.M.; et al. IGF2 gene variants and risk of hypertension in obese children and adolescents. Pediatr. Res. 2010, 67, 340–344. [Google Scholar] [CrossRef]
- O’Dell, S.D.; Miller, G.J.; Cooper, J.A.; Hindmarsh, P.C.; Pringle, P.J.; Ford, H.; Humphries, S.E.; Day, I.N. Apal polymorphism in insulin-like growth factor II (IGF2) gene and weight in middle-aged males. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 822–825. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, S.; Gaunt, T.R.; O’Dell, S.D.; Chen, X.H.; Gu, D.; Hawe, E.; Miller, G.J.; Humphries, S.E.; Day, I.N. Haplotypic analyses of the IGF2-INS-TH gene cluster in relation to cardiovascular risk traits. Hum. Mol. Genet. 2004, 13, 715–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, S.M.; Schrager, M.A.; Metter, E.J.; Riechman, S.E.; Fleg, J.L.; Hurley, B.F.; Ferrell, R.E. IGF2 genotype and obesity in men and women across the adult age span. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 585–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, T.; Chagnon, Y.C.; Perusse, L.; Borecki, I.B.; Ukkola, O.; Rankinen, T.; Gagnon, J.; Leon, A.S.; Skinner, J.S.; Wilmore, J.H.; et al. A genomewide linkage scan for abdominal subcutaneous and visceral fat in black and white families: The HERITAGE Family Study. Diabetes 2002, 51, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Hoyo, C.; Fortner, K.; Murtha, A.P.; Schildkraut, J.M.; Soubry, A.; Demark-Wahnefried, W.; Jirtle, R.L.; Kurtzberg, J.; Forman, M.R.; Overcash, F.; et al. Association of cord blood methylation fractions at imprinted insulin-like growth factor 2 (IGF2), plasma IGF2, and birth weight. Cancer Causes Control. 2012, 23, 635–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, A.; Heeson, S.; Cooper, W.N.; Douglas, E.; Apostolidou, S.; Moore, G.E.; Maher, E.R.; Reik, W. An association between variants in the IGF2 gene and Beckwith-Wiedemann syndrome: Interaction between genotype and epigenotype. Hum. Mol. Genet. 2004, 13, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Mercader, J.M.; Liao, R.G.; Bell, A.D.; Dymek, Z.; Estrada, K.; Tukiainen, T.; Huerta-Chagoya, A.; Moreno-Macias, H.; Jablonski, K.A.; Hanson, R.L.; et al. A Loss-of-Function Splice Acceptor Variant in IGF2 Is Protective for Type 2 Diabetes. Diabetes 2017, 66, 2903–2914. [Google Scholar] [CrossRef] [PubMed]
- Heald, A.H.; Karvestedt, L.; Anderson, S.G.; McLaughlin, J.; Knowles, A.; Wong, L.; Grill, V.; Cruickshank, J.K.; White, A.; Gibson, J.M.; et al. Low insulin-like growth factor-II levels predict weight gain in normal weight subjects with type 2 diabetes. Am. J. Med. 2006, 119, e169-115. [Google Scholar] [CrossRef]
- Martin, R.M.; Holly, J.M.; Davey Smith, G.; Gunnell, D. Associations of adiposity from childhood into adulthood with insulin resistance and the insulin-like growth factor system: 65-year follow-up of the Boyd Orr Cohort. J. Clin. Endocrinol. Metab. 2006, 91, 3287–3295. [Google Scholar] [CrossRef]
- Sandhu, M.S.; Gibson, J.M.; Heald, A.H.; Dunger, D.B.; Wareham, N.J. Low circulating IGF-II concentrations predict weight gain and obesity in humans. Diabetes 2003, 52, 1403–1408. [Google Scholar] [CrossRef]
- Bann, D.; Holly, J.M.; Lashen, H.; Hardy, R.; Adams, J.; Kuh, D.; Ong, K.K.; Ben-Shlomo, Y. Changes in insulin-like growth factor-I and -II associated with fat but not lean mass in early old age. Obesity 2015, 23, 692–698. [Google Scholar] [CrossRef] [Green Version]
- Murphy, R.; Baptista, J.; Holly, J.; Umpleby, A.M.; Ellard, S.; Harries, L.W.; Crolla, J.; Cundy, T.; Hattersley, A.T. Severe intrauterine growth retardation and atypical diabetes associated with a translocation breakpoint disrupting regulation of the insulin-like growth factor 2 gene. J. Clin. Endocrinol. Metab. 2008, 93, 4373–4380. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, Y.; Timurkan, E.S.; Aydin, S.; Sahin, I.; Timurkan, M.; Citil, C.; Kalayci, M.; Yilmaz, M.; Aksoy, A.; Catak, Z. Acylated and desacylated ghrelin, preptin, leptin, and nesfatin-1 Peptide changes related to the body mass index. Int. J. Endocrinol. 2013, 2013, 236085. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Li, L.; Chen, W.; Liu, H.; Boden, G.; Li, K. Circulating preptin levels in normal, impaired glucose tolerance, and type 2 diabetic subjects. Ann. Med. 2009, 41, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.C.; Li, Y.X.; Asakawa, A.; Ushikai, M.; Kato, I.; Sato, Y.; Cheng, J.T.; Inui, A. Characterization of preptin-induced insulin secretion in pancreatic beta-cells. J. Endocrinol. 2012, 215, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Nunez Lopez, Y.O.; Retnakaran, R.; Zinman, B.; Pratley, R.E.; Seyhan, A.A. Predicting and understanding the response to short-term intensive insulin therapy in people with early type 2 diabetes. Mol. Metab. 2019, 20, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Gallo, W.; Esguerra, J.L.S.; Eliasson, L.; Melander, O. miR-483-5p associates with obesity and insulin resistance and independently associates with new onset diabetes mellitus and cardiovascular disease. PLoS ONE 2018, 13, e0206974. [Google Scholar] [CrossRef]
- Ghaedi, H.; Zare, A.; Omrani, M.D.; Doustimotlagh, A.H.; Meshkani, R.; Alipoor, S.; Alipoor, B. Genetic variants in long noncoding RNA H19 and MEG3 confer risk of type 2 diabetes in an Iranian population. Gene 2018, 675, 265–271. [Google Scholar] [CrossRef]
- Diabetes Genetics Initiative of Broad Institute of, H.; Mit, L.U.; Novartis Institutes of BioMedical, R.; Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar] [CrossRef]
- Jia, H.; Yu, L.; Jiang, Z.; Ji, Q. Association between IGF2BP2 rs4402960 polymorphism and risk of type 2 diabetes mellitus: A meta-analysis. Arch. Med. Res. 2011, 42, 361–367. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, Y.S.; Fang, Y.; Liu, L.; Wu, S.D.; Fu, D.; Wang, X.F. IGF2BP2 genetic variation and type 2 diabetes: A global meta-analysis. DNA Cell Biol. 2012, 31, 713–720. [Google Scholar] [CrossRef]
- Rodriguez, S.; Eiriksdottir, G.; Gaunt, T.R.; Harris, T.B.; Launer, L.J.; Gudnason, V.; Day, I.N. IGF2BP1, IGF2BP2 and IGF2BP3 genotype, haplotype and genetic model studies in metabolic syndrome traits and diabetes. Growth Horm. Igf. Res. 2010, 20, 310–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heald, A.H.; Kaushal, K.; Siddals, K.W.; Rudenski, A.S.; Anderson, S.G.; Gibson, J.M. Insulin-like growth factor binding protein-2 (IGFBP-2) is a marker for the metabolic syndrome. Exp. Clin. Endocrinol. Diabetes 2006, 114, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Jeyaratnaganthan, N.; Hojlund, K.; Kroustrup, J.P.; Larsen, J.F.; Bjerre, M.; Levin, K.; Beck-Nielsen, H.; Frago, S.; Hassan, A.B.; Flyvbjerg, A.; et al. Circulating levels of insulin-like growth factor-II/mannose-6-phosphate receptor in obesity and type 2 diabetes. Growth Horm. Igf. Res. 2010, 20, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Bodkin, N.L.; Ortmeyer, H.K.; Zenilman, M.E.; Webster, N.; Hansen, B.C.; Shuldiner, A.R. Altered insulin receptor messenger ribonucleic acid splicing in liver is associated with deterioration of glucose tolerance in the spontaneously obese and diabetic rhesus monkey: Analysis of controversy between monkey and human studies. J. Clin. Endocrinol. Metab. 1996, 81, 1552–1556. [Google Scholar] [PubMed]
- Kaminska, D.; Hamalainen, M.; Cederberg, H.; Kakela, P.; Venesmaa, S.; Miettinen, P.; Ilves, I.; Herzig, K.H.; Kolehmainen, M.; Karhunen, L.; et al. Adipose tissue INSR splicing in humans associates with fasting insulin level and is regulated by weight loss. Diabetologia 2014, 57, 347–351. [Google Scholar] [CrossRef]
- Besic, V.; Shi, H.; Stubbs, R.S.; Hayes, M.T. Aberrant liver insulin receptor isoform a expression normalises with remission of type 2 diabetes after gastric bypass surgery. PLoS ONE 2015, 10, e0119270. [Google Scholar] [CrossRef]
- Escribano, O.; Beneit, N.; Rubio-Longas, C.; Lopez-Pastor, A.R.; Gomez-Hernandez, A. The Role of Insulin Receptor Isoforms in Diabetes and Its Metabolic and Vascular Complications. J. Diabetes Res. 2017, 2017, 1403206. [Google Scholar] [CrossRef]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef]
- Froesch, E.R.; Buergi, H.; Ramseier, E.B.; Bally, P.; Labhart, A. Antibody-Suppressible and Nonsuppressible Insulin-Like Activities in Human Serum and Their Physiologic Significance. An Insulin Assay with Adipose Tissue of Increased Precision and Specificity. J. Clin. Investig. 1963, 42, 1816–1834. [Google Scholar] [CrossRef]
- Rinderknecht, E.; Humbel, R.E. Amino-terminal sequences of two polypeptides from human serum with nonsuppressible insulin-like and cell-growth-promoting activities: Evidence for structural homology with insulin B chain. Proc. Natl. Acad. Sci. USA 1976, 73, 4379–4381. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Pinal, C.S. Regulation of fetal growth by the somatotrophic axis. J. Nutr. 2003, 133, 1741S–1746S. [Google Scholar] [CrossRef] [PubMed]
- Holly, J.; Sabin, M.; Perks, C.; Shield, J. Adipogenesis and IGF-1. Metab. Syndr. Relat. Disord. 2006, 4, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, F.M.; Smas, C.M.; Sul, H.S. Understanding adipocyte differentiation. Physiol. Rev. 1998, 78, 783–809. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.T.; Hausman, G.J. Insulinlike growth factor-1 (IGF-1)-induced stimulation of porcine preadipocyte replication. In Vitro. Cell Dev. Biol. Anim. 1995, 31, 404–408. [Google Scholar] [CrossRef]
- Suryawan, A.; Swanson, L.V.; Hu, C.Y. Insulin and hydrocortisone, but not triiodothyronine, are required for the differentiation of pig preadipocytes in primary culture. J. Anim. Sci. 1997, 75, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Back, K.; Brannmark, C.; Stralfors, P.; Arnqvist, H.J. Differential effects of IGF-I, IGF-II and insulin in human preadipocytes and adipocytes--role of insulin and IGF-I receptors. Mol. Cell Endocrinol. 2011, 339, 130–135. [Google Scholar] [CrossRef]
- Despres, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef]
- Grohmann, M.; Sabin, M.; Holly, J.; Shield, J.; Crowne, E.; Stewart, C. Characterization of differentiated subcutaneous and visceral adipose tissue from children: The influences of TNF-alpha and IGF-I. J. Lipid Res. 2005, 46, 93–103. [Google Scholar] [CrossRef]
- Lundgren, M.; Buren, J.; Ruge, T.; Myrnas, T.; Eriksson, J.W. Glucocorticoids down-regulate glucose uptake capacity and insulin-signaling proteins in omental but not subcutaneous human adipocytes. J. Clin. Endocrinol. Metab. 2004, 89, 2989–2997. [Google Scholar] [CrossRef]
- Alfares, M.N.; Perks, C.M.; Hamilton-Shield, J.P.; Holly, J.M.P. Insulin-like growth factor-II in adipocyte regulation: Depot-specific actions suggest a potential role limiting excess visceral adiposity. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E1098–E1107. [Google Scholar] [CrossRef]
- Florini, J.R.; Magri, K.A.; Ewton, D.Z.; James, P.L.; Grindstaff, K.; Rotwein, P.S. “Spontaneous” differentiation of skeletal myoblasts is dependent upon autocrine secretion of insulin-like growth factor-II. J. Biol. Chem. 1991, 266, 15917–15923. [Google Scholar] [PubMed]
- Wilson, E.M.; Hsieh, M.M.; Rotwein, P. Autocrine growth factor signaling by insulin-like growth factor-II mediates MyoD-stimulated myocyte maturation. J. Biol. Chem. 2003, 278, 41109–41113. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.M.; Rotwein, P. Control of MyoD function during initiation of muscle differentiation by an autocrine signaling pathway activated by insulin-like growth factor-II. J. Biol. Chem. 2006, 281, 29962–29971. [Google Scholar] [CrossRef] [PubMed]
- Zierath, J.R.; Bang, P.; Galuska, D.; Hall, K.; Wallberg-Henriksson, H. Insulin-like growth factor II stimulates glucose transport in human skeletal muscle. FEBS Lett. 1992, 307, 379–382. [Google Scholar] [CrossRef] [Green Version]
- Hiden, U.; Glitzner, E.; Hartmann, M.; Desoye, G. Insulin and the IGF system in the human placenta of normal and diabetic pregnancies. J. Anat 2009, 215, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Fu, Z.; Jiang, H.; Chen, L.; Wu, X.; Ding, H.; Xia, Y.; Wang, X.; Tang, Q.; Wu, W. IGF2-derived miR-483-3p contributes to macrosomia through regulating trophoblast proliferation by targeting RB1CC1. Mol. Hum. Reprod. 2018, 24, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Chen, F.; Lv, P.P.; Zhang, D.; Ding, G.L.; Hu, X.L.; Feng, C.; Sheng, J.Z.; Huang, H.F. Maternal high estradiol exposure alters CDKN1C and IGF2 expression in human placenta. Placenta 2018, 61, 72–79. [Google Scholar] [CrossRef]
- Song, C.; Yang, Z.; Dong, D.; Xu, J.; Wang, J.; Li, H.; Huang, Y.; Lan, X.; Lei, C.; Ma, Y.; et al. miR-483 inhibits bovine myoblast cell proliferation and differentiation via IGF1/PI3K/AKT signal pathway. J. Cell Physiol. 2019, 234, 9839–9848. [Google Scholar] [CrossRef]
- Ferland-McCollough, D.; Fernandez-Twinn, D.S.; Cannell, I.G.; David, H.; Warner, M.; Vaag, A.A.; Bork-Jensen, J.; Brons, C.; Gant, T.W.; Willis, A.E.; et al. Programming of adipose tissue miR-483-3p and GDF-3 expression by maternal diet in type 2 diabetes. Cell Death Differ. 2012, 19, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; He, H.; Xie, Y.; Zhao, L.; Zhao, S.; Wan, X.; Yang, W.; Mo, Z. miR-125a-3p and miR-483-5p promote adipogenesis via suppressing the RhoA/ROCK1/ERK1/2 pathway in multiple symmetric lipomatosis. Sci. Rep. 2015, 5, 11909. [Google Scholar] [CrossRef]
- Escribano, O.; Gomez-Hernandez, A.; Diaz-Castroverde, S.; Nevado, C.; Garcia, G.; Otero, Y.F.; Perdomo, L.; Beneit, N.; Benito, M. Insulin receptor isoform A confers a higher proliferative capability to pancreatic beta cells enabling glucose availability and IGF-I signaling. Mol. Cell Endocrinol. 2015, 409, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.; Sivadas, A.; Shamsudheen, K.V.; Jayarajan, R.; Verma, A.; Sivasubbu, S.; Scaria, V.; Datta, M. RNA sequencing of db/db mice liver identifies lncRNA H19 as a key regulator of gluconeogenesis and hepatic glucose output. Sci. Rep. 2017, 7, 8312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Geng, T.; Wang, Z.; Zhang, R.; Cao, T.; Camporez, J.P.; Cai, S.Y.; Liu, Y.; Dandolo, L.; Shulman, G.I.; et al. Elevated hepatic expression of H19 long noncoding RNA contributes to diabetic hyperglycemia. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, T.; Liu, Y.; Xu, Y.; Jiang, Y.; Zhang, N.; Wang, Z.; Carmichael, G.G.; Taylor, H.S.; Li, D.; Huang, Y. H19 lncRNA Promotes Skeletal Muscle Insulin Sensitivity in Part by Targeting AMPK. Diabetes 2018, 67, 2183–2198. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S. A Regulator of Metabolic Reprogramming: MicroRNA Let-7. Transl. Oncol. 2019, 12, 1005–1013. [Google Scholar] [CrossRef]
- Dey, B.K.; Pfeifer, K.; Dutta, A. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev. 2014, 28, 491–501. [Google Scholar] [CrossRef]
- Lewis, A.; Lee, J.Y.; Donaldson, A.V.; Natanek, S.A.; Vaidyanathan, S.; Man, W.D.; Hopkinson, N.S.; Sayer, A.A.; Patel, H.P.; Cooper, C.; et al. Increased expression of H19/miR-675 is associated with a low fat-free mass index in patients with COPD. J. Cachexia Sarcopenia Muscle 2016, 7, 330–344. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Wang, J.; Liu, D.; Zang, S.; Ma, N.; Zhao, L.; Zhang, L.; Zhang, X.; Qiao, C. The lncRNA H19/miR-675 axis regulates myocardial ischemic and reperfusion injury by targeting PPARalpha. Mol. Immunol. 2019, 105, 46–54. [Google Scholar] [CrossRef]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef]
- Yi, X.; Liu, J.; Wu, P.; Gong, Y.; Xu, X.; Li, W. The key microRNA on lipid droplet formation during adipogenesis from human mesenchymal stem cells. J. Cell Physiol. 2019. [Google Scholar] [CrossRef]
- Nevado, C.; Valverde, A.M.; Benito, M. Role of insulin receptor in the regulation of glucose uptake in neonatal hepatocytes. Endocrinology 2006, 147, 3709–3718. [Google Scholar] [CrossRef]
- Diaz-Castroverde, S.; Gomez-Hernandez, A.; Fernandez, S.; Garcia-Gomez, G.; Di Scala, M.; Gonzalez-Aseguinolaza, G.; Fernandez-Millan, E.; Gonzalez-Rodriguez, A.; Garcia-Bravo, M.; Chambon, P.; et al. Insulin receptor isoform A ameliorates long-term glucose intolerance in diabetic mice. Dis. Model. Mech. 2016, 9, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Nica, A.C.; Ongen, H.; Irminger, J.C.; Bosco, D.; Berney, T.; Antonarakis, S.E.; Halban, P.A.; Dermitzakis, E.T. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res. 2013, 23, 1554–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, D.J.; Strutt, B.; Arany, E.; Zaina, S.; Coukell, S.; Graham, C.F. Increased and persistent circulating insulin-like growth factor II in neonatal transgenic mice suppresses developmental apoptosis in the pancreatic islets. Endocrinology 2000, 141, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Cornu, M.; Yang, J.Y.; Jaccard, E.; Poussin, C.; Widmann, C.; Thorens, B. Glucagon-like peptide-1 protects beta-cells against apoptosis by increasing the activity of an IGF-2/IGF-1 receptor autocrine loop. Diabetes 2009, 58, 1816–1825. [Google Scholar] [CrossRef] [PubMed]
- Modi, H.; Jacovetti, C.; Tarussio, D.; Metref, S.; Madsen, O.D.; Zhang, F.P.; Rantakari, P.; Poutanen, M.; Nef, S.; Gorman, T.; et al. Autocrine Action of IGF2 Regulates Adult beta-Cell Mass and Function. Diabetes 2015, 64, 4148–4157. [Google Scholar] [CrossRef] [PubMed]
- Casellas, A.; Mallol, C.; Salavert, A.; Jimenez, V.; Garcia, M.; Agudo, J.; Obach, M.; Haurigot, V.; Vila, L.; Molas, M.; et al. Insulin-like Growth Factor 2 Overexpression Induces beta-Cell Dysfunction and Increases Beta-cell Susceptibility to Damage. J. Biol. Chem. 2015, 290, 16772–16785. [Google Scholar] [CrossRef]
- Sanchez-Parra, C.; Jacovetti, C.; Dumortier, O.; Lee, K.; Peyot, M.L.; Guay, C.; Prentki, M.; Laybutt, D.R.; Van Obberghen, E.; Regazzi, R. Contribution of the Long Noncoding RNA H19 to beta-Cell Mass Expansion in Neonatal and Adult Rodents. Diabetes 2018, 67, 2254–2267. [Google Scholar] [CrossRef]
- Mohan, R.; Mao, Y.; Zhang, S.; Zhang, Y.W.; Xu, C.R.; Gradwohl, G.; Tang, X. Differentially Expressed MicroRNA-483 Confers Distinct Functions in Pancreatic beta- and alpha-Cells. J. Biol. Chem. 2015, 290, 19955–19966. [Google Scholar] [CrossRef]
- Durante, F. Nesso fisio-pathologico tra la struttura dei nei materni e la genesi di alcuni tumori maligni. Arch. Mem. Ed. Oss. Di Chirugia Pract. 1874, 11, 217–226. [Google Scholar]
- Liu, J. The dualistic origin of human tumors. Semin. Cancer Biol. 2018, 53, 1–16. [Google Scholar] [CrossRef]
- Milanovic, M.; Yu, Y.; Schmitt, C.A. The Senescence-Stemness Alliance—A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018, 28, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Vinnitsky, V. The development of a malignant tumor is due to a desperate asexual self-cloning process in which cancer stem cells develop the ability to mimic the genetic program of germline cells. Intrinsically Disord. Proteins 2014, 2, e29997. [Google Scholar] [CrossRef] [Green Version]
- Brouwer-Visser, J.; Huang, G.S. IGF2 signaling and regulation in cancer. Cytokine Growth Factor Rev. 2015, 26, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, C. IGF2 and cancer. Endocr Relat Cancer. 2013, 20, R321–R339. [Google Scholar] [CrossRef] [Green Version]
- Halje, M.; Nordin, M.; Bergman, D.; Engström, W. Review: The effect of insulin-like growth factor II in the regulation of tumour cell growth in vitro and tumourigenesis in vivo. In Vivo 2012, 26, 519–526. [Google Scholar]
- Rainier, S.; Johnson, L.A.; Dobry, C.J.; Ping, A.J.; Grundy, P.E.; Feinberg, A.P. Relaxation of imprinted genes in human cancer. Nature 1993, 362, 747–749. [Google Scholar] [CrossRef]
- Ward, A. Beck-Wiedemann syndrome and Wilms’ tumour. Mol. Hum. Reprod. 1997, 3, 157–168. [Google Scholar] [CrossRef]
- Leick, M.B.; Shoff, C.J.; Wang, E.C.; Congress, J.L.; Gallicano, G.I. Loss of imprinting of IGF2 and the epigenetic progenitor model of cancer. Am. J. Stem Cells 2012, 1, 59–74. [Google Scholar]
- Grbesa, I.; Marinkovic, M.; Ivkic, M.; Kruslin, B.; Novak-Kujundzic, R.; Pegan, B.; Bogdanovic, O.; Bedekovic, V.; Gall-Troselj, K. Loss of imprinting of IGF2 and H19, loss of heterozygosity of IGF2R and CTCF, and Helicobacter pylori infection in laryngeal squamous cell carcinoma. J. Mol. Med. (Berl.) 2008, 86, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Maenaka, S.; Hikichi, T.; Imai, M.A.; Minamoto, T.; Kawahara, E. Loss of imprinting in IGF2 in colorectal carcinoma assessed by microdissection. Oncol. Rep. 2006, 15, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Chadwick, R.B.; Peltomaki, P.; Plass, C.; Nakamura, Y.; de La Chapelle, A. Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Bhusari, S.; Yang, B.; Kueck, J.; Huang, W.; Jarrard, D.F. Insulin-like growth factor-2 (IGF2) loss of imprinting marks a field defect within human prostates containing cancer. Prostate 2011, 71, 1621–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, V.X.; Dobosy, J.R.; Desotelle, J.A.; Almassi, N.; Ewald, J.A.; Srinivasan, R.; Berres, M.; Svaren, J.; Weindruch, R.; Jarrard, D.F. Aging and cancer-related loss of insulin-like growth factor 2 imprinting in the mouse and human prostate. Cancer Res. 2008, 68, 6797–6802. [Google Scholar] [CrossRef]
- Holly, J.M.; Zeng, L.; Perks, C.M. Epithelial cancers in the post-genomic era: Should we reconsider our lifestyle? Cancer Metastasis Rev. 2013, 32, 673–705. [Google Scholar] [CrossRef]
- Sakatani, T.; Kaneda, A.; Iacobuzio-Donahue, C.A.; Carter, M.G.; de Boom Witzel, S.; Okano, H.; Ko, M.S.; Ohlsson, R.; Longo, D.L.; Feinberg, A.P. Loss of imprinting of Igf2 alters intestinal maturation and tumorigenesis in mice. Science 2005, 307, 1976–1978. [Google Scholar] [CrossRef]
- Damaschke, N.A.; Yang, B.; Bhusari, S.; Avilla, M.; Zhong, W.; Blute, M.L., Jr.; Huang, W.; Jarrard, D.F. Loss of Igf2 Gene Imprinting in Murine Prostate Promotes Widespread Neoplastic Growth. Cancer Res. 2017, 77, 5236–5247. [Google Scholar] [CrossRef]
- Cui, H.; Cruz-Correa, M.; Giardiello, F.M.; Hutcheon, D.F.; Kafonek, D.R.; Brandenburg, S.; Wu, Y.; He, X.; Powe, N.R.; Feinberg, A.P. Loss of IGF2 imprinting: A potential marker of colorectal cancer risk. Science 2003, 299, 1753–1755. [Google Scholar] [CrossRef]
- Woodson, K.; Flood, A.; Green, L.; Tangrea, J.A.; Hanson, J.; Cash, B.; Schatzkin, A.; Schoenfeld, P. Loss of insulin-like growth factor-II imprinting and the presence of screen-detected colorectal adenomas in women. J. Natl. Cancer Inst. 2004, 96, 407–410. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Belfiore, A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front. Endocrinol. (Lausanne) 2014, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Shin, D.M.; Schneider, G.; Ratajczak, J.; Kucia, M. Parental imprinting regulates insulin-like growth factor signaling: A Rosetta Stone for understanding the biology of pluripotent stem cells, aging and cancerogenesis. Leukemia 2013, 27, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Nosho, K.; Shima, K.; Huttenhower, C.; Tanaka, N.; Hazra, A.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. Hypomethylation of the IGF2 DMR in colorectal tumors, detected by bisulfite pyrosequencing, is associated with poor prognosis. Gastroenterology 2010, 139, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Katsaros, D.; Canuto, E.M.; Biglia, N.; Risch, H.A.; Yu, H. LIN-28B/let-7a/IGF-II axis molecular subtypes are associated with epithelial ovarian cancer prognosis. Gynecol. Oncol. 2016, 141, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Tahara, S.; Horiguchi, N.; Kawamura, T.; Okubo, M.; Yamada, H.; Yoshida, D.; Ohmori, T.; Maeda, K.; Komura, N.; et al. Methylation status of IGF2 DMR and LINE1 in leukocyte DNA provides distinct clinicopathological features of gastric cancer patients. Clin. Exp. Med. 2018, 18, 215–220. [Google Scholar] [CrossRef]
- Van Arsdale, A.R.; Arend, R.C.; Cossio, M.J.; Erickson, B.K.; Wang, Y.; Doo, D.W.; Leath, C.A.; Goldberg, G.L.; Huang, G.S. Insulin-like growth factor 2: A poor prognostic biomarker linked to racial disparity in women with uterine carcinosarcoma. Cancer Med. 2018, 7, 616–625. [Google Scholar] [CrossRef]
- Gao, T.; Liu, X.; He, B.; Nie, Z.; Zhu, C.; Zhang, P.; Wang, S. Exosomal lncRNA 91H is associated with poor development in colorectal cancer by modifying HNRNPK expression. Cancer Cell Int. 2018, 18, 11. [Google Scholar] [CrossRef]
- Han, D.; Gao, X.; Wang, M.; Qiao, Y.; Xu, Y.; Yang, J.; Dong, N.; He, J.; Sun, Q.; Lv, G.; et al. Long noncoding RNA H19 indicates a poor prognosis of colorectal cancer and promotes tumor growth by recruiting and binding to eIF4A3. Oncotarget 2016, 7, 22159–22173. [Google Scholar] [CrossRef]
- Li, H.; Yu, B.; Li, J.; Su, L.; Yan, M.; Zhu, Z.; Liu, B. Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget 2014, 5, 2318–2329. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Jin, J.; Rong, K.; Zhuo, L.; Li, P. MicroRNA675 inhibits cell proliferation and invasion in melanoma by directly targeting metadherin. Mol. Med. Rep. 2018, 17, 3372–3379. [Google Scholar] [CrossRef]
- Lv, M.; Zhong, Z.; Huang, M.; Tian, Q.; Jiang, R.; Chen, J. lncRNA H19 regulates epithelial-mesenchymal transition and metastasis of bladder cancer by miR-29b-3p as competing endogenous RNA. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1887–1899. [Google Scholar] [CrossRef] [PubMed]
- Matouk, I.J.; Raveh, E.; Abu-lail, R.; Mezan, S.; Gilon, M.; Gershtain, E.; Birman, T.; Gallula, J.; Schneider, T.; Barkali, M.; et al. Oncofetal H19 RNA promotes tumor metastasis. Biochim. Biophys. Acta 2014, 1843, 1414–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvianti, F.; Canu, L.; Poli, G.; Armignacco, R.; Scatena, C.; Cantini, G.; Di Franco, A.; Gelmini, S.; Ercolino, T.; Pazzagli, M.; et al. New insights in the clinical and translational relevance of miR483-5p in adrenocortical cancer. Oncotarget 2017, 8, 65525–65533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Wang, Y.; Luan, W.; Wang, P.; Tao, T.; Zhang, J.; Qian, J.; Liu, N.; You, Y. Long non-coding RNA H19 promotes glioma cell invasion by deriving miR-675. PLoS ONE 2014, 9, e86295. [Google Scholar] [CrossRef] [PubMed]
- Si, X.; Zang, R.; Zhang, E.; Liu, Y.; Shi, X.; Zhang, E.; Shao, L.; Li, A.; Yang, N.; Han, X.; et al. LncRNA H19 confers chemoresistance in ERalpha-positive breast cancer through epigenetic silencing of the pro-apoptotic gene BIK. Oncotarget 2016, 7, 81452–81462. [Google Scholar] [CrossRef]
- Tan, D.; Wu, Y.; Hu, L.; He, P.; Xiong, G.; Bai, Y.; Yang, K. Long noncoding RNA H19 is up-regulated in esophageal squamous cell carcinoma and promotes cell proliferation and metastasis. Dis. Esophagus 2017, 30, 1–9. [Google Scholar] [CrossRef]
- Vennin, C.; Spruyt, N.; Dahmani, F.; Julien, S.; Bertucci, F.; Finetti, P.; Chassat, T.; Bourette, R.P.; Le Bourhis, X.; Adriaenssens, E. H19 non coding RNA-derived miR-675 enhances tumorigenesis and metastasis of breast cancer cells by downregulating c-Cbl and Cbl-b. Oncotarget 2015, 6, 29209–29223. [Google Scholar] [CrossRef]
- Wang, Q.; Cheng, N.; Li, X.; Pan, H.; Li, C.; Ren, S.; Su, C.; Cai, W.; Zhao, C.; Zhang, L.; et al. Correlation of long non-coding RNA H19 expression with cisplatin-resistance and clinical outcome in lung adenocarcinoma. Oncotarget 2017, 8, 2558–2567. [Google Scholar] [CrossRef]
- Yang, X.; Lou, Y.; Wang, M.; Liu, C.; Liu, Y.; Huang, W. miR675 promotes colorectal cancer cell growth dependent on tumor suppressor DMTF1. Mol. Med. Rep. 2019, 19, 1481–1490. [Google Scholar] [CrossRef]
- Yang, Y.; Meng, Q.; Wang, C.; Li, X.; Lu, Y.; Xin, X.; Zheng, Q.; Lu, D. MicroRNA 675 cooperates PKM2 to aggravate progression of human liver cancer stem cells induced from embryonic stem cells. J. Mol. Med. (Berl.) 2018, 96, 1119–1130. [Google Scholar] [CrossRef]
- Zhang, D.-M.; Lin, Z.-Y.; Yang, Z.-H.; Wang, Y.-Y.; Wan, D.; Zhong, J.-L.; Zhuang, P.-L.; Huang, Z.-Q.; Zhou, B.; Chen, W.-L. IncRNA H19 promotes tongue squamous cell carcinoma progression through β-catenin/GSK3β/EMT signaling via association with EZH2. Am. J. Transl. Res. 2017, 9, 3474–3486. [Google Scholar] [PubMed]
- Zheng, Z.G.; Xu, H.; Suo, S.S.; Xu, X.L.; Ni, M.W.; Gu, L.H.; Chen, W.; Wang, L.Y.; Zhao, Y.; Tian, B.; et al. The Essential Role of H19 Contributing to Cisplatin Resistance by Regulating Glutathione Metabolism in High-Grade Serous Ovarian Cancer. Sci. Rep. 2016, 6, 26093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.W.; Zhang, H.; Duan, C.J.; Gao, Y.; Cheng, Y.D.; He, D.; Li, R.; Zhang, C.F. miR-675-5p enhances tumorigenesis and metastasis of esophageal squamous cell carcinoma by targeting REPS2. Oncotarget 2016, 7, 30730–30747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Wang, N.; Yi, X.; Tang, C.; Wang, D. Long non-coding RNA H19 regulates E2F1 expression by competitively sponging endogenous miR-29a-3p in clear cell renal cell carcinoma. Cell Biosci. 2017. [Google Scholar] [CrossRef]
- Abue, M.; Yokoyama, M.; Shibuya, R.; Tamai, K.; Yamaguchi, K.; Sato, I.; Tanaka, N.; Hamada, S.; Shimosegawa, T.; Sugamura, K.; et al. Circulating miR-483-3p and miR-21 is highly expressed in plasma of pancreatic cancer. Int. J. Oncol. 2015, 46, 539–547. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, J.F.; Wang, H.; Cui, J.; Gao, S.; Hoffman, A.R.; Li, W. CRISPR Cas9-guided chromatin immunoprecipitation identifies miR483 as an epigenetic modulator of IGF2 imprinting in tumors. Oncotarget 2017, 8, 34177–34190. [Google Scholar] [CrossRef]
- Lecerf, C.; Le Bourhis, X.; Adriaenssens, E. The long non-coding RNA H19: An active player with multiple facets to sustain the hallmarks of cancer. Cell Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Raveh, E.; Matouk, I.J.; Gilon, M.; Hochberg, A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis—A proposed unifying theory. Mol. Cancer 2015, 14, 184. [Google Scholar] [CrossRef]
- Yoshimura, H.; Matsuda, Y.; Yamamoto, M.; Kamiya, S.; Ishiwata, T. Expression and role of long non-coding RNA H19 in carcinogenesis. Front. Biosci. (Landmark Ed.) 2018, 23, 614–625. [Google Scholar]
- Lu, Y.; Song, S.; Jiang, X.; Meng, Q.; Wang, C.; Li, X.; Yang, Y.; Xin, X.; Zheng, Q.; Wang, L.; et al. miR675 Accelerates Malignant Transformation of Mesenchymal Stem Cells by Blocking DNA Mismatch Repair. Mol. Nucleic Acids 2019, 14, 171–183. [Google Scholar] [CrossRef]
- Kim, H.Y.; Ha Thi, H.T.; Hong, S. IMP2 and IMP3 cooperate to promote the metastasis of triple-negative breast cancer through destabilization of progesterone receptor. Cancer Lett. 2018, 415, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.X.; Hong, F.; Zhang, Y.; Ansa-Addo, E.; Li, Z. GRP94/gp96 in Cancer: Biology, Structure, Immunology, and Drug Development. Adv. Cancer Res. 2016, 129, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Arner, P. Human fat cell lipolysis: Biochemistry, regulation and clinical role. Best. Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 471–482. [Google Scholar] [CrossRef]
- Klein, S.; Fontana, L.; Young, V.L.; Coggan, A.R.; Kilo, C.; Patterson, B.W.; Mohammed, B.S. Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N. Engl. J. Med. 2004, 350, 2549–2557. [Google Scholar] [CrossRef]
- Andersson, D.P.; Thorell, A.; Löfgren, P.; Wirén, M.; Toft, E.; Qvisth, V.; Riserus, U.; Berglund, L.; Näslund, E.; Bringman, S.; et al. Omentectomy in addition to gastric bypass surgery and influence on insulin sensitivity: A randomized double blind controlled trial. Clin. Nutr. 2014, 33, 991–996. [Google Scholar] [CrossRef] [Green Version]
- Parks, E.J.; Parks, E.J. Changes in fat synthesis influenced by dietary macronutrient content. Proc. Nutr. Soc. 2002, 61, 281–286. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holly, J.M.P.; Biernacka, K.; Perks, C.M. The Neglected Insulin: IGF-II, a Metabolic Regulator with Implications for Diabetes, Obesity, and Cancer. Cells 2019, 8, 1207. https://doi.org/10.3390/cells8101207
Holly JMP, Biernacka K, Perks CM. The Neglected Insulin: IGF-II, a Metabolic Regulator with Implications for Diabetes, Obesity, and Cancer. Cells. 2019; 8(10):1207. https://doi.org/10.3390/cells8101207
Chicago/Turabian StyleHolly, Jeff M. P., Kalina Biernacka, and Claire M. Perks. 2019. "The Neglected Insulin: IGF-II, a Metabolic Regulator with Implications for Diabetes, Obesity, and Cancer" Cells 8, no. 10: 1207. https://doi.org/10.3390/cells8101207
APA StyleHolly, J. M. P., Biernacka, K., & Perks, C. M. (2019). The Neglected Insulin: IGF-II, a Metabolic Regulator with Implications for Diabetes, Obesity, and Cancer. Cells, 8(10), 1207. https://doi.org/10.3390/cells8101207