Contextual Regulation of TGF-β Signaling in Liver Cancer


Abstract
:1. Introduction
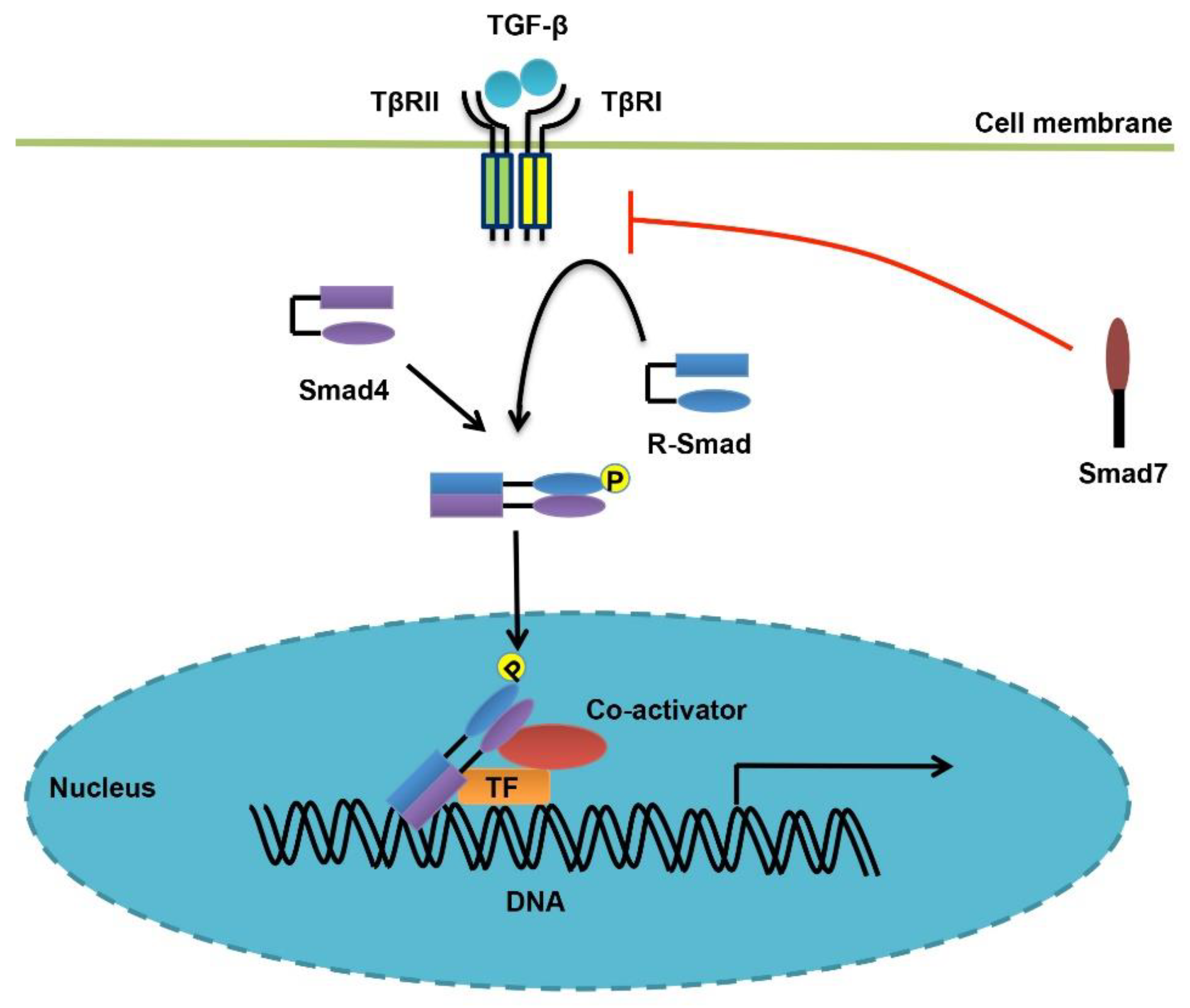
2. Overview of TGF-β Signaling
3. Alterations of TGF-β Signaling in Liver Cancer Revealed by High-Throughput Studies
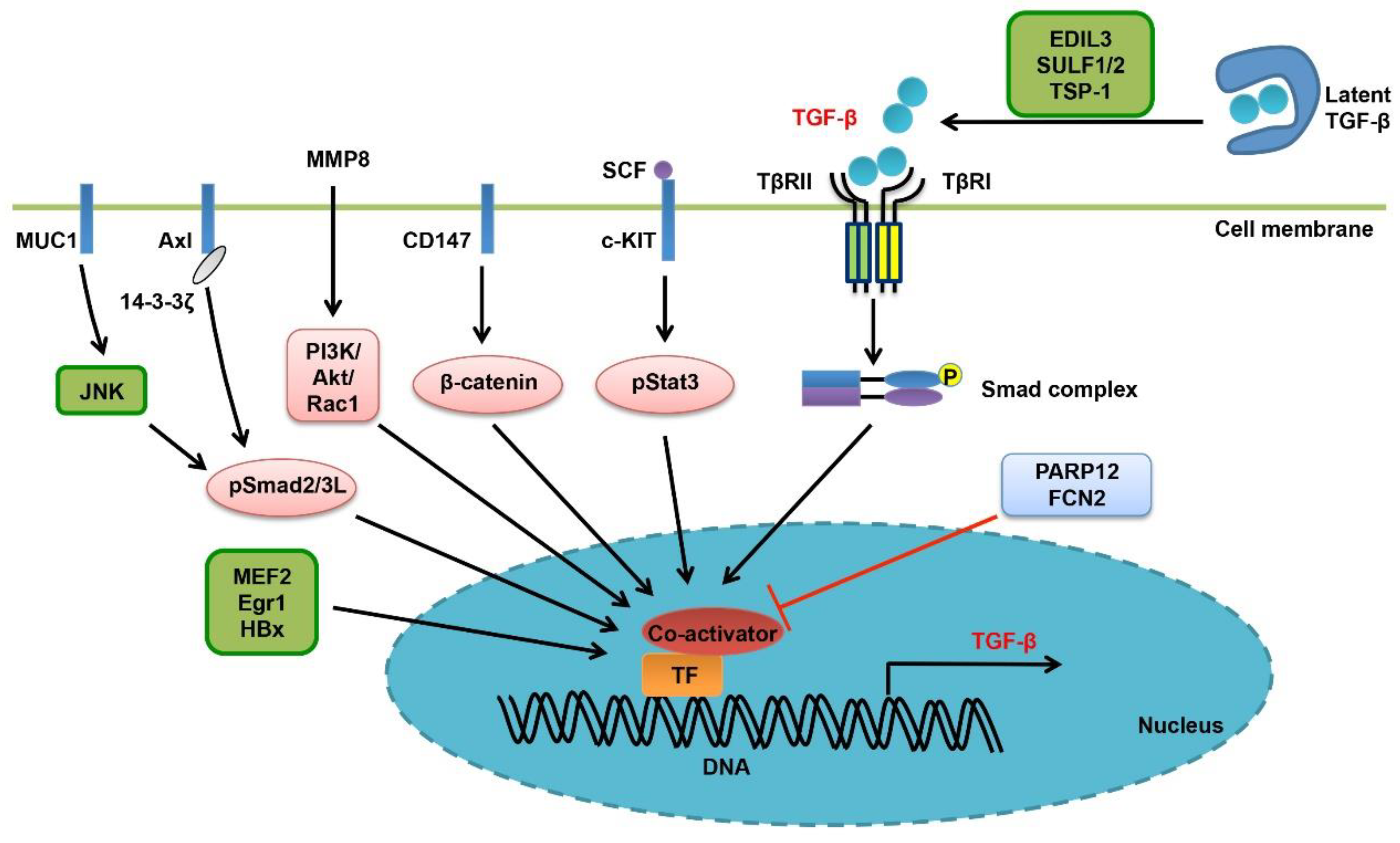
4. Enhanced Bioavailability of TGF-β Ligand in Liver Cancer
5. Deregulation of TGF-β Receptors
6. Regulation of Smad Proteins
7. Altering the Transcriptional Activity of Smads
8. Conclusions and Outlooks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC: | hepatocellular carcinoma |
| TGF-β: | Transforming growth factor beta |
| BMP: | bone morphogenetic protein |
| GDF: | growth and differentiation factor |
| R-Smad: | receptor-regulated Smad protein |
| Co-Smad: | common Smad |
| I-Smad: | inhibitory Smad |
| pSmad3L: | the linker-phosphorylated form of Smad3 |
| pSmad3C: | the C-terminus-phosphorylated form of Smad3 |
| MAPK: | mitogen-activated protein kinase |
| ERK: | extracellular signal-regulated kinase |
| PTM: | post-translational modification |
| HBx: | hepatitis B virus X protein |
| HCV NS3: | HCV nonstructural protein 3 |
| MMP: | matrix metalloproteinase |
| MEF2: | myocyte enhancer factors 2 |
| Egr1: | Early growth response factor 1 |
| HSPG: | heparan sulfate proteoglycan |
| HSC: | hepatic stellate cell |
| IQGAP: | IQ motif-containing GTPase activating protein |
| XIST: | X-inactive specific transcript |
| FHL: | four-and-a-half LIM protein |
| NEK6: | NIMA-related kinases 6 |
| NCX1: | Na+/Ca2+ exchanger 1 |
| TRPC6: | transient receptor potential channel 6 |
| SRF: | serum response factor |
| cPLA2α: | cytosolic phospholipase A2α |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, A. Hepatocellular Carcinoma. N Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Marquardt, J.U.; Andersen, J.B.; Thorgeirsson, S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 2015, 15, 653–667. [Google Scholar] [CrossRef]
- Katz, L.H.; Likhter, M.; Jogunoori, W.; Belkin, M.; Ohshiro, K.; Mishra, L. TGF-beta signaling in liver and gastrointestinal cancers. Cancer Lett. 2016, 379, 166–172. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Diaz, D. Transforming Growth Factor-beta-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef]
- Dituri, F.; Mancarella, S.; Cigliano, A.; Chieti, A.; Giannelli, G. TGF-beta as Multifaceted Orchestrator in HCC Progression: Signaling, EMT, Immune Microenvironment, and Novel Therapeutic Perspectives. Semin. Liver Dis. 2019, 39, 53–69. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Seki, T.; Okazaki, K. TGF-beta signal shifting between tumor suppression and fibro-carcinogenesis in human chronic liver diseases. J. Gastroenterol. 2014, 49, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Moreno-Caceres, J.; Sanchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P. TGF-beta signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Bottinger, E.P.; Jakowlew, S.B.; Bagnall, K.M.; Mariano, J.; Anver, M.R.; Letterio, J.J.; Wakefield, L.M. Transforming growth factor-beta1 is a new form of tumor suppressor with true haploid insufficiency. Nat. Med. 1998, 4, 802–807. [Google Scholar] [CrossRef]
- Im, Y.H.; Kim, H.T.; Kim, I.Y.; Factor, V.M.; Hahm, K.B.; Anzano, M.; Jang, J.J.; Flanders, K.; Haines, D.C.; Thorgeirsson, S.S.; et al. Heterozygous mice for the transforming growth factor-beta type II receptor gene have increased susceptibility to hepatocellular carcinogenesis. Cancer Res. 2001, 61, 6665–6668. [Google Scholar]
- Yang, Y.A.; Zhang, G.M.; Feigenbaum, L.; Zhang, Y.E. Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through downregulation of Bcl-2. Cancer Cell 2006, 9, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Dooley, S.; ten Dijke, P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef]
- Giannelli, G.; Mikulits, W.; Dooley, S.; Fabregat, I.; Moustakas, A.; ten Dijke, P.; Portincasa, P.; Winter, P.; Janssen, R.; Leporatti, S.; et al. The rationale for targeting TGF-beta in chronic liver diseases. Eur. J. Clin. Investig. 2016, 46, 349–361. [Google Scholar] [CrossRef]
- Abou-Shady, M.; Baer, H.U.; Friess, H.; Berberat, P.; Zimmermann, A.; Graber, H.; Gold, L.I.; Korc, M.; Buchler, M.W. Transforming growth factor betas and their signaling receptors in human hepatocellular carcinoma. Am. J. Surg. 1999, 177, 209–215. [Google Scholar] [CrossRef]
- Song, B.C.; Chung, Y.H.; Kim, J.A.; Choi, W.B.; Suh, D.D.; Pyo, S.I.; Shin, J.W.; Lee, H.C.; Lee, Y.S.; Suh, D.J. Transforming growth factor-beta1 as a useful serologic marker of small hepatocellular carcinoma. Cancer 2002, 94, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.H.; Shao, Y.Y.; Chan, S.Y.; Huang, C.Y.; Hsu, C.H.; Cheng, A.L. High Serum Transforming Growth Factor-beta1 Levels Predict Outcome in Hepatocellular Carcinoma Patients Treated with Sorafenib. Clin. Cancer Res. 2015, 21, 3678–3684. [Google Scholar] [CrossRef]
- Watanabe, Y.; Iwamura, A.; Shimada, Y.J.; Wakai, K.; Tamakoshi, A.; Iso, H.; Group, J.S. Transforming Growth Factor-beta1 as a Predictor for the Development of Hepatocellular Carcinoma: A Nested Case-Controlled Study. EBioMedicine 2016, 12, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Factor, V.M.; Loi, R.; Thorgeirsson, S.S. Activation of beta-catenin during hepatocarcinogenesis in transgenic mouse models: Relationship to phenotype and tumor grade. Cancer Res. 2001, 61, 2085–2091. [Google Scholar] [PubMed]
- Deane, N.G.; Lee, H.; Hamaamen, J.; Ruley, A.; Washington, M.K.; LaFleur, B.; Thorgeirsson, S.S.; Price, R.; Beauchamp, R.D. Enhanced tumor formation in cyclin D1 x transforming growth factor beta1 double transgenic mice with characterization by magnetic resonance imaging. Cancer Res. 2004, 64, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Ikushima, H.; Miyazono, K. Cellular context-dependent “colors” of transforming growth factor-beta signaling. Cancer Sci. 2010, 101, 306–312. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Padgett, R.W. Matters of context guide future research in TGFbeta superfamily signaling. Sci. Signal. 2015, 8, re10. [Google Scholar] [CrossRef]
- Zhang, Y.E. Mechanistic insight into contextual TGF-beta signaling. Curr. Opin. Cell Biol. 2018, 51, 1–7. [Google Scholar] [CrossRef]
- Yan, X.; Xiong, X.; Chen, Y.G. Feedback regulation of TGF-beta signaling. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 37–50. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Moses, H.L.; Roberts, A.B.; Derynck, R. The Discovery and Early Days of TGF-beta: A Historical Perspective. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-beta and TGF-beta-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Chang, C. Agonists and Antagonists of TGF-beta Family Ligands. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-beta Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.G. TGF-beta Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Ramel, M.C.; Hill, C.S. Spatial regulation of BMP activity. FEBS Lett. 2012, 586, 1929–1941. [Google Scholar] [CrossRef]
- Katagiri, T.; Watabe, T. Bone Morphogenetic Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Cai, J.; Pardali, E.; Sanchez-Duffhues, G.; ten Dijke, P. BMP signaling in vascular diseases. FEBS Lett. 2012, 586, 1993–2002. [Google Scholar] [CrossRef]
- Xu, P.; Lin, X.; Feng, X.H. Posttranslational Regulation of Smads. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.J.; Martin-Malpartida, P.; Massague, J. Structural determinants of Smad function in TGF-beta signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-beta Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Chen, Y.G. Smad7: Not only a regulator, but also a cross-talk mediator of TGF-beta signalling. Biochem. J. 2011, 434, 1–10. [Google Scholar] [CrossRef]
- Yan, X.; Liao, H.; Cheng, M.; Shi, X.; Lin, X.; Feng, X.H.; Chen, Y.G. Smad7 Protein Interacts with Receptor-regulated Smads (R-Smads) to Inhibit Transforming Growth Factor-beta (TGF-beta)/Smad Signaling. J. Biol. Chem. 2016, 291, 382–392. [Google Scholar] [CrossRef]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef]
- Galliher-Beckley, A.J.; Schiemann, W.P. Grb2 binding to Tyr284 in TbetaR-II is essential for mammary tumor growth and metastasis stimulated by TGF-beta. Carcinogenesis 2008, 29, 244–251. [Google Scholar] [CrossRef]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; Garcia de Vinuesa, A.; de Kruijf, E.M.; Mesker, W.E.; Hui, L.; Drabsch, Y.; Li, Y.; Bauer, A.; Rousseau, A.; et al. TRAF4 promotes TGF-beta receptor signaling and drives breast cancer metastasis. Mol. Cell 2013, 51, 559–572. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; ten Dijke, P. Signaling interplay between transforming growth factor-beta receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 2013, 38, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.Y.; Shin, I.; Arteaga, C.L. Type I transforming growth factor beta receptor binds to and activates phosphatidylinositol 3-kinase. J. Biol. Chem. 2005, 280, 10870–10876. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, A.; Song, J.; Thakur, N.; Itoh, S.; Marcusson, A.; Bergh, A.; Heldin, C.H.; Landstrom, M. TGF-beta promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85alpha. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Muthusamy, B.P.; Saeteurn, K.Y. Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2014, 31, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, M.C.; Murphy, S.J.; Garamszegi, N.; Leof, E.B. Cell-type-specific activation of PAK2 by transforming growth factor beta independent of Smad2 and Smad3. Mol. Cell Biol. 2003, 23, 8878–8889. [Google Scholar] [CrossRef] [PubMed]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science (New York, N.Y.) 2005, 307, 1603–1609. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Konigsrainer, A.; Weng, H.; Dooley, S.; ten Dijke, P. Transforming growth factor-beta (TGF-beta)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, J.; Sun, Q.; Tuazon, P.T.; Wu, X.; Traugh, J.A.; Chen, Y.G. p21-Activated kinase 2 (PAK2) inhibits TGF-beta signaling in Madin-Darby canine kidney (MDCK) epithelial cells by interfering with the receptor-Smad interaction. J. Biol. Chem. 2012, 287, 13705–13712. [Google Scholar] [CrossRef]
- Gingold, J.A.; Zhu, D.; Lee, D.F.; Kaseb, A.; Chen, J. Genomic Profiling and Metabolic Homeostasis in Primary Liver Cancers. Trends Mol. Med. 2018, 24, 395–411. [Google Scholar] [CrossRef]
- Nakagawa, H.; Fujita, M.; Fujimoto, A. Genome sequencing analysis of liver cancer for precision medicine. Semin. Cancer Biol. 2019, 55, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Korkut, A.; Zaidi, S.; Kanchi, R.S.; Rao, S.; Gough, N.R.; Schultz, A.; Li, X.; Lorenzi, P.L.; Berger, A.C.; Robertson, G.; et al. A Pan-Cancer Analysis Reveals High-Frequency Genetic Alterations in Mediators of Signaling by the TGF-beta Superfamily. Cell Syst. 2018, 7, 422–437.e7. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zaidi, S.; Rao, S.; Chen, J.S.; Phan, L.; Farci, P.; Su, X.; Shetty, K.; White, J.; Zamboni, F.; et al. Analysis of Genomes and Transcriptomes of Hepatocellular Carcinomas Identifies Mutations and Gene Expression Changes in the Transforming Growth Factor-beta Pathway. Gastroenterology 2018, 154, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology 2008, 47, 2059–2067. [Google Scholar] [CrossRef]
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, A.; Zhao, Y.; Ying, W.; Sun, H.; Yang, X.; Xing, B.; Sun, W.; Ren, L.; Hu, B.; et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567, 257–261. [Google Scholar] [CrossRef]
- Luo, J.; Chen, X.Q.; Li, P. The Role of TGF-beta and Its Receptors in Gastrointestinal Cancers. Transl. Oncol. 2019, 12, 475–484. [Google Scholar] [CrossRef]
- Doyle, J.J.; Gerber, E.E.; Dietz, H.C. Matrix-dependent perturbation of TGFbeta signaling and disease. FEBS Lett. 2012, 586, 2003–2015. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-beta1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Ito, N.; Kawata, S.; Tamura, S.; Takaishi, K.; Shirai, Y.; Kiso, S.; Yabuuchi, I.; Matsuda, Y.; Nishioka, M.; Tarui, S. Elevated levels of transforming growth factor beta messenger RNA and its polypeptide in human hepatocellular carcinoma. Cancer Res. 1991, 51, 4080–4083. [Google Scholar] [PubMed]
- Shirai, Y.; Kawata, S.; Tamura, S.; Ito, N.; Tsushima, H.; Takaishi, K.; Kiso, S.; Matsuzawa, Y. Plasma transforming growth factor-beta 1 in patients with hepatocellular carcinoma. Comparison with chronic liver diseases. Cancer 1994, 73, 2275–2279. [Google Scholar] [CrossRef]
- Giannelli, G.; Fransvea, E.; Marinosci, F.; Bergamini, C.; Colucci, S.; Schiraldi, O.; Antonaci, S. Transforming growth factor-beta1 triggers hepatocellular carcinoma invasiveness via alpha3beta1 integrin. Am. J. Pathol. 2002, 161, 183–193. [Google Scholar] [CrossRef]
- Sugano, Y.; Matsuzaki, K.; Tahashi, Y.; Furukawa, F.; Mori, S.; Yamagata, H.; Yoshida, K.; Matsushita, M.; Nishizawa, M.; Fujisawa, J.; et al. Distortion of autocrine transforming growth factor beta signal accelerates malignant potential by enhancing cell growth as well as PAI-1 and VEGF production in human hepatocellular carcinoma cells. Oncogene 2003, 22, 2309–2321. [Google Scholar] [CrossRef] [PubMed]
- Reichl, P.; Dengler, M.; van Zijl, F.; Huber, H.; Fuhrlinger, G.; Reichel, C.; Sieghart, W.; Peck-Radosavljevic, M.; Grubinger, M.; Mikulits, W. Axl activates autocrine transforming growth factor-beta signaling in hepatocellular carcinoma. Hepatology 2015, 61, 930–941. [Google Scholar] [CrossRef]
- Qin, G.; Luo, M.; Chen, J.; Dang, Y.; Chen, G.; Li, L.; Zeng, J.; Lu, Y.; Yang, J. Reciprocal activation between MMP-8 and TGF-beta1 stimulates EMT and malignant progression of hepatocellular carcinoma. Cancer Lett. 2016, 374, 85–95. [Google Scholar] [CrossRef]
- Yu, W.; Huang, C.; Wang, Q.; Huang, T.; Ding, Y.; Ma, C.; Ma, H.; Chen, W. MEF2 transcription factors promotes EMT and invasiveness of hepatocellular carcinoma through TGF-beta1 autoregulation circuitry. Tumour Biol. 2014, 35, 10943–10951. [Google Scholar] [CrossRef]
- Rojas, A.; Zhang, P.; Wang, Y.; Foo, W.C.; Munoz, N.M.; Xiao, L.; Wang, J.; Gores, G.J.; Hung, M.C.; Blechacz, B. A Positive TGF-beta/c-KIT Feedback Loop Drives Tumor Progression in Advanced Primary Liver Cancer. Neoplasia 2016, 18, 371–386. [Google Scholar] [CrossRef]
- Wu, J.; Lu, M.; Li, Y.; Shang, Y.K.; Wang, S.J.; Meng, Y.; Wang, Z.; Li, Z.S.; Chen, H.; Chen, Z.N.; et al. Regulation of a TGF-beta1-CD147 self-sustaining network in the differentiation plasticity of hepatocellular carcinoma cells. Oncogene 2016, 35, 5468–5479. [Google Scholar] [CrossRef]
- Bi, J.G.; Zheng, J.F.; Li, Q.; Bao, S.Y.; Yu, X.F.; Xu, P.; Liao, C.X. MicroRNA-181a-5p suppresses cell proliferation by targeting Egr1 and inhibiting Egr1/TGF-beta/Smad pathway in hepatocellular carcinoma. Int. J. Biochem. Cell Biol. 2019, 106, 107–116. [Google Scholar] [CrossRef]
- Shao, C.; Qiu, Y.; Liu, J.; Feng, H.; Shen, S.; Saiyin, H.; Yu, W.; Wei, Y.; Yu, L.; Su, W.; et al. PARP12 (ARTD12) suppresses hepatocellular carcinoma metastasis through interacting with FHL2 and regulating its stability. Cell Death Dis. 2018, 9, 856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Li, Q.J.; Feng, Y.; Zhang, Y.; Markowitz, G.J.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, G.; Shao, D.; Wang, J.; Yuan, H.; Chen, T.; Zhai, R.; Ni, W.; Tai, G. Mucin1 mediates autocrine transforming growth factor beta signaling through activating the c-Jun N-terminal kinase/activator protein 1 pathway in human hepatocellular carcinoma cells. Int. J. Biochem. Cell Biol. 2015, 59, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, G.; Li, Q.; Wang, F.; Xie, F.; Zhai, R.; Guo, Y.; Chen, T.; Zhang, N.; Ni, W.; et al. Mucin1 promotes the migration and invasion of hepatocellular carcinoma cells via JNK-mediated phosphorylation of Smad2 at the C-terminal and linker regions. Oncotarget 2015, 6, 19264–19278. [Google Scholar] [CrossRef]
- Ozaki, I.; Hamajima, H.; Matsuhashi, S.; Mizuta, T. Regulation of TGF-beta1-Induced Pro-Apoptotic Signaling by Growth Factor Receptors and Extracellular Matrix Receptor Integrins in the Liver. Front. Physiol. 2011, 2, 78. [Google Scholar] [CrossRef]
- Xia, H.; Chen, J.; Shi, M.; Gao, H.; Sekar, K.; Seshachalam, V.P.; Ooi, L.L.; Hui, K.M. EDIL3 is a novel regulator of epithelial-mesenchymal transition controlling early recurrence of hepatocellular carcinoma. J. Hepatol. 2015, 63, 863–873. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Nakamura, I.; Hu, C.; Chen, G.; Oseini, A.M.; Seven, E.S.; Miamen, A.G.; Moser, C.D.; Zhou, W.; van Kuppevelt, T.H.; et al. Activation of the transforming growth factor-beta/SMAD transcriptional pathway underlies a novel tumor-promoting role of sulfatase 1 in hepatocellular carcinoma. Hepatology 2015, 61, 1269–1283. [Google Scholar] [CrossRef]
- Benzoubir, N.; Lejamtel, C.; Battaglia, S.; Testoni, B.; Benassi, B.; Gondeau, C.; Perrin-Cocon, L.; Desterke, C.; Thiers, V.; Samuel, D.; et al. HCV core-mediated activation of latent TGF-beta via thrombospondin drives the crosstalk between hepatocytes and stromal environment. J. Hepatol. 2013, 59, 1160–1168. [Google Scholar] [CrossRef]
- Hosui, A.; Kimura, A.; Yamaji, D.; Zhu, B.M.; Na, R.; Hennighausen, L. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-{beta} and STAT3 activation. J. Exp. Med. 2009, 206, 819–831. [Google Scholar] [CrossRef]
- Yang, G.; Liang, Y.; Zheng, T.; Song, R.; Wang, J.; Shi, H.; Sun, B.; Xie, C.; Li, Y.; Han, J.; et al. FCN2 inhibits epithelial-mesenchymal transition-induced metastasis of hepatocellular carcinoma via TGF-beta/Smad signaling. Cancer Lett. 2016, 378, 80–86. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Y.; Ma, H.; Wang, B.; Xu, L.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Hepatitis B virus X protein amplifies TGF-beta promotion on HCC motility through down-regulating PPM1a. Oncotarget 2016, 7, 33125–33135. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Murata, M.; Yoshida, K.; Sekimoto, G.; Uemura, Y.; Sakaida, N.; Kaibori, M.; Kamiyama, Y.; Nishizawa, M.; Fujisawa, J.; et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007, 46, 48–57. [Google Scholar] [CrossRef]
- Murata, M.; Matsuzaki, K.; Yoshida, K.; Sekimoto, G.; Tahashi, Y.; Mori, S.; Uemura, Y.; Sakaida, N.; Fujisawa, J.; Seki, T.; et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology 2009, 49, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Jiao, T.; Huang, Y.; Liu, W.; Li, Z.; Ye, X. Hepatitis B virus regulates apoptosis and tumorigenesis through the microRNA-15a-Smad7-transforming growth factor beta pathway. J. Virol. 2015, 89, 2739–2749. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Li, Y. HCV core protein promotes hepatocyte proliferation and chemoresistance by inhibiting NR4A1. Biochem. Biophys. Res. Commun. 2015, 466, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Matsuzaki, K.; Inokuchi, R.; Kawamura, R.; Yoshida, K.; Murata, M.; Fujisawa, J.; Fukushima, N.; Sata, M.; Kage, M.; et al. Phosphorylated Smad2 and Smad3 signaling: Shifting between tumor suppression and fibro-carcinogenesis in chronic hepatitis C. Hepatol. Res. 2013, 43, 1327–1342. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, Y.; Gao, Y.; Hu, W.; Qu, Y.; Lou, N.; Zhu, Y.; Zhang, X.; Yang, H. Hepatitis C virus NS3 protein enhances hepatocellular carcinoma cell invasion by promoting PPM1A ubiquitination and degradation. J. Exp. Clin. Cancer Res. 2017, 36, 42. [Google Scholar] [CrossRef] [Green Version]
- Verga-Gerard, A.; Porcherot, M.; Meyniel-Schicklin, L.; Andre, P.; Lotteau, V.; Perrin-Cocon, L. Hepatitis C virus/human interactome identifies SMURF2 and the viral protease as critical elements for the control of TGF-beta signaling. FASEB J. 2013, 27, 4027–4040. [Google Scholar] [CrossRef]
- Cheng, P.L.; Chang, M.H.; Chao, C.H.; Lee, Y.H. Hepatitis C viral proteins interact with Smad3 and differentially regulate TGF-beta/Smad3-mediated transcriptional activation. Oncogene 2004, 23, 7821–7838. [Google Scholar] [CrossRef]
- Yakymovych, I.; Yakymovych, M.; Heldin, C.H. Intracellular trafficking of transforming growth factor beta receptors. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 3–11. [Google Scholar] [CrossRef]
- Reisenbichler, H.; Chari, R.S.; Boyer, I.J.; Jirtle, R.L. Transforming growth factor-beta receptors type I, II and III in phenobarbital-promoted rat liver tumors. Carcinogenesis 1994, 15, 2763–2767. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Peltier, E.; Terris, B.; Franco, D.; Poynard, T. Transforming growth factor-beta 1 (TGF-beta 1) and TGF-beta 1 receptors in normal, cirrhotic, and neoplastic human livers. Hepatology 1995, 21, 760–766. [Google Scholar] [PubMed]
- Mamiya, T.; Yamazaki, K.; Masugi, Y.; Mori, T.; Effendi, K.; Du, W.; Hibi, T.; Tanabe, M.; Ueda, M.; Takayama, T.; et al. Reduced transforming growth factor-beta receptor II expression in hepatocellular carcinoma correlates with intrahepatic metastasis. Lab. Investig. 2010, 90, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Oe, S.; Lemmer, E.R.; Conner, E.A.; Factor, V.M.; Leveen, P.; Larsson, J.; Karlsson, S.; Thorgeirsson, S.S. Intact signaling by transforming growth factor beta is not required for termination of liver regeneration in mice. Hepatology 2004, 40, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Romero-Gallo, J.; Sozmen, E.G.; Chytil, A.; Russell, W.E.; Whitehead, R.; Parks, W.T.; Holdren, M.S.; Her, M.F.; Gautam, S.; Magnuson, M.; et al. Inactivation of TGF-beta signaling in hepatocytes results in an increased proliferative response after partial hepatectomy. Oncogene 2005, 24, 3028–3041. [Google Scholar] [CrossRef] [PubMed]
- Kanzler, S.; Meyer, E.; Lohse, A.W.; Schirmacher, P.; Henninger, J.; Galle, P.R.; Blessing, M. Hepatocellular expression of a dominant-negative mutant TGF-beta type II receptor accelerates chemically induced hepatocarcinogenesis. Oncogene 2001, 20, 5015–5024. [Google Scholar] [CrossRef]
- Zhao, W.; Kobayashi, M.; Ding, W.; Yuan, L.; Seth, P.; Cornain, S.; Wang, J.; Okada, F.; Hosokawa, M. Suppression of in vivo tumorigenicity of rat hepatoma cell line KDH-8 cells by soluble TGF-beta receptor type II. Cancer Immunol. Immunother. 2002, 51, 381–388. [Google Scholar] [CrossRef]
- Maass, T.; Thieringer, F.R.; Mann, A.; Longerich, T.; Schirmacher, P.; Strand, D.; Hansen, T.; Galle, P.R.; Teufel, A.; Kanzler, S. Liver specific overexpression of platelet-derived growth factor-B accelerates liver cancer development in chemically induced liver carcinogenesis. Int. J. Cancer 2011, 128, 1259–1268. [Google Scholar] [CrossRef]
- Shi, Y.; Qin, N.; Zhou, Q.; Chen, Y.; Huang, S.; Chen, B.; Shen, G.; Jia, H. Role of IQGAP3 in metastasis and epithelial-mesenchymal transition in human hepatocellular carcinoma. J. Transl. Med. 2017, 15, 176. [Google Scholar] [CrossRef]
- Liu, C.; Billadeau, D.D.; Abdelhakim, H.; Leof, E.; Kaibuchi, K.; Bernabeu, C.; Bloom, G.S.; Yang, L.; Boardman, L.; Shah, V.H.; et al. IQGAP1 suppresses TbetaRII-mediated myofibroblastic activation and metastatic growth in liver. J. Clin. Investig. 2013, 123, 1138–1156. [Google Scholar] [CrossRef]
- Zhu, L.; Chen, S.; Chen, Y. Unraveling the biological functions of Smad7 with mouse models. Cell Biosci 2011, 1, 44. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Kuratomi, G.; Komuro, A.; Goto, K.; Shinozaki, M.; Miyazawa, K.; Miyazono, K.; Imamura, T. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. Biochem. J. 2005, 386, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Imamura, T.; Saitoh, M.; Yoshida, Y.; Yamori, T.; Miyazono, K.; Miyazawa, K. Negative regulation of transforming growth factor-beta (TGF-beta) signaling by WW domain-containing protein 1 (WWP1). Oncogene 2004, 23, 6914–6923. [Google Scholar] [CrossRef]
- Shi, W.; Sun, C.; He, B.; Xiong, W.; Shi, X.; Yao, D.; Cao, X. GADD34-PP1c recruited by Smad7 dephosphorylates TGFbeta type I receptor. J. Cell Biol. 2004, 164, 291–300. [Google Scholar] [CrossRef]
- Moren, A.; Imamura, T.; Miyazono, K.; Heldin, C.H.; Moustakas, A. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J. Biol. Chem. 2005, 280, 22115–22123. [Google Scholar] [CrossRef]
- Zhang, S.; Fei, T.; Zhang, L.; Zhang, R.; Chen, F.; Ning, Y.; Han, Y.; Feng, X.H.; Meng, A.; Chen, Y.G. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol. Cell Biol. 2007, 27, 4488–4499. [Google Scholar] [CrossRef]
- Yan, X.; Pan, J.; Xiong, W.; Cheng, M.; Sun, Y.; Zhang, S.; Chen, Y. Yin Yang 1 (YY1) synergizes with Smad7 to inhibit TGF-beta signaling in the nucleus. Sci. China Life Sci. 2014, 57, 128–136. [Google Scholar] [CrossRef]
- Yan, X.; Lin, Z.; Chen, F.; Zhao, X.; Chen, H.; Ning, Y.; Chen, Y.G. Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-beta signaling. J. Biol. Chem. 2009, 284, 30097–30104. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, J.; Pan, L.; Wang, P.; Xue, H.; Zhang, L.; Gao, X.; Zhao, X.; Ning, Y.; Chen, Y.G. TSC-22 promotes transforming growth factor beta-mediated cardiac myofibroblast differentiation by antagonizing Smad7 activity. Mol. Cell Biol. 2011, 31, 3700–3709. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.K.; Moses, H.L. STRAP and Smad7 synergize in the inhibition of transforming growth factor beta signaling. Mol. Cell Biol. 2000, 20, 3157–3167. [Google Scholar] [CrossRef] [PubMed]
- Lonn, P.; Vanlandewijck, M.; Raja, E.; Kowanetz, M.; Watanabe, Y.; Kowanetz, K.; Vasilaki, E.; Heldin, C.H.; Moustakas, A. Transcriptional induction of salt-inducible kinase 1 by transforming growth factor beta leads to negative regulation of type I receptor signaling in cooperation with the Smurf2 ubiquitin ligase. J. Biol. Chem. 2012, 287, 12867–12878. [Google Scholar] [CrossRef] [PubMed]
- Elkouris, M.; Kontaki, H.; Stavropoulos, A.; Antonoglou, A.; Nikolaou, K.C.; Samiotaki, M.; Szantai, E.; Saviolaki, D.; Brown, P.J.; Sideras, P.; et al. SET9-Mediated Regulation of TGF-beta Signaling Links Protein Methylation to Pulmonary Fibrosis. Cell Rep. 2016, 15, 2733–2744. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Date, M.; Furukawa, F.; Tahashi, Y.; Matsushita, M.; Sugano, Y.; Yamashiki, N.; Nakagawa, T.; Seki, T.; Nishizawa, M.; et al. Regulatory mechanisms for transforming growth factor beta as an autocrine inhibitor in human hepatocellular carcinoma: Implications for roles of smads in its growth. Hepatology 2000, 32, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Dzieran, J.; Gu, X.; Marhenke, S.; Vogel, A.; Machida, K.; Weiss, T.S.; Ruemmele, P.; Kollmar, O.; Hoffmann, P.; et al. Smad7 regulates compensatory hepatocyte proliferation in damaged mouse liver and positively relates to better clinical outcome in human hepatocellular carcinoma. Clin. Sci. (Lond.) 2015, 128, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Ju, H.L.; Chung, S.I.; Cho, K.J.; Eun, J.W.; Nam, S.W.; Han, K.H.; Calvisi, D.F.; Ro, S.W. Transforming Growth Factor-beta Promotes Liver Tumorigenesis in Mice via Up-regulation of Snail. Gastroenterology 2017, 153, 1378–1391.e6. [Google Scholar] [CrossRef]
- Wang, R.; Fu, T.; You, K.; Li, S.; Zhao, N.; Yang, J.; Zhuang, S.M. Identification of a TGF-beta-miR-195 positive feedback loop in hepatocytes and its deregulation in hepatoma cells. FASEB J. 2018, 32, 3936–3945. [Google Scholar] [CrossRef]
- Chang, L.; Li, C.; Guo, T.; Wang, H.; Ma, W.; Yuan, Y.; Liu, Q.; Ye, Q.; Liu, Z. The human RNA surveillance factor UPF1 regulates tumorigenesis by targeting Smad7 in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 8. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhao, J.; Chu, E.S.; Mok, M.T.; Go, M.Y.; Man, K.; Heuchel, R.; Lan, H.Y.; Chang, Z.; Sung, J.J.; et al. Inhibitory role of Smad7 in hepatocarcinogenesis in mice and in vitro. J. Pathol. 2013, 230, 441–452. [Google Scholar] [CrossRef]
- Feng, T.; Dzieran, J.; Yuan, X.; Dropmann, A.; Maass, T.; Teufel, A.; Marhenke, S.; Gaiser, T.; Ruckert, F.; Kleiter, I.; et al. Hepatocyte-specific Smad7 deletion accelerates DEN-induced HCC via activation of STAT3 signaling in mice. Oncogenesis 2017, 6, e294. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Choi, S.; Jeong, S.J.; Lee, S.A.; Kwak, T.K.; Kim, H.; Jung, O.; Lee, M.S.; Ko, Y.; Ryu, J.; et al. Cross-talk between TGFbeta1 and EGFR signalling pathways induces TM4SF5 expression and epithelial-mesenchymal transition. Biochem. J. 2012, 443, 691–700. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Ding, W.; Rountree, C.B. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology 2010, 51, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.K.; Yang, Y.T.; Ma, X.; Han, B.; Wang, Z.S.; Zhao, Q.Y.; Wu, L.Q.; Qu, Z.Q. MicroRNA-92b promotes hepatocellular carcinoma progression by targeting Smad7 and is mediated by long non-coding RNA XIST. Cell Death Dis. 2016, 7, e2203. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Ooi, L.L.; Hui, K.M. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology 2013, 58, 629–641. [Google Scholar] [CrossRef]
- Kan, H.; Guo, W.; Huang, Y.; Liu, D. MicroRNA-520g induces epithelial-mesenchymal transition and promotes metastasis of hepatocellular carcinoma by targeting SMAD7. FEBS Lett. 2015, 589, 102–109. [Google Scholar] [CrossRef]
- Chang, L.; Yuan, Y.; Li, C.; Guo, T.; Qi, H.; Xiao, Y.; Dong, X.; Liu, Z.; Liu, Q. Upregulation of SNHG6 regulates ZEB1 expression by competitively binding miR-101-3p and interacting with UPF1 in hepatocellular carcinoma. Cancer Lett. 2016, 383, 183–194. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, M.; Lu, P.; Li, X.; Zhu, S.; Yue, S. NEDD9 may regulate hepatocellular carcinoma cell metastasis by promoting epithelial-mesenchymal-transition and stemness via repressing Smad7. Oncotarget 2017, 8, 1714–1724. [Google Scholar] [CrossRef]
- Sun, H.; Peng, Z.; Tang, H.; Xie, D.; Jia, Z.; Zhong, L.; Zhao, S.; Ma, Z.; Gao, Y.; Zeng, L.; et al. Loss of KLF4 and consequential downregulation of Smad7 exacerbate oncogenic TGF-beta signaling in and promote progression of hepatocellular carcinoma. Oncogene 2017, 36, 2957–2968. [Google Scholar] [CrossRef]
- Hujie, G.; Zhou, S.H.; Zhang, H.; Qu, J.; Xiong, X.W.; Hujie, O.; Liao, C.G.; Yang, S.E. MicroRNA-10b regulates epithelial-mesenchymal transition by modulating KLF4/KLF11/Smads in hepatocellular carcinoma. Cancer Cell Int. 2018, 18, 10. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; Drabsch, Y.; Gao, R.; Snaar-Jagalska, B.E.; Mickanin, C.; Huang, H.; Sheppard, K.A.; Porter, J.A.; Lu, C.X.; et al. USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-beta type I receptor. Nat. Cell Biol. 2012, 14, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Liu, Y.; Mei, Y.; Zou, M.; Zhao, Z.; Ye, M.; Wu, X. Ubiquitin-speci fi c protease 4 promotes metastasis of hepatocellular carcinoma by increasing TGF-beta signaling-induced epithelial-mesenchymal transition. Aging (Albany NY) 2018, 10, 2783–2799. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xu, X.; Yang, Z.; Zhang, L.; Liu, Y.; Ma, A.; Xu, G.; Tang, M.; Jing, T.; Wu, L.; et al. POH1 contributes to hyperactivation of TGF-beta signaling and facilitates hepatocellular carcinoma metastasis through deubiquitinating TGF-beta receptors and caveolin-1. EBioMedicine 2019, 41, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, W.; Lu, Y.; Jiang, S.; Li, C.; Chen, J.; Tao, D.; Liu, Y.; Yang, Y.; Ma, Y. A TALEN-based specific transcript knock-down of PIWIL2 suppresses cell growth in HepG2 tumor cell. Cell Prolif. 2014, 47, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Zhang, P.F.; Yang, X.; Wei, C.Y.; Huang, X.Y.; Cai, J.B.; Lu, J.C.; Gao, C.; Sun, H.X.; Gao, Q.; et al. Overexpression of RNF38 facilitates TGF-beta signaling by Ubiquitinating and degrading AHNAK in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 113. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, T.; Adachi, S.; Kohu, K.; Yamada, T.; Higuchi, O.; Furukawa, Y.; Nakamura, Y.; Nakamura, T.; Tashiro, K.; Kuhara, S.; et al. Identification of BMP and activin membrane-bound inhibitor (BAMBI), an inhibitor of transforming growth factor-beta signaling, as a target of the beta-catenin pathway in colorectal tumor cells. J. Biol. Chem. 2004, 279, 6840–6846. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Katuri, V.; Dillner, A.; Mishra, B.; Deng, C.X.; Mishra, L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science 2003, 299, 574–577. [Google Scholar] [CrossRef]
- Kitisin, K.; Ganesan, N.; Tang, Y.; Jogunoori, W.; Volpe, E.A.; Kim, S.S.; Katuri, V.; Kallakury, B.; Pishvaian, M.; Albanese, C.; et al. Disruption of transforming growth factor-beta signaling through beta-spectrin ELF leads to hepatocellular cancer through cyclin D1 activation. Oncogene 2007, 26, 7103–7110. [Google Scholar] [CrossRef]
- Baek, H.J.; Pishvaian, M.J.; Tang, Y.; Kim, T.H.; Yang, S.; Zouhairi, M.E.; Mendelson, J.; Shetty, K.; Kallakury, B.; Berry, D.L.; et al. Transforming growth factor-beta adaptor, beta2-spectrin, modulates cyclin dependent kinase 4 to reduce development of hepatocellular cancer. Hepatology 2011, 53, 1676–1684. [Google Scholar] [CrossRef]
- Saha, T.; Vardhini, D.; Tang, Y.; Katuri, V.; Jogunoori, W.; Volpe, E.A.; Haines, D.; Sidawy, A.; Zhou, X.; Gallicano, I.; et al. RING finger-dependent ubiquitination by PRAJA is dependent on TGF-beta and potentially defines the functional status of the tumor suppressor ELF. Oncogene 2006, 25, 693–705. [Google Scholar] [CrossRef]
- Ho, J.; Cocolakis, E.; Dumas, V.M.; Posner, B.I.; Laporte, S.A.; Lebrun, J.J. The G protein-coupled receptor kinase-2 is a TGFbeta-inducible antagonist of TGFbeta signal transduction. EMBO J. 2005, 24, 3247–3258. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wang, Z.; Yan, J.; Yang, X.; Liu, A.; Qiu, W.; Zhu, J.; Han, J.; Zhang, H.; Lin, J.; et al. Human four-and-a-half LIM family members suppress tumor cell growth through a TGF-beta-like signaling pathway. J. Clin. Investig. 2009, 119, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Ma, H.; Cai, H.; Wu, Y.; Jiang, W.; Yu, L. An inhibitory role of NEK6 in TGFbeta/Smad signaling pathway. BMB Rep. 2015, 48, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Matsuzaki, K.; Murata, M.; Yamaguchi, T.; Suwa, K.; Okazaki, K. Clinico-Pathological Importance of TGF-beta/Phospho-Smad Signaling during Human Hepatic Fibrocarcinogenesis. Cancers (Basel) 2018, 10, 183. [Google Scholar] [CrossRef]
- Nagata, H.; Hatano, E.; Tada, M.; Murata, M.; Kitamura, K.; Asechi, H.; Narita, M.; Yanagida, A.; Tamaki, N.; Yagi, S.; et al. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor-suppression in rat hepatocellular carcinoma. Hepatology 2009, 49, 1944–1953. [Google Scholar] [CrossRef]
- Li, Q.; Liu, G.; Yuan, H.; Wang, J.; Guo, Y.; Chen, T.; Zhai, R.; Shao, D.; Ni, W.; Tai, G. Mucin1 shifts Smad3 signaling from the tumor-suppressive pSmad3C/p21(WAF1) pathway to the oncogenic pSmad3L/c-Myc pathway by activating JNK in human hepatocellular carcinoma cells. Oncotarget 2015, 6, 4253–4265. [Google Scholar] [CrossRef]
- Liu, R.; Tang, C.; Shen, A.; Luo, H.; Wei, X.; Zheng, D.; Sun, C.; Li, Z.; Zhu, D.; Li, T.; et al. IL-37 suppresses hepatocellular carcinoma growth by converting pSmad3 signaling from JNK/pSmad3L/c-Myc oncogenic signaling to pSmad3C/P21 tumor-suppressive signaling. Oncotarget 2016, 7, 85079–85096. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Duan, X.; Liang, Y.Y.; Su, Y.; Wrighton, K.H.; Long, J.; Hu, M.; Davis, C.M.; Wang, J.; Brunicardi, F.C.; et al. PPM1A functions as a Smad phosphatase to terminate TGFbeta signaling. Cell 2006, 125, 915–928. [Google Scholar] [CrossRef]
- Zhang, Y.; Tao, R.; Wu, S.S.; Xu, C.C.; Wang, J.L.; Chen, J.; Yu, Y.S.; Tang, Z.H.; Chen, X.H.; Zang, G.Q. TRIM52 up-regulation in hepatocellular carcinoma cells promotes proliferation, migration and invasion through the ubiquitination of PPM1A. J. Exp. Clin. Cancer Res. 2018, 37, 116. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.Y.; Jin, G.N.; Wang, W.; Chen, W.X.; Wu, Y.H.; Ai, X.; Chen, L.; Zhang, W.G.; Liang, H.F.; Laurence, A.; et al. Reduced expression of transcriptional intermediary factor 1 gamma promotes metastasis and indicates poor prognosis of hepatocellular carcinoma. Hepatology 2014, 60, 1620–1636. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yang, Y.; Xie, R.; Liu, J.; Nie, X.; An, J.; Wen, G.; Liu, X.; Jin, H.; Tuo, B. The NCX1/TRPC6 Complex Mediates TGFbeta-Driven Migration and Invasion of Human Hepatocellular Carcinoma Cells. Cancer Res. 2018, 78, 2564–2576. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Peng, C. Nodal enhances the activity of FoxO3a and its synergistic interaction with Smads to regulate cyclin G2 transcription in ovarian cancer cells. Oncogene 2011, 30, 3953–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Liu, Y.; Du, L.; He, L.; Ni, B.; Hu, J.; Zhu, D.; Chen, Q. Threonine 32 (Thr32) of FoxO3 is critical for TGF-beta-induced apoptosis via Bim in hepatocarcinoma cells. Protein Cell 2015, 6, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.S.; Tsai, W.W.; Schumacher, M.A.; Barton, M.C. Chromatin-bound p53 anchors activated Smads and the mSin3A corepressor to confer transforming-growth-factor-beta-mediated transcription repression. Mol. Cell Biol. 2008, 28, 1988–1998. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Mishra, L. Targeting Transforming Growth Factor Beta Signaling in Liver Cancer. Hepatology 2019, 69, 1375–1378. [Google Scholar] [CrossRef]
- Morris, S.M.; Baek, J.Y.; Koszarek, A.; Kanngurn, S.; Knoblaugh, S.E.; Grady, W.M. Transforming growth factor-beta signaling promotes hepatocarcinogenesis induced by p53 loss. Hepatology 2012, 55, 121–131. [Google Scholar] [CrossRef]
- Lee, H.J.; Yun, C.H.; Lim, S.H.; Kim, B.C.; Baik, K.G.; Kim, J.M.; Kim, W.H.; Kim, S.J. SRF is a nuclear repressor of Smad3-mediated TGF-beta signaling. Oncogene 2007, 26, 173–185. [Google Scholar] [CrossRef]
- Yan, X.; Wu, J.; Jiang, Q.; Cheng, H.; Han, J.J.; Chen, Y.G. CXXC5 suppresses hepatocellular carcinoma by promoting TGF-beta-induced cell cycle arrest and apoptosis. J. Mol. Cell Biol. 2018, 10, 48–59. [Google Scholar] [CrossRef]
- Xiong, X.; Tu, S.; Wang, J.; Luo, S.; Yan, X. CXXC5: A novel regulator and coordinator of TGF-beta, BMP and Wnt signaling. J. Cell Mol. Med. 2019, 23, 740–749. [Google Scholar] [CrossRef]
- Ali, A.; Zhang, P.; Liangfang, Y.; Wenshe, S.; Wang, H.; Lin, X.; Dai, Y.; Feng, X.H.; Moses, R.; Wang, D.; et al. KLF17 empowers TGF-beta/Smad signaling by targeting Smad3-dependent pathway to suppress tumor growth and metastasis during cancer progression. Cell Death Dis. 2015, 6, e1681. [Google Scholar] [CrossRef] [PubMed]
- Yasui, K.; Konishi, C.; Gen, Y.; Endo, M.; Dohi, O.; Tomie, A.; Kitaichi, T.; Yamada, N.; Iwai, N.; Nishikawa, T.; et al. EVI1, a target gene for amplification at 3q26, antagonizes transforming growth factor-beta-mediated growth inhibition in hepatocellular carcinoma. Cancer Sci. 2015, 106, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Bowen, W.C.; Li, G.; Demetris, A.J.; Michalopoulos, G.K.; Wu, T. Cytosolic phospholipase A2alpha and peroxisome proliferator-activated receptor gamma signaling pathway counteracts transforming growth factor beta-mediated inhibition of primary and transformed hepatocyte growth. Hepatology 2010, 52, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Zhang, J.; Zhu, X.; Myles, D.E.; Willson, T.M.; Liu, X.; Chen, Y.E. Peroxisome proliferator-activated receptor gamma inhibits transforming growth factor beta-induced connective tissue growth factor expression in human aortic smooth muscle cells by interfering with Smad3. J. Biol. Chem. 2001, 276, 45888–45894. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; He, Y.; Qi, L.; Chen, L.; Luo, Y.; Chen, L.; Li, Y.; Zhang, N.; Guo, H. cPLA2alpha activates PI3K/AKT and inhibits Smad2/3 during epithelial-mesenchymal transition of hepatocellular carcinoma cells. Cancer Lett. 2017, 403, 260–270. [Google Scholar] [CrossRef]
- Xie, F.; Ling, L.; van Dam, H.; Zhou, F.; Zhang, L. TGF-beta signaling in cancer metastasis. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 121–132. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, L. A special issue on TGF-beta signaling: Regulation, crosstalk, and biology. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 1–2. [Google Scholar] [CrossRef]
- Achyut, B.R.; Yang, L. Transforming growth factor-beta in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology 2011, 141, 1167–1178. [Google Scholar] [CrossRef]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-beta and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci 2018, 19, 1294. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulators | Functions | References |
|---|---|---|
| HBV | HBV X protein (HBx) could enhance TGF-β expression in liver and drive liver cancer progression. | [82] |
| HBx promotes PPM1A degradation to enhance the tumor-promoting Smad signaling. | [91] | |
| HBx alters the phosphorylation pattern of Smad3, averting its activity from tumor-suppressive to oncogenic. | [92,93] | |
| Attenuates TGF-β-induced apoptosis in HCC cells by enhancing Smad7 expression. | [94] | |
| HCV | HCV core protein increases TGF-β level in transgenic mice, by engaging thrombospondin-1. | [88] |
| HCV core protein promotes HepG2 cell proliferation and chemoresistance by enhancing Smad7 expression and subsequently attenuating the cytostatic effects of TGF-β. | [95] | |
| Shifts the TβRI/pSmad3C/p21 tumor-suppressive pathway to the JNK/pSmad3L/c-Myc oncogenic signaling. | [92] | |
| Induces JNK-mediated linker phosphorylation of Smad2 and generates pSmad2L/C, contributing to HCC development. | [96] | |
| The HCV nonstructural protein 3 (NS3) promotes ubiquitination and degradation of PPM1A, thereby promoting Smad signaling and HCC progression. | [97] | |
| HCV-encoded protease NS3-4A strengthens and prolongs TGF-β-induced phosphorylation levels of Smad2/3. | [98] | |
| HCV core protein disrupts the Smad3/Sp1 complex and blocks their binding to the promoters of CDK inhibitors. | [99] |
| Regulators | Functions | Alternation of the Regulators | References |
|---|---|---|---|
| FHL1 | Exerts a tumor-suppressive role in liver cancer by triggering CK1δ-mediated and TGF-β receptor-independent phosphorylation of R-Smads. | Decreased in HCC tissues | [152] |
| NEK6 | Exerts a tumor-promoting effect by interacting with Smad4 and attenuating the anti-proliferative effect of TGF-β. | Undetermined | [153] |
| IL-37 | Inhibits Smad3-mediated oncogenic effects by inducing a conversion from the pSmad3L/c-Myc signaling to the pSmad3C/p21 signaling. | Decreased in human HCC tissues and cell lines | [157] |
| ELF | Acts as an adaptor protein for Smad3 and Smad4, therefore promoting the cytostatic effects of TGF-β in liver cancer. ELF+/- mice spontaneously developed HCC. | Reduced in liver cancer tissues | [147,148,149] |
| PRAJA | Alleviates the tumor-inhibitory effect of TGF-β by associating with both ELF and Smad3 upon ligand stimulation. | Elevated in liver cancer tissues | [150] |
| GRK2 | Interferes with Smad-mediated cytostatic effects by inhibiting TGF-β-induced C-terminal phosphorylation of Smad2/3. | Undetermined | [151] |
| TNF-α/ IL-1β | Promotes hepatocyte proliferation by inducing JNK-mediated linker phosphorylation of Smad3 and enhancing c-Myc expression. | Undetermined | [154] |
| MUC1 | Converts the pSmad2/3C/p21 signaling to the pSmad2/3L/c-Myc oncogenic signaling by inducing JNK-mediated linker phosphorylation of Smad2/3. | Overexpressed in HCC cell lines | [83,84,156] |
| TRIM52 | Promotes liver cancer progression by inducing PPM1A degradation and enhancing the pSmad2/3C levels. | Highly expressed in HCC tissues | [158,159] |
| TIF1γ | Antagonizes TGF-β-mediated cytostatic effects in the early stage and inhibiting EMT in the late stage by mono-ubiquitynating Smad4 and inhibiting its oligomerization with R-Smads. | Reduced in advanced HCCs | [160] |
| NCX1/ TRPC6 | Be required for TGF-β-induced R-Smad activation and contribute to TGF-β-mediated EMT, invasion and intrahepatic metastasis. | Elevated in human HCC cells | [161] |
| CXXC5 | Removes the histone deacetylase HDAC1 from activated Smad2/3 to potentiate TGF-β-mediated growth inhibition of HCC cells. | Decreased in HCC tissues | [169,170] |
| KLF17 | Interacts with and enhances the transcriptional activity of Smad3, thereby facilitating TGF-β-mediated cytostatic effects in HCC. | Decreased in advanced HCCs | [171] |
| SRF | Attenuates the cytostatic functions of TGF-β by inhibiting Smad-DNA binding in HCC cells. | Undetermined | [168] |
| EVI | Acts as a transcriptional repressor for Smad3 to blunt TGF-β-mediated growth inhibition of HCC cells. | Elevated in a subset of primary HCCs | [172] |
| FoxO3 | Interact with TGF-β-activated Smad2/3 and mediates TGF-β-induced apoptosis of liver cancer cells. | Undetermined | [163,164] |
| cPLA2α | Counteracts TGF-β-induced cytostasis by activating PPARγ and inhibiting R-Smad activity, and promotes HCC cell proliferation, EMT, migration and invasion by activating PI3K/Akt signaling. | Highly expressed in metastatic HCC cell lines and at the invasive edge in HCC tissues | [173,174] |
| p53 | Cooperates with Smads to enhance the expression levels of cell cycle- and apoptosis-related genes like p21, p15, Bim and DAPK, and meanwhile to inhibit those of AFP and Snail. | Reduced or mutated in liver cancer | [127,165,166,167] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, S.; Huang, W.; Huang, C.; Luo, Z.; Yan, X. Contextual Regulation of TGF-β Signaling in Liver Cancer. Cells 2019, 8, 1235. https://doi.org/10.3390/cells8101235
Tu S, Huang W, Huang C, Luo Z, Yan X. Contextual Regulation of TGF-β Signaling in Liver Cancer. Cells. 2019; 8(10):1235. https://doi.org/10.3390/cells8101235
Chicago/Turabian StyleTu, Shuo, Wei Huang, Chunhong Huang, Zhijun Luo, and Xiaohua Yan. 2019. "Contextual Regulation of TGF-β Signaling in Liver Cancer" Cells 8, no. 10: 1235. https://doi.org/10.3390/cells8101235
APA StyleTu, S., Huang, W., Huang, C., Luo, Z., & Yan, X. (2019). Contextual Regulation of TGF-β Signaling in Liver Cancer. Cells, 8(10), 1235. https://doi.org/10.3390/cells8101235