
TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Liver Fibrosis and Hepatic Stellate Cell (HSC) Activation
1.2. TGF-β Family
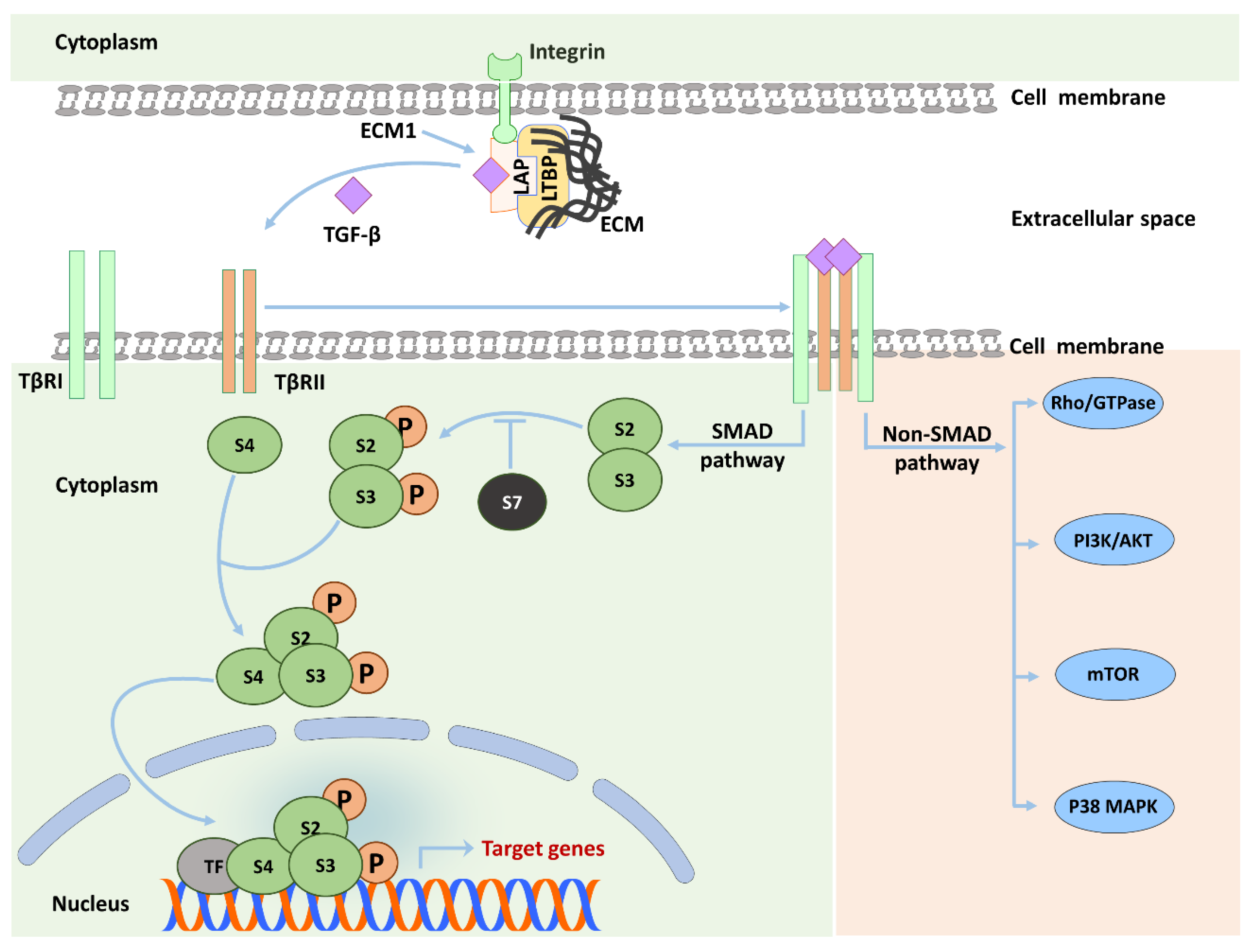
1.3. TGF-β Signaling
1.3.1. SMAD2 vs. SMAD3
1.3.2. SMAD Phosphorylation Dynamics
2. Regulation of the TGF-β Pathway
New Targets and Regulators of the TGF-β Pathway in Liver Fibrosis
3. TGF-β Activity and the Microenvironment in Liver Fibrosis
3.1. Composition of the ECM
3.2. Matrix Stiffness
3.3. TGF-β and Inflammatory Cells
3.4. TGF-β and Pathophysiological Blood Flow in Liver Fibrosis
3.5. Dynamics of TGF-β Ligand Availability
4. TGF-β Signaling, Cell Damage and Oxidative Stress in Liver Fibrosis
5. TGF-β Signaling and Epigenetics in Liver Fibrosis
6. TGF-β and Mesenchymal Transition in Liver Fibrosis
7. TGF-β and Metabolic Fate Changes in Liver Fibrosis
8. Circadian Rhythm, TGF-β Signaling, and Liver Fibrosis
9. TGF-β, Autophagy, and Senescence in Liver Fibrosis
10. Targeting TGF-β in Liver Fibrosis
11. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, J.Y.; Thakar, D.; Chang, T.T. Liver Fibrosis: Current Approaches and Future Directions for Diagnosis and Treatment. In Fibrosis in Disease: An Organ-Based Guide to Disease Pathophysiology and Therapeutic Considerations; Willis, M.S., Yates, C.C., Schisler, J.C., Eds.; Springer: Cham, Germany, 2019; pp. 387–417. [Google Scholar]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Li, J.-M.; Liu, M.-K.; Zhang, T.-T.; Wang, D.-P.; Zhou, W.-H.; Hu, L.-Z.; Lv, W.-L. Pathological process of liver sinusoidal endothelial cells in liver diseases. World J. Gastroenterol. 2017, 23, 7666–7677. [Google Scholar] [CrossRef] [PubMed]
- Elpek, G.Ö. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Brenner, D.A. Recent advancement of molecular mechanisms of liver fibrosis. J. Hepato-Biliary-Pancreat. Sci. 2015, 22, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-L.; Zhu, R.-T.; Sun, Y.-L. Epithelial-mesenchymal transition in liver fibrosis. Biomed. Rep. 2016, 4, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Caballero-Díaz, D. Transforming growth factor-β-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Soukupova, J.; Fabregat, I.; Dooley, S. TGF-β in hepatic stellate cell activation and liver fibrogenesis: Updated. Curr. Pathobiol. Rep. 2015, 3, 291–305. [Google Scholar] [CrossRef]
- Schon, H.-T.; Weiskirchen, R. Immunomodulatory effects of transforming growth factor-β in the liver. Hepatobiliary Surg. Nutr. 2014, 3, 386–406. [Google Scholar] [PubMed]
- Ghafoory, S.; Varshney, R.; Robison, T.; Kouzbari, K.; Woolington, S.; Murphy, B.; Xia, L.; Ahamed, J. Platelet TGF-β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018, 2, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.C.; Martin, J.S.; Cousins, F.M.; Kulkarni, A.B.; Karlsson, S.; Akhurst, R.J. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 1995, 121, 1845–1854. [Google Scholar] [PubMed]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warburton, D.; Bu, D.; Heisterkamp, N.; Groffen, J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P. IT-LIVER Consortium TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Dropmann, A.; Dediulia, T.; Breitkopf-Heinlein, K.; Korhonen, H.; Janicot, M.; Weber, S.N.; Thomas, M.; Piiper, A.; Bertran, E.; Fabregat, I.; et al. TGF-β1 and TGF-β2 abundance in liver diseases of mice and men. Oncotarget 2016, 7, 19499–19518. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Meguid, M.; Dawood, R.M.; Mokhles, M.A.; El Awady, M.K. Extrahepatic upregulation of transforming growth factor beta 2 in HCV genotype 4-induced liver fibrosis. J. Int. Soc. Interferon Cytokine Res. 2018, 38, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Chida, T.; Ito, M.; Nakashima, K.; Kanegae, Y.; Aoshima, T.; Takabayashi, S.; Kawata, K.; Nakagawa, Y.; Yamamoto, M.; Shimano, H.; et al. Critical role of CREBH-mediated induction of transforming growth factor β2 by hepatitis C virus infection in fibrogenic responses in hepatic stellate cells. Hepatology. 2017, 66, 1430–1443. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Marshall, J.F. The role of integrins in TGFβ activation in the tumour stroma. Cell Tissue Res. 2016, 365, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.L. Integrin-mediated transforming growth factor-beta activation, a potential therapeutic target in fibrogenic disorders. Am. J. Pathol. 2009, 175, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Liu, T.; Chen, W.; Hammad, S.; Longerich, T.; Fu, Y.; Li, N.; He, Y.; Liu, C.; Zhang, Y.; et al. ECM1 Prevents activation of transforming growth factor beta, hepatic stellate cells, and fibrogenesis in mice. Gastroenterology 2019, 157, 1352–1367. [Google Scholar] [CrossRef] [PubMed]
- Carthy, J.M. TGFβ signaling and the control of myofibroblast differentiation: Implications for chronic inflammatory disorders. J. Cell. Physiol. 2018, 233, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H. Transforming Growth Factor-β Signaling. In TGF-β in Human Disease; Moustakas, A., Miyazawa, K., Eds.; Springer: Tokyo, Japan, 2013; pp. 3–32. [Google Scholar]
- Levy, L.; Hill, C.S. Smad4 dependency defines two classes of transforming growth factor β (TGF-β) target genes and distinguishes TGF-β-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol. Cell. Biol. 2005, 25, 8108–8125. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.-H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Ko, S.-G.; Kim, H.-P.; Kim, Y.-B.; Lee, S.-Y.; Kim, S.-G.; Jong, H.-S.; Kim, T.-Y.; Lee, J.W.; Bang, Y.-J. Smad2 mediates Erk1/2 activation by TGF-beta1 in suspended, but not in adherent, gastric carcinoma cells. Int. J. Oncol. 2004, 24, 1229–1234. [Google Scholar] [PubMed]
- Zhang, L.; Duan, C.J.; Binkley, C.; Li, G.; Uhler, M.D.; Logsdon, C.D.; Simeone, D.M. A transforming growth factor beta-induced Smad3/Smad4 complex directly activates protein kinase A. Mol. Cell. Biol. 2004, 24, 2169–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlman, R.; Schiemann, W.P.; Brooks, M.W.; Lodish, H.F.; Weinberg, R.A. TGF-beta-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat. Cell Biol. 2001, 3, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Massagué, J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 1997, 389, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moses, H.L. Transforming growth factor β: Tumor suppressor or promoter? Are host immune cells the answer? Cancer Res. 2008, 68, 9107–9111. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Matsuzaki, K.; Murata, M.; Yamaguchi, T.; Suwa, K.; Okazaki, K. Clinico-Pathological importance of TGF-β/phospho-smad signaling during human hepatic fibrocarcinogenesis. Cancers 2018, 10, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeone, D.M.; Zhang, L.; Graziano, K.; Nicke, B.; Pham, T.; Schaefer, C.; Logsdon, C.D. Smad4 mediates activation of mitogen-activated protein kinases by TGF-beta in pancreatic acinar cells. Am. J. Physiol. Cell Physiol. 2001, 281, C311–C319. [Google Scholar] [CrossRef] [PubMed]
- Olsson, N.; Piek, E.; Sundström, M.; ten Dijke, P.; Nilsson, G. Transforming growth factor-beta-mediated mast cell migration depends on mitogen-activated protein kinase activity. Cell. Signal. 2001, 13, 483–490. [Google Scholar] [CrossRef]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. Embo J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zhou, D.; Meng, X.; Wang, X.; Liu, C.; Huang, C.; Li, J.; Zhang, L. Smad2 increases the apoptosis of activated human hepatic stellate cells induced by TRAIL. Int. Immunopharmacol. 2016, 32, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.-J.; et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Carthy, J.M.; Sundqvist, A.; Heldin, A.; van Dam, H.; Kletsas, D.; Heldin, C.-H.; Moustakas, A. Tamoxifen inhibits TGF-β-mediated activation of myofibroblasts by blocking non-smad signaling through ERK1/2. J. Cell. Physiol. 2015, 230, 3084–3092. [Google Scholar] [CrossRef] [PubMed]
- Ard, S.; Reed, E.B.; Smolyaninova, L.V.; Orlov, S.N.; Mutlu, G.M.; Guzy, R.D.; Dulin, N.O. Sustained smad2 phosphorylation is required for myofibroblast transformation in response to TGF-β. Am. J. Respir. Cell Mol. Biol. 2019, 60, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H. Fine tuning and cross-talking of TGF-beta signal by inhibitory Smads. J. Biochem. Mol. Biol. 2005, 38, 9–16. [Google Scholar] [PubMed] [Green Version]
- Goto, K.; Kamiya, Y.; Imamura, T.; Miyazono, K.; Miyazawa, K. Selective inhibitory effects of Smad6 on bone morphogenetic protein type I receptors. J. Biol. Chem. 2007, 282, 20603–20611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Sun, C.; He, B.; Xiong, W.; Shi, X.; Yao, D.; Cao, X. GADD34–PP1c recruited by Smad7 dephosphorylates TGFβ type I receptor. J. Cell Biol. 2004, 164, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Malonis, R.J.; Fu, W.; Jelcic, M.J.; Thompson, M.; Canter, B.S.; Tsikitis, M.; Esteva, F.J.; Sánchez, I. RNF11 sequestration of the E3 ligase SMURF2 on membranes antagonizes SMAD7 down-regulation of transforming growth factor β signaling. J. Biol. Chem. 2017, 292, 7435–7451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Fan, Y.; Xie, F.; Zhou, H.; Jin, K.; Shao, L.; Shi, W.; Fang, P.; Yang, B.; van Dam, H.; et al. Breast cancer metastasis suppressor OTUD1 deubiquitinates SMAD7. Nat. Commun. 2017, 8, 2116. [Google Scholar] [CrossRef] [PubMed]
- Chandhoke, A.S.; Karve, K.; Dadakhujaev, S.; Netherton, S.; Deng, L.; Bonni, S. The ubiquitin ligase Smurf2 suppresses TGFβ-induced epithelial–mesenchymal transition in a sumoylation-regulated manner. Cell Death Differ. 2016, 23, 876–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihata, W.A.; Putra, M.R.A.; Chin-Dusting, J.P.F. Is there a potential therapeutic role for caveolin-1 in fibrosis? Front. Pharm. 2017, 8, 567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Roy, C.; Wrana, J.L. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat. Rev. Mol. Cell Biol. 2005, 6, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-G. Endocytic regulation of TGF-beta signaling. Cell Res. 2009, 19, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Godoy, P.; Bachmann, A.; Liu, Y.; Barzan, D.; Ilkavets, I.; Maier, P.; Herskind, C.; Hengstler, J.G.; Dooley, S. Distinct role of endocytosis for Smad and non-Smad TGF-β signaling regulation in hepatocytes. J. Hepatol. 2011, 55, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.-G.; Zhang, Y.; Yao, S.-M.; Zhai, X.-J.; Zhang, L.-R.; Zhang, Y.-Z.; Li, H. Cav-1 deficiency promotes liver fibrosis in carbon tetrachloride (CCl4)-induced mice by regulation of oxidative stress and inflammation responses. Biomed. Pharm. Biomed. Pharm. 2018, 102, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhang, J.; Wang, Y.; Sun, Q. Caveolin-1 scaffolding domain peptides alleviate liver fibrosis by inhibiting tgf-β1/smad signaling in mice. Int. J. Mol. Sci. 2018, 19, 1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.; Liu, Y.; Kaul, A.; Peipe, I.; Dooley, S. Caveolin-1 abrogates TGF-β mediated hepatocyte apoptosis. Cell Death Dis. 2013, 4, e466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.; Dzieran, J.; Liu, Y.; Schindler, F.; Munker, S.; Müller, A.; Coulouarn, C.; Dooley, S. Distinct dedifferentiation processes affect caveolin-1 expression in hepatocytes. Cell Commun. Signal. 2013, 11, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Chen, D.; Huang, H.; Wang, J.; Wan, X.; Xu, C.; Li, C.; Ma, H.; Yu, C.; Li, Y. Caveolin1 protects against diet induced hepatic lipid accumulation in mice. PLoS ONE 2017, 12, e0178748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.M.; Noureddin, M.; Liu, C.; Ohashi, K.; Kim, S.Y.; Ramnath, D.; Powell, E.E.; Sweet, M.J.; Roh, Y.S.; Hsin, I.-F.; et al. Hyaluronan synthase 2-mediated hyaluronan production mediates Notch1 activation and liver fibrosis. Sci. Transl. Med. 2019, 11, eaat9284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-W.; Zhao, Y.-X.; Wei, D.; Li, Y.-L.; Zhang, Y.; Wu, J.; Xu, J.; Chen, C.; Tang, H.; Zhang, W.; et al. HAb18G/CD147 promotes activation of hepatic stellate cells and is a target for antibody therapy of liver fibrosis. J. Hepatol. 2012, 57, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-Y.; Ju, D.; Zhang, D.-W.; Li, H.; Kong, L.-M.; Guo, Y.; Li, C.; Wang, X.-L.; Chen, Z.-N.; Bian, H. Activation of TGF-β1-CD147 positive feedback loop in hepatic stellate cells promotes liver fibrosis. Sci. Rep. 2015, 5, 16552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibanuma, M.; Mashimo, J.; Kuroki, T.; Nose, K. Characterization of the TGF beta 1-inducible hic-5 gene that encodes a putative novel zinc finger protein and its possible involvement in cellular senescence. J. Biol. Chem. 1994, 269, 26767–26774. [Google Scholar] [PubMed]
- Varney, S.D.; Betts, C.B.; Zheng, R.; Wu, L.; Hinz, B.; Zhou, J.; Van De Water, L. Hic-5 is required for myofibroblast differentiation by regulating mechanically dependent MRTF-A nuclear accumulation. J. Cell Sci. 2016, 129, 774–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, X.-F.; Fu, W.; Kim-Kaneyama, J.-R.; Omoto, T.; Miyazaki, T.; Li, B.; Miyazaki, A. Hic-5 deficiency attenuates the activation of hepatic stellate cells and liver fibrosis through upregulation of Smad7 in mice. J. Hepatol. 2016, 64, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Kladney, R.D.; Cui, X.; Bulla, G.A.; Brunt, E.M.; Fimmel, C.J. Expression of GP73, a resident Golgi membrane protein, in viral and nonviral liver disease. Hepatology 2002, 35, 1431–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Wei, C.; Liu, N.; Wu, F.; Chen, J.; Wang, C.; Sun, Z.; Wang, Y.; Liu, L.; Zhang, X.; et al. GP73, a novel TGF-β target gene, provides selective regulation on Smad and non-Smad signaling pathways. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Earl, L.A.; Bi, S.; Baum, L.G. Galectin multimerization and lattice formation are regulated by linker region structure. Glycobiology 2011, 21, 6–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.-H.; Hong, H.-C.; Hong, T.-M.; Chiang, W.-F.; Jin, Y.-T.; Chen, Y.-L. Targeting galectin-1 in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis by downregulating MCP-1/CCL2 expression. Clin. Cancer Res. 2011, 17, 1306–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsumi, T.; Suzuki, T.; Moriya, K.; Shintani, Y.; Fujie, H.; Miyoshi, H.; Matsuura, Y.; Koike, K.; Miyamura, T. Hepatitis C virus core protein activates ERK and p38 MAPK in cooperation with ethanol in transgenic mice. Hepatology 2003, 38, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-H.; Chen, Y.-L.; Lee, K.-H.; Chang, C.-C.; Cheng, T.-M.; Wu, S.-Y.; Tu, C.-C.; Tsui, W.-L. Glycosylation-dependent galectin-1/neuropilin-1 interactions promote liver fibrosis through activation of TGF-β- and PDGF-like signals in hepatic stellate cells. Sci. Rep. 2017, 7, 11006. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Rui, B.-B.; Tang, L.-Y.; Hu, C.-M. Lipin family proteins—Key regulators in lipid metabolism. Ann. Nutr. Metab. 2015, 66, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Gropler, M.C.; Chen, Z.; Leone, T.C.; Croce, M.A.; Harris, T.E.; Lawrence, J.C.; Kelly, D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006, 4, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, C.H.; Kim, K.M.; Yang, J.H.; Cho, S.S.; Kim, S.J.; Shin, S.M.; Cho, I.J.; Ki, S.H. The Role of Lipin-1 in the Regulation of Fibrogenesis and TGF-β Signaling in Hepatic Stellate Cells. Toxicol. Sci. 2016, 153, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, R.-W.; Han, B.; Li, Z.; Xiong, L.; Zhang, F.-Y.; Cong, B.-B.; Zhang, B. Notch signaling mediated by TGF-β/Smad pathway in concanavalin A-induced liver fibrosis in rats. World J. Gastroenterol. 2017, 23, 2330–2336. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Liu, Y.; Dewidar, B.; Hu, J.; Park, O.; Feng, T.; Xu, C.; Yu, C.; Li, Q.; Meyer, C.; et al. Delta-like ligand 4 modulates liver damage by down-regulating chemokine expression. Am. J. Pathol. 2016, 186, 1874–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, P.; Zhang, J.; Xu, D.; Zhu, J.; Li, W.; Liu, J.; Liu, F. Positive feedback loop of YB-1 interacting with Smad2 promotes liver fibrosis. Biochem. Biophys. Res. Commun. 2017, 484, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Kolliopoulos, C.; Raja, E.; Razmara, M.; Heldin, P.; Heldin, C.-H.; Moustakas, A.; van der Heide, L.P. Transforming growth factor β (TGFβ) induces NUAK kinase expression to fine-tune its signaling output. J. Biol. Chem. 2019, 294, 4119–4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Königsrainer, A.; Weng, H.; Dooley, S.; ten Dijke, P. Transforming growth factor-β (TGF-β)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.-Y.; Heller, M.; Meng, Z.; Yu, L.-R.; Tang, Y.; Zhou, M.; Zhang, Y.E. Transforming growth factor-β (TGF-β) Directly activates the JAK1-STAT3 axis to induce hepatic fibrosis in coordination with the SMAD pathway. J. Biol. Chem. 2017, 292, 4302–4312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Yu, Y.; Sun, C.; Liu, T.; Liang, T.; Zhan, L.; Lin, X.; Feng, X.-H. STAT3 selectively interacts with Smad3 to antagonize TGF-β signalling. Oncogene 2016, 35, 4388–4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.-H.; Sudo, K.; Kuroda, M.; Kato, M.; Lee, I.-K.; Han, J.S.; Nakae, S.; Imamura, T.; Kim, J.; Ju, J.H.; et al. Phosphorylation status determines the opposing functions of Smad2/Smad3 as STAT3 cofactors in TH17 differentiation. Nat. Commun. 2015, 6, 7600. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Ham, S.; Lee, Y.; Suh, G.Y.; Lee, Y.-S. TTC3 contributes to TGF-β1-induced epithelial-mesenchymal transition and myofibroblast differentiation, potentially through SMURF2 ubiquitylation and degradation. Cell Death Dis. 2019, 10, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.-H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3297–E3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastan, I.; Hassan, R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. 2014, 74, 2907–2912. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Wang, P.; Liang, S.; Iwaisako, K.; Liu, X.; Xu, J.; Zhang, M.; Sun, M.; Cong, M.; Karin, D.; et al. Mesothelin/mucin 16 signaling in activated portal fibroblasts regulates cholestatic liver fibrosis. J. Clin. Investig. 2017, 127, 1254–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, B.; Addante, A.; Sánchez, A. BMP Signalling at the crossroad of liver fibrosis and regeneration. Int. J. Mol. Sci. 2017, 19, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitkopf-Heinlein, K.; Meyer, C.; König, C.; Gaitantzi, H.; Addante, A.; Thomas, M.; Wiercinska, E.; Cai, C.; Li, Q.; Wan, F.; et al. BMP-9 interferes with liver regeneration and promotes liver fibrosis. Gut 2017, 66, 939–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.-H.; Huang, Y.-H.; Chu, T.-H.; Chen, C.-L.; Lin, P.-R.; Huang, S.-C.; Wu, D.-C.; Huang, C.-C.; Hu, T.-H.; Kao, Y.-H.; et al. BMP-2 restoration aids in recovery from liver fibrosis by attenuating TGF-β1 signaling. Lab. Investig. J. Tech. Methods Pathol. 2018, 98, 999–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-Q.; Wan, L.-Y.; He, X.-M.; Ni, Y.-R.; Wang, C.; Liu, C.-B.; Wu, J.-F. Gremlin1 Accelerates Hepatic Stellate Cell Activation Through Upregulation of TGF-Beta Expression. DNA Cell Biol. 2017, 36, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Corcuera, A.; López-Zabalza, M.J.; Martínez-Irujo, J.J.; Álvarez-Sola, G.; Ávila, M.A.; Iraburu, M.J.; Ansorena, E.; Montiel-Duarte, C. Role of AGAP2 in the profibrogenic effects induced by TGFβ in LX-2 hepatic stellate cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Römisch, J. Factor VII activating protease (FSAP): A novel protease in hemostasis. Biol. Chem. 2002, 383, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Leiting, S.; Seidl, S.; Martinez-Palacian, A.; Muhl, L.; Kanse, S.M. Transforming growth factor-β (TGF-β) inhibits the expression of factor vii-activating protease (FSAP) in hepatocytes. J. Biol. Chem. 2016, 291, 21020–21028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkham-Kamphorst, E.; Zimmermann, H.W.; Gassler, N.; Bissels, U.; Bosio, A.; Tacke, F.; Weiskirchen, R.; Kanse, S.M. Factor VII activating protease (FSAP) exerts anti-inflammatory and anti-fibrotic effects in liver fibrosis in mice and men. J. Hepatol. 2013, 58, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Wasmuth, H.E.; Tag, C.G.; Van de Leur, E.; Hellerbrand, C.; Mueller, T.; Berg, T.; Puhl, G.; Neuhaus, P.; Samuel, D.; Trautwein, C.; et al. The Marburg I variant (G534E) of the factor VII-activating protease determines liver fibrosis in hepatitis C infection by reduced proteolysis of platelet-derived growth factor BB. Hepatology 2009, 49, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Spanjer, A.I.R.; Baarsma, H.A.; Oostenbrink, L.M.; Jansen, S.R.; Kuipers, C.C.; Lindner, M.; Postma, D.S.; Meurs, H.; Heijink, I.H.; Gosens, R.; et al. TGF-β-induced profibrotic signaling is regulated in part by the WNT receptor Frizzled-8. FASEB J. 2016, 30, 1823–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beljaars, L.; Daliri, S.; Dijkhuizen, C.; Poelstra, K.; Gosens, R. WNT-5A regulates TGF-β-related activities in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G219–G227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustin, H.G.; Koh, G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017, 357, eaal2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, D.; Koyama, Y.; Brenner, D.; Kisseleva, T. The characteristics of activated portal fibroblasts/myofibroblasts in liver fibrosis. Differ. Res. Biol. Divers. 2016, 92, 84–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Kisseleva, T. Reversibility of liver fibrosis. Clin. Res. Hepatol. Gastroenterol. 2015, 39, S60–S63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar] [PubMed]
- Heymann, F.; Trautwein, C.; Tacke, F. Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm. Allergy Drug Targets 2009, 8, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Ruehl, M.; Somasundaram, R.; Hahn, E.G. Matrix as a modulator of hepatic fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaso, E.; Ikeda, K.; Eng, F.J.; Xu, L.; Wang, L.H.; Lin, H.C.; Friedman, S.L. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J. Clin. Investig. 2001, 108, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Nakagawa, S.; Yazdani, S.; van Baarlen, J.; Venkatesh, A.; Koh, A.P.; Song, W.-M.; Goossens, N.; Watanabe, H.; Beasley, M.B.; et al. Integrin alpha 11 in the regulation of the myofibroblast phenotype: Implications for fibrotic diseases. Exp. Mol. Med. 2017, 49, e396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhalgh, S.N.; Matchett, K.P.; Taylor, R.S.; Huang, K.; Li, J.T.; Saeteurn, K.; Donnelly, M.C.; Simpson, E.E.M.; Pollack, J.L.; Atakilit, A.; et al. Loss of integrin αvβ8 in murine hepatocytes accelerates liver regeneration. Am. J. Pathol. 2019, 189, 258–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rognoni, E.; Ruppert, R.; Fässler, R. The kindlin family: Functions, signaling properties and implications for human disease. J. Cell Sci. 2016, 129, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Hu, Y.; Gao, Y.; Li, Q.; Zeng, Z.; Li, Y.; Chen, H. Kindlin-2 regulates hepatic stellate cells activation and liver fibrogenesis. Cell Death Discov. 2018, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Zollinger, A.J.; Smith, M.L. Fibronectin, the extracellular glue. Matrix Biol. J. Int. Soc. Matrix Biol. 2017, 60–61, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Chau, G.; Walraven, M.; Boo, S.; Koehler, A.; Chow, M.L.; Olsen, A.L.; Im, M.; Lodyga, M.; Wells, R.G.; et al. The fibronectin ED-A domain enhances recruitment of latent TGF-β-binding protein-1 to the fibroblast matrix. J. Cell Sci. 2018, 131, jcs201293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J.H.; Hynes, R.O. Fibronectin isoform distribution in the mouse. I. The alternatively spliced EIIIB, EIIIA, and V segments show widespread codistribution in the developing mouse embryo. Cell Adhes. Commun. 1996, 4, 103–125. [Google Scholar] [CrossRef] [PubMed]
- Zent, J.; Guo, L.-W. Signaling Mechanisms of Myofibroblastic Activation: Outside-in and Inside-Out. Cell. Physiol. Biochem. 2018, 49, 848–868. [Google Scholar] [CrossRef] [PubMed]
- Muro, A.F.; Moretti, F.A.; Moore, B.B.; Yan, M.; Atrasz, R.G.; Wilke, C.A.; Flaherty, K.R.; Martinez, F.J.; Tsui, J.L.; Sheppard, D.; et al. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 638–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawelke, N.; Vasel, M.; Sens, C.; von Au, A.; Dooley, S.; Nakchbandi, I.A. Fibronectin protects from excessive liver fibrosis by modulating the availability of and responsiveness of stellate cells to active TGF-β. PLoS ONE 2011, 6, e28181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altrock, E.; Sens, C.; Wuerfel, C.; Vasel, M.; Kawelke, N.; Dooley, S.; Sottile, J.; Nakchbandi, I.A. Inhibition of fibronectin deposition improves experimental liver fibrosis. J. Hepatol. 2015, 62, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Phanish, M.K.; Heidebrecht, F.; Nabi, M.E.; Shah, N.; Niculescu-Duvaz, I.; Dockrell, M.E.C. The regulation of TGFβ1 Induced fibronectin eda exon alternative splicing in human renal proximal tubule epithelial cells. J. Cell. Physiol. 2015, 230, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Ma, M.; Jiang, S.; Zhang, X.; Zhang, Y.; Yang, X.; Xu, C.; Tian, G.; Li, Q.; et al. Autocrine CTHRC1 activates hepatic stellate cells and promotes liver fibrosis by activating TGF-β signaling. EBioMedicine 2019, 40, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.; Lagares, D. Matrix stiffness: The conductor of organ fibrosis. Curr. Rheumatol. Rep. 2018, 20, 2. [Google Scholar] [CrossRef] [PubMed]
- Lampi, M.C.; Reinhart-King, C.A. Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials. Sci. Transl. Med. 2018, 10, eaao0475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caliari, S.R.; Perepelyuk, M.; Soulas, E.M.; Lee, G.Y.; Wells, R.G.; Burdick, J.A. Gradually softening hydrogels for modeling hepatic stellate cell behavior during fibrosis regression. Integr. Biol. Quant. Biosci. Nano Macro 2016, 8, 720–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterová, E.; Mrkvicová, A.; Podmolíková, L.; Řezáčová, M.; Kanta, J. The role of cytokines TGF-beta1 and FGF-1 in the expression of characteristic markers of rat liver myofibroblasts cultured in three-dimensional collagen gel. Physiol. Res. 2016, 65, 661–672. [Google Scholar] [PubMed]
- Siegel, R.C.; Pinnell, S.R.; Martin, G.R. Cross-linking of collagen and elastin. Properties of lysyl oxidase. Biochemistry 1970, 9, 4486–4492. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.B.; Ikenaga, N.; Peng, Z.-W.; Sverdlov, D.Y.; Greenstein, A.; Smith, V.; Schuppan, D.; Popov, Y. Lysyl oxidase activity contributes to collagen stabilization during liver fibrosis progression and limits spontaneous fibrosis reversal in mice. FASEB J. 2016, 30, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zeng, Y.; Wei, J.; Yang, D.; Ding, G.; Liu, J.; Shang, J.; Kang, Y.; Ji, X. Knockdown of LOXL1 inhibits TGF-β1-induced proliferation and fibrogenesis of hepatic stellate cells by inhibition of Smad2/3 phosphorylation. Biomed. Pharm. Biomed. Pharm. 2018, 107, 1728–1735. [Google Scholar] [CrossRef] [PubMed]
- Mesarwi, O.A.; Shin, M.-K.; Drager, L.F.; Bevans-Fonti, S.; Jun, J.C.; Putcha, N.; Torbenson, M.S.; Pedrosa, R.P.; Lorenzi-Filho, G.; Steele, K.E.; et al. Lysyl Oxidase as a serum biomarker of liver fibrosis in patients with severe obesity and obstructive sleep apnea. Sleep 2015, 38, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Perez, M.; Lee, E.-S.; Kojima, S.; Griffin, M. The functional relationship between transglutaminase 2 and transforming growth factor β1 in the regulation of angiogenesis and endothelial-mesenchymal transition. Cell Death Dis. 2017, 8, e3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verderio, E.; Gaudry, C.; Gross, S.; Smith, C.; Downes, S.; Griffin, M. Regulation of cell surface tissue transglutaminase: Effects on matrix storage of latent transforming growth factor-beta binding protein-1. J. Histochem. Cytochem. 1999, 47, 1417–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskirchen, R.; Tacke, F. Liver fibrosis: From pathogenesis to novel therapies. Dig. Dis. 2016, 34, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Lodyga, M.; Cambridge, E.; Karvonen, H.M.; Pakshir, P.; Wu, B.; Boo, S.; Kiebalo, M.; Kaarteenaho, R.; Glogauer, M.; Kapoor, M.; et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β. Sci. Signal. 2019, 12, e3469. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Li, Z.; Zhang, Q.; Qu, Y.; Xu, M.; Wan, X.; Lu, L. CXCL6-EGFR-induced Kupffer cells secrete TGF-β1 promoting hepatic stellate cell activation via the SMAD2/BRD4/C-MYC/EZH2 pathway in liver fibrosis. J. Cell. Mol. Med. 2018, 22, 5050–5061. [Google Scholar] [CrossRef] [PubMed]
- Fabre, T.; Molina, M.F.; Soucy, G.; Goulet, J.-P.; Willems, B.; Villeneuve, J.-P.; Bilodeau, M.; Shoukry, N.H. Type 3 cytokines IL-17A and IL-22 drive TGF-β-dependent liver fibrosis. Sci. Immunol. 2018, 3, e7754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Zhao, J.; Zhang, X.; Cheng, Y.; Hu, J.; Li, Y.; Zhao, X.; Shang, Q.; Sun, Y.; Tu, B.; et al. Activated hepatic stellate cells impair NK cell anti-fibrosis capacity through a TGF-β-dependent emperipolesis in HBV cirrhotic patients. Sci. Rep. 2017, 7, 44544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, C.P.; Hinz, B.; Swartz, M.A. Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. J. Cell Sci. 2005, 118, 4731–4739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nithiananthan, S.; Crawford, A.; Knock, J.C.; Lambert, D.W.; Whawell, S.A. Physiological fluid flow moderates fibroblast responses to TGF-β1. J. Cell. Biochem. 2017, 118, 878–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansorge, M.; Sapudom, J.; Chkolnikov, M.; Wilde, M.; Anderegg, U.; Möller, S.; Schnabelrauch, M.; Pompe, T. Mimicking paracrine TGFβ1 Signals during myofibroblast differentiation in 3D Collagen networks. Sci. Rep. 2017, 7, 5664. [Google Scholar] [CrossRef] [PubMed]
- Zi, Z.; Feng, Z.; Chapnick, D.A.; Dahl, M.; Deng, D.; Klipp, E.; Moustakas, A.; Liu, X. Quantitative analysis of transient and sustained transforming growth factor-β signaling dynamics. Mol. Syst. Biol. 2011, 7, 492. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zi, Z.; Liu, X. Measuring TGF-β ligand dynamics in culture medium. Methods Mol. Biol. 2016, 1344, 379–389. [Google Scholar] [PubMed]
- Hara, M.; Kirita, A.; Kondo, W.; Matsuura, T.; Nagatsuma, K.; Dohmae, N.; Ogawa, S.; Imajoh-Ohmi, S.; Friedman, S.L.; Rifkin, D.B.; et al. LAP degradation product reflects plasma kallikrein-dependent TGF-β activation in patients with hepatic fibrosis. SpringerPlus 2014, 3, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, M.; Inoue, I.; Yamazaki, Y.; Kirita, A.; Matsuura, T.; Friedman, S.L.; Rifkin, D.B.; Kojima, S. L59 TGF-β LAP degradation products serve as a promising blood biomarker for liver fibrogenesis in mice. Fibrogenesis Tissue Repair 2015, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novo, E.; Parola, M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenesis Tissue Repair 2008, 1, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, K.; Kietzmann, T. Reactive oxygen species and fibrosis: Further evidence of a significant liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.-M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Wang, W.; Peng, X.-M.; He, Y.; Xiong, Y.-X.; Liang, H.-F.; Chu, L.; Zhang, B.-X.; Ding, Z.-Y.; Chen, X.-P. Rapamycin Upregulates connective tissue growth factor expression in hepatic progenitor cells through TGF-β-Smad2 dependent signaling. Front. Pharm. 2018, 9, 877. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, Y.-H.; Iwaisako, K.; Seki, E.; Inokuchi, S.; Schnabl, B.; Osterreicher, C.H.; Kisseleva, T.; Brenner, D.A. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011, 53, 1730–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Zhang, J.; Zhang, X.; Liu, J.; Jiang, J.X.; Yamaguchi, K.; Taruno, A.; Katsuyama, M.; Iwata, K.; Ibi, M.; et al. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2018, 115, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernández-Rodriguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andueza, A.; Garde, N.; García-Garzón, A.; Ansorena, E.; López-Zabalza, M.J.; Iraburu, M.J.; Zalba, G.; Martínez-Irujo, J.J. NADPH oxidase 5 promotes proliferation and fibrosis in human hepatic stellate cells. Free Radic. Biol. Med. 2018, 126, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Latella, G. Redox Imbalance in intestinal fibrosis: Beware of the TGFβ-1, ROS, and Nrf2 connection. Dig. Dis. Sci. 2018, 63, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Hellerbrand, C.; Köhler, U.A.; Bugnon, P.; Kan, Y.-W.; Werner, S.; Beyer, T.A. The Nrf2 transcription factor protects from toxin-induced liver injury and fibrosis. Lab. Investig. J. Tech. Methods Pathol. 2008, 88, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.J.; Kim, J.-Y.; Min, A.-K.; Park, K.-G.; Harris, R.A.; Kim, H.-J.; Lee, I.-K. Sulforaphane attenuates hepatic fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-β/Smad signaling. Free Radic. Biol. Med. 2012, 52, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Prestigiacomo, V.; Suter-Dick, L. Nrf2 protects stellate cells from Smad-dependent cell activation. PLoS ONE 2018, 13, e0201044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Tan, Y.; Liu, W.; Yang, J.; Wang, D.; Pan, D.; Sun, Y.; Zheng, C. NF-E2-related factor 2 suppresses intestinal fibrosis by inhibiting reactive oxygen species-dependent TGF-β1/SMADs pathway. Dig. Dis. Sci. 2018, 63, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, H.; Cao, Y.; Li, Y.; Sun, S.; Zhang, J.; Zhang, G. Schisandrin B attenuates CCl4-induced liver fibrosis in rats by regulation of Nrf2-ARE and TGF-β/Smad signaling pathways. Drug Des. Dev. 2017, 11, 2179–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piersma, B.; Wouters, O.Y.; de Rond, S.; Boersema, M.; Gjaltema, R.A.F.; Bank, R.A. Ascorbic acid promotes a TGFβ1-induced myofibroblast phenotype switch. Physiol. Rep. 2017, 5, e13324. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.-J.; Yoo, H.-S.; Shin, S.; Park, Y.-J.; Jeon, S.-M. Dysregulation of NRF2 in cancer: From Molecular mechanisms to therapeutic opportunities. Biomology 2018, 26, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camarena, V.; Wang, G. The epigenetic role of vitamin C in health and disease. Cell. Mol. Life Sci. 2016, 73, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argemi, J.; Latasa, M.U.; Atkinson, S.R.; Blokhin, I.O.; Massey, V.; Gue, J.P.; Cabezas, J.; Lozano, J.J.; Van Booven, D.; Bell, A.; et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat. Commun. 2019, 10, 3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Chen, B.; Fan, X.; Li, G.; Dong, P.; Zheng, J. Epigenetically-regulated MicroRNA-9-5p suppresses the activation of hepatic stellate cells via TGFBR1 and TGFBR2. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharm. 2017, 43, 2242–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davoodian, P.; Ravanshad, M.; Hosseini, S.Y.; Khanizadeh, S.; Almasian, M.; Nejati Zadeh, A.; Esmaiili Lashgarian, H. Effect of TGF-β/smad signaling pathway blocking on expression profiles of miR-335, miR-150, miR-194, miR-27a, and miR-199a of hepatic stellate cells (HSCs). Gastroenterol. Hepatol. Bed Bench 2017, 10, 112–117. [Google Scholar] [PubMed]
- Bowen, T.; Jenkins, R.H.; Fraser, D.J. MicroRNAs, transforming growth factor beta-1, and tissue fibrosis. J. Pathol. 2013, 229, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.; El Taghdouini, A.; Perea, L.; Mannaerts, I.; Vila-Casadesús, M.; Blaya, D.; Rodrigo-Torres, D.; Affò, S.; Morales-Ibanez, O.; Graupera, I.; et al. Integrative miRNA and Gene expression profiling analysis of human quiescent hepatic stellate cells. Sci. Rep. 2015, 5, 11549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roderburg, C.; Luedde, M.; Vargas Cardenas, D.; Vucur, M.; Mollnow, T.; Zimmermann, H.W.; Koch, A.; Hellerbrand, C.; Weiskirchen, R.; Frey, N.; et al. miR-133a mediates TGF-β-dependent derepression of collagen synthesis in hepatic stellate cells during liver fibrosis. J. Hepatol. 2013, 58, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Benz, F.; Vargas Cardenas, D.; Vucur, M.; Gautheron, J.; Schneider, A.; Hellerbrand, C.; Pottier, N.; Alder, J.; Tacke, F.; et al. miR-30c and miR-193 are a part of the TGF-β-dependent regulatory network controlling extracellular matrix genes in liver fibrosis. J. Dig. Dis. 2015, 16, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Brandon-Warner, E.; Benbow, J.H.; Swet, J.H.; Feilen, N.A.; Culberson, C.R.; McKillop, I.H.; deLemos, A.S.; Russo, M.W.; Schrum, L.W. Adeno-associated virus serotype 2 vector-mediated reintroduction of microrna-19b attenuates hepatic fibrosis. Hum. Gene Ther. 2018, 29, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Feili, X.; Wu, S.; Ye, W.; Tu, J.; Lou, L. MicroRNA-34a-5p inhibits liver fibrosis by regulating TGF-β1/Smad3 pathway in hepatic stellate cells. Cell Biol. Int. 2018, 42, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, S.; Li, Z.; Song, D.; Zhang, S.; Yao, Q. MiR-146a attenuates liver fibrosis by inhibiting transforming growth factor-β1 mediated epithelial-mesenchymal transition in hepatocytes. Cell. Signal. 2019, 58, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Cai, Y.; Lu, D.; Zhou, Y.; Yao, Q.; Zhang, S. MicroRNA-146a-5p attenuates liver fibrosis by suppressing profibrogenic effects of TGFβ1 and lipopolysaccharide. Cell. Signal. 2017, 39, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lei, S.; Wang, X.; Xu, W.; Hu, P.; Chen, F.; Zhang, X.; Yin, C.; Xie, W. MicroRNA-134 deactivates hepatic stellate cells by targeting tgf-β activated kinase 1-binding protein 1. Biochem. Cell Biol. 2019, 97, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Xue, D.; Shen, D.; Ma, W.; Zhang, J.; Wang, X.; Zhang, W.; Wu, L.; Pan, K.; Yang, Y.; et al. MicroRNA-942 mediates hepatic stellate cell activation by regulating BAMBI expression in human liver fibrosis. Arch. Toxicol. 2018, 92, 2935–2946. [Google Scholar] [CrossRef] [PubMed]
- You, K.; Li, S.-Y.; Gong, J.; Fang, J.-H.; Zhang, C.; Zhang, M.; Yuan, Y.; Yang, J.; Zhuang, S.-M. MicroRNA-125b promotes hepatic stellate cell activation and liver fibrosis by activating rhoa signaling. Mol. Nucleic Acids 2018, 12, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Guo, Y.; Chen, B.; Dong, P.; Zheng, J. MicroRNA-17-5p activates hepatic stellate cells through targeting of Smad7. Lab. Investig. J. Tech. Methods Pathol. 2015, 95, 781–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Zhang, Z.; Zhang, Y.; Li, W.; Zheng, W.; Yu, J.; Wang, B.; Chen, L.; Zhuo, Q.; Chen, L.; et al. MicroRNA-212 activates hepatic stellate cells and promotes liver fibrosis via targeting SMAD7. Biochem. Biophys. Res. Commun. 2018, 496, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Zheng, X.; Li, H.; Cao, Z.; Chang, H.; Luan, S.; Zhu, J.; Chen, J.; Zang, Y.; Zhang, J. MicroRNA-30 Protects against carbon tetrachloride-induced liver fibrosis by attenuating transforming growth factor beta signaling in hepatic stellate cells. Toxicol. Sci. 2015, 146, 157–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.; Sata, T.N.; Yadav, A.K.; Mishra, A.; Vats, N.; Hossain, M.M.; Sanal, M.G.; Venugopal, S.K. TGF-β induces liver fibrosis via miRNA-181a-mediated down regulation of augmenter of liver regeneration in hepatic stellate cells. PLoS ONE 2019, 14, e0214534. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Dan, X.; Men, R.; Ma, L.; Wen, M.; Peng, Y.; Yang, L. MiR-142-3p blocks TGF-β-induced activation of hepatic stellate cells through targeting TGFβRI. Life Sci. 2017, 187, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Wang, Q.; Zhou, H.; Qiu, J.; Li, C.; Shi, C.; Zhou, S.; Liu, R.; Lu, L. miR-455-3p Alleviates Hepatic Stellate Cell Activation and Liver Fibrosis by Suppressing HSF1 Expression. Mol. Nucleic Acids 2019, 16, 758–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Hao, C.; Li, M.; Dai, X.; Qin, H.; Li, J.; Xu, H.; Wu, X.; Zhang, L.; Fang, M.; et al. MKL1 is an epigenetic modulator of TGF-β induced fibrogenesis. Biochim. Biophys. Acta 2015, 1849, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, C.; Liu, Q.; Liu, B.; Song, C.; Zhu, S.; Wu, C.; Liu, S.; Yu, H.; Yao, D.; et al. The role of the miR-31/FIH1 pathway in TGF-β-induced liver fibrosis. Clin. Sci. 2015, 129, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Genz, B.; Coleman, M.A.; Irvine, K.M.; Kutasovic, J.R.; Miranda, M.; Gratte, F.D.; Tirnitz-Parker, J.E.E.; Olynyk, J.K.; Calvopina, D.A.; Weis, A.; et al. Overexpression of miRNA-25-3p inhibits Notch1 signaling and TGF-β-induced collagen expression in hepatic stellate cells. Sci. Rep. 2019, 9, 8541. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Wan, L.-Y.; Liang, J.-J.; Zhang, Y.-Q.; Ai, W.-B.; Wu, J.-F. The roles of lncRNA in hepatic fibrosis. Cell Biosci. 2018, 8, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, P.M.-K.; Zhang, Y.-Y.; Lan, H.-Y. LncRNAs in TGF-β-driven tissue fibrosis. Non-Coding RNA 2018, 4, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, X.; Zhang, Y.; Zheng, X.; Deng, J.; Li, H.; Kang, Z.; Cao, Z.; Huang, Z.; Ding, Z.; Dong, L.; et al. TGF-β-induced hepatocyte lincRNA-p21 contributes to liver fibrosis in mice. Sci. Rep. 2017, 7, 2957. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, C.; Barbier, O.; Smalling, R.; Tsuchiya, H.; Lee, S.; Delker, D.; Zou, A.; Hagedorn, C.H.; Wang, L. Bcl2 is a critical regulator of bile acid homeostasis by dictating Shp and lncRNA H19 function. Sci. Rep. 2016, 6, 20559. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yamamoto, M.; Fujii, K.; Nagahama, Y.; Ooshio, T.; Xin, B.; Okada, Y.; Furukawa, H.; Nishikawa, Y. Differential reactivation of fetal/neonatal genes in mouse liver tumors induced in cirrhotic and non-cirrhotic conditions. Cancer Sci. 2015, 106, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Luo, Z.; Pan, Y.; Zheng, W.; Li, W.; Zhang, Z.; Xiong, P.; Xu, D.; Du, M.; Wang, B.; et al. H19/miR-148a/USP4 axis facilitates liver fibrosis by enhancing TGF-β signaling in both hepatic stellate cells and hepatocytes. J. Cell. Physiol. 2019, 234, 9698–9710. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Xia, L.; Sun, M.; Yang, C.; Wang, F. Circular RNA in Liver: Health and diseases. Adv. Exp. Med. Biol. 2018, 1087, 245–257. [Google Scholar] [PubMed]
- Chen, Y.; Yuan, B.; Wu, Z.; Dong, Y.; Zhang, L.; Zeng, Z. Microarray profiling of circular RNAs and the potential regulatory role of hsa_circ_0071410 in the activated human hepatic stellate cell induced by irradiation. Gene 2017, 629, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lv, X.; Qu, H.; Zhao, K.; Fu, L.; Zhu, L.; Ye, G.; Guo, J. Preliminary screening and functional analysis of circular RNAs associated with hepatic stellate cell activation. Gene 2018, 677, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jung, Y.; Hyun, J.; Friedersdorf, M.; Oh, S.-H.; Kim, J.; Premont, R.T.; Keene, J.D.; Diehl, A.M. RNA binding proteins control transdifferentiation of hepatic stellate cells into myofibroblasts. Cell. Physiol. Biochem. 2018, 48, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-J.; Huang, C.; Meng, X.-M.; Li, J. Epigenetic modifications by histone deacetylases: Biological implications and therapeutic potential in liver fibrosis. Biochimie 2015, 116, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Hardy, T.; Mann, D.A. Epigenetics in liver disease: From biology to therapeutics. Gut 2016, 65, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zeng, S.; Li, L.; Fan, Z.; Tian, W.; Li, M.; Xu, H.; Wu, X.; Fang, M.; Xu, Y. Angiogenic factor with G patch and FHA domains 1 (Aggf1) regulates liver fibrosis by modulating TGF-β signaling. Biochim. Biophys. Acta 2016, 1862, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Zeybel, M.; Luli, S.; Sabater, L.; Hardy, T.; Oakley, F.; Leslie, J.; Page, A.; Moran Salvador, E.; Sharkey, V.; Tsukamoto, H.; et al. A proof-of-concept for epigenetic therapy of tissue fibrosis: Inhibition of liver fibrosis progression by 3-deazaneplanocin A. Mol. J. Am. Soc. Gene 2017, 25, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Martin-Mateos, R.; De Assuncao, T.M.; Arab, J.P.; Jalan-Sakrikar, N.; Yaqoob, U.; Greuter, T.; Verma, V.K.; Mathison, A.J.; Cao, S.; Lomberk, G.; et al. Enhancer of zeste homologue 2 inhibition attenuates TGF-β dependent hepatic stellate cell activation and liver fibrosis. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.; Wang, S.; Shao, C.; Yuan, X.; Wandrer, F.; Bantel, H.; Marx, A.; Ebert, M.; Ding, H.; Dooley, S.; et al. THU-375-Transcription factor TRIM33 controls liver progenitor cell towards hepatocyte differentiation through synergizing with SMAD2/3 following massive parenchymal loss. J. Hepatol. 2019, 70, e318–e319. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, L.; Jiao, F.-Z.; Zhang, W.-B.; Chen, Q.; Gong, Z.-J. Histone deacetylase inhibitor suberoylanilide hydroxamic acid alleviates liver fibrosis by suppressing the transforming growth factor-β1 signal pathway. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Tu, A.W.; Luo, K. Acetylation of Smad2 by the co-activator p300 regulates activin and transforming growth factor beta response. J. Biol. Chem. 2007, 282, 21187–21196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, Y.; Itoh, Y.; Abe, K.; Okamoto, T.; Daitoku, H.; Fukamizu, A.; Onozaki, K.; Hayashi, H. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene 2007, 26, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janknecht, R.; Wells, N.J.; Hunter, T. TGF-beta-stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes Dev. 1998, 12, 2114–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tu, K.; Liu, D.; Guo, L.; Chen, Y.; Li, Q.; Maiers, J.L.; Liu, Z.; Shah, V.H.; Dou, C.; et al. p300 acetyltransferase is a cytoplasm-to-nucleus shuttle for SMAD2/3 and TAZ nuclear transport in transforming growth factor β-stimulated hepatic stellate cells. Hepatology 2019, 70, 1409–1423. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Zhou, Y.; Wang, S.; Pang, N.; Huang, Y.; Ye, M.; Wan, T.; Qiu, Y.; Pei, L.; Jiang, X.; et al. Nicotinamide riboside protects against liver fibrosis induced by CCl4 via regulating the acetylation of Smads signaling pathway. Life Sci. 2019, 225, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Merino, E.; Martín Orozco, R.; Ruíz-Llorente, L.; Martínez-Iglesias, O.A.; Velasco-Martín, J.P.; Montero-Pedrazuela, A.; Fanjul-Rodríguez, L.; Contreras-Jurado, C.; Regadera, J.; Aranda, A. Thyroid hormones inhibit TGF-β signaling and attenuate fibrotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E3451–E3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munker, S.; Wu, Y.-L.; Ding, H.-G.; Liebe, R.; Weng, H.-L. Can a fibrotic liver afford epithelial-mesenchymal transition? World J. Gastroenterol. 2017, 23, 4661–4668. [Google Scholar] [CrossRef] [PubMed]
- Taura, K.; Iwaisako, K.; Hatano, E.; Uemoto, S. Controversies over the epithelial-to-mesenchymal transition in liver fibrosis. J. Clin. Med. 2016, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Diehl, A.M. Epithelial-to-mesenchymal transitions in the liver. Hepatology 2009, 50, 2007–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisseleva, T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017, 65, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Kalluri, R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am. J. Physiol. Cell Physiol. 2013, 304, C216–C225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Ding, Z.-Y.; Jin, G.-N.; Xiong, Y.-X.; Yu, B.; Sun, Y.-M.; Wang, W.; Liang, H.-F.; Zhang, B.; Chen, X.-P. Autocrine transforming growth factor-β/activin A-Smad signaling induces hepatic progenitor cells undergoing partial epithelial-mesenchymal transition states. Biochimie 2018, 148, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Meyer, D.S.; Zhang, Z.; Shokat, K.M.; Akhurst, R.J.; Miyazono, K.; Derynck, R. Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal. 2019, 12, eaau8544. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Xie, F.; Zhang, Q.; Cui, Z.; Cheng, X.; Zhong, F.; He, K.; Zhou, J. Advanced oxidation protein products induce hepatocyte epithelial-mesenchymal transition via a ROS-dependent, TGF-β/Smad signaling pathway. Cell Biol. Int. 2017, 41, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Asahina, K.; Zhou, B.; Pu, W.T.; Tsukamoto, H. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology 2011, 53, 983–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Lua, I.; French, S.W.; Asahina, K. Role of TGF-β signaling in differentiation of mesothelial cells to vitamin A-poor hepatic stellate cells in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G262–G272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.H.; Shepherd, E.L.; Lalor, P.F. Could endothelial TGFβ signaling be a promising new target for liver disease? Expert Rev. Gastroenterol. Hepatol. 2018, 12, 637–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribera, J.; Pauta, M.; Melgar-Lesmes, P.; Córdoba, B.; Bosch, A.; Calvo, M.; Rodrigo-Torres, D.; Sancho-Bru, P.; Mira, A.; Jiménez, W.; et al. A small population of liver endothelial cells undergoes endothelial-to-mesenchymal transition in response to chronic liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G492–G504. [Google Scholar] [CrossRef] [PubMed]
- Dufton, N.P.; Peghaire, C.R.; Osuna-Almagro, L.; Raimondi, C.; Kalna, V.; Chuahan, A.; Webb, G.; Yang, Y.; Birdsey, G.M.; Lalor, P.; et al. Dynamic regulation of canonical TGFβ signalling by endothelial transcription factor ERG protects from liver fibrogenesis. Nat. Commun. 2017, 8, 895. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Thermodynamic Aspects and Reprogramming Cellular Energy Metabolism during the Fibrosis Process. Int. J. Mol. Sci. 2017, 18, 2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottmann, R.M.; Kulkarni, A.A.; Smolnycki, K.A.; Lyda, E.; Dahanayake, T.; Salibi, R.; Honnons, S.; Jones, C.; Isern, N.G.; Hu, J.Z.; et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am. J. Respir. Crit. Care Med. 2012, 186, 740–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ghazwani, M.; Liu, K.; Huang, Y.; Chang, N.; Fan, J.; He, F.; Li, L.; Bu, S.; Xie, W.; et al. Regulation of hepatic stellate cell proliferation and activation by glutamine metabolism. PLoS ONE 2017, 12, e0182679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, K.; Logsdon, N.J.; Benavides, G.A.; Sanders, Y.; Zhang, J.; Darley-Usmar, V.M.; Thannickal, V.J. Glutaminolysis is required for transforming growth factor-β1-induced myofibroblast differentiation and activation. J. Biol. Chem. 2018, 293, 1218–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Alves, C.R.R.; Stanford, K.I.; Middelbeek, R.J.W.; Nigro, P.; Ryan, R.E.; Xue, R.; Sakaguchi, M.; Lynes, M.D.; So, K.; et al. TGF-β2 is an exercise-induced adipokine that regulates glucose and fatty acid metabolism. Nat. Metab. 2019, 1, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Starling, S. A new metabolic role for TGFβ2. Nat. Rev. Endocrinol. 2019, 15, 191. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Wang, G.Z.; Guo, D.D.; Bai, R.X.; Wang, M.; Du, S.Y. Deletion of Smad4 reduces hepatic inflammation and fibrogenesis during nonalcoholic steatohepatitis progression. J. Dig. Dis. 2018, 19, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.H.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 2006, 15, R271–R277. [Google Scholar] [CrossRef] [PubMed]
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998, 280, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Kakan, X.; Wang, S.; Dong, W.; Jia, A.; Cai, C.; Zhang, J. Deletion of clock gene Per2 exacerbates cholestatic liver injury and fibrosis in mice. Exp. Toxicol. Pathol. 2013, 65, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Han, Z.; Yang, P.; Zhu, L.; Hua, Z.; Zhang, J. Loss of clock gene mPer2 promotes liver fibrosis induced by carbon tetrachloride. Hepatol. Res. 2010, 40, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Janich, P.; Pascual, G.; Merlos-Suárez, A.; Batlle, E.; Ripperger, J.; Albrecht, U.; Cheng, H.-Y.M.; Obrietan, K.; Di Croce, L.; Benitah, S.A. The circadian molecular clock creates epidermal stem cell heterogeneity. Nature 2011, 480, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gast, H.; Gordic, S.; Petrzilka, S.; Lopez, M.; Müller, A.; Gietl, A.; Hock, C.; Birchler, T.; Fontana, A. Transforming growth factor-beta inhibits the expression of clock genes. Ann. N. Y. Acad. Sci. 2012, 1261, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Gongora, R.; Sosulski, M.L.; Luo, F.; Sanchez, C.G. Regulation of transforming growth factor-beta1 (TGF-β1)-induced pro-fibrotic activities by circadian clock gene BMAL1. Respir. Res. 2016, 17, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, J.; Ince, L.; Matthews, L.; Mei, J.; Bell, T.; Yang, N.; Saer, B.; Begley, N.; Poolman, T.; Pariollaud, M.; et al. An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat. Med. 2014, 20, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.-W.; Sundar, I.K.; Yao, H.; Sellix, M.T.; Rahman, I. Circadian clock function is disrupted by environmental tobacco/cigarette smoke, leading to lung inflammation and injury via a SIRT1-BMAL1 pathway. FASEB J. 2014, 28, 176–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Eheim, A.L.; Klein, S.; Uschner, F.E.; Smith, A.C.; Brandon-Warner, E.; Ghosh, S.; Bonkovsky, H.L.; Trebicka, J.; Schrum, L.W. Novel role of nuclear receptor Rev-erbα in hepatic stellate cell activation: Potential therapeutic target for liver injury. Hepatology 2014, 59, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Kojetin, D.J.; Burris, T.P. REV-ERB and ROR nuclear receptors as drug targets. Nat. Rev. Drug Discov. 2014, 13, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomes, P.G.; Brandon-Warner, E.; Li, T.; Donohue, T.M.; Schrum, L.W. Rev-erb agonist and TGF-β similarly affect autophagy but differentially regulate hepatic stellate cell fibrogenic phenotype. Int. J. Biochem. Cell Biol. 2016, 81, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.; Fallowfield, J.; Iredale, J.P. Senescence of activated stellate cells: Not just early retirement. Hepatology 2009, 49, 1045–1047. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiemann, S.U.; Satyanarayana, A.; Tsahuridu, M.; Tillmann, H.L.; Zender, L.; Klempnauer, J.; Flemming, P.; Franco, S.; Blasco, M.A.; Manns, M.P.; et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J. 2002, 16, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Razdan, N.; Vasilopoulos, T.; Herbig, U. Telomere dysfunction promotes transdifferentiation of human fibroblasts into myofibroblasts. Aging Cell 2018, 17, e12838. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.G.; Müller, M.; Boulter, L.; Vincent, D.F.; Ridgway, R.A.; Lopez-Guadamillas, E.; Lu, W.-Y.; Jamieson, T.; Govaere, O.; Campbell, A.D.; et al. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl. Med. 2018, 10, eaan1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira-Gonzalez, S.; Lu, W.-Y.; Raven, A.; Dwyer, B.; Man, T.Y.; O’Duibhir, E.; Lewis, P.J.S.; Campana, L.; Kendall, T.J.; Bird, T.G.; et al. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat. Commun. 2018, 9, 1020. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Aguilar Schall, R.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, Y.; Li, X.; Yan, L.; Guan, H.; Han, R.; Han, Y.; Gui, J.; Xu, X.; Dong, Y.; et al. Expression, purification, and evaluation of in vivo anti-fibrotic activity for soluble truncated TGF-β receptor II as a cleavable His-SUMO fusion protein. World J. Microbiol. Biotechnol. 2018, 34, 181. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Mancarella, S.; Cigliano, A.; Chieti, A.; Giannelli, G. TGF-β as Multifaceted Orchestrator in HCC Progression: Signaling, EMT, Immune Microenvironment, and Novel Therapeutic Perspectives. Semin. Liver Dis. 2019, 39, 53–69. [Google Scholar] [PubMed]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luangmonkong, T.; Suriguga, S.; Bigaeva, E.; Boersema, M.; Oosterhuis, D.; de Jong, K.P.; Schuppan, D.; Mutsaers, H.A.M.; Olinga, P. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br. J. Pharm. 2017, 174, 3107–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammad, S.; Cavalcanti, E.; Werle, J.; Caruso, M.L.; Dropmann, A.; Ignazzi, A.; Ebert, M.P.; Dooley, S.; Giannelli, G. Galunisertib modifies the liver fibrotic composition in the Abcb4Ko mouse model. Arch. Toxicol. 2018, 92, 2297–2309. [Google Scholar] [CrossRef] [PubMed]
- Ikenaga, N.; Peng, Z.-W.; Vaid, K.A.; Liu, S.B.; Yoshida, S.; Sverdlov, D.Y.; Mikels-Vigdal, A.; Smith, V.; Schuppan, D.; Popov, Y.V. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 2017, 66, 1697–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Son, M.K.; Jung, K.H.; Kim, K.; Kim, G.J.; Lee, S.-H.; Hong, S.-S.; Park, S.G. Aminoacyl-tRNA synthetase interacting multi-functional protein 1 attenuates liver fibrosis by inhibiting TGFβ signaling. Int. J. Oncol. 2016, 48, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ju, B.; Zhang, X.; Zhu, Y.; Nie, Y.; Xu, Y.; Lei, Q. Magnolol attenuates concanavalin a-induced hepatic fibrosis, inhibits cd4+ t helper 17 (Th17) cell differentiation and suppresses hepatic stellate cell activation: Blockade of smad3/smad4 signalling. Basic Clin. Pharm. Toxicol. 2017, 120, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wang, H.; Wu, Z.; Zhong, X.; Zhu, M.; Zhang, Y.; Tan, R.; Liu, Y.; Li, J.; Wang, L. Specific Inhibitor of smad3 (SIS3) attenuates fibrosis, apoptosis, and inflammation in unilateral ureteral obstruction kidneys by inhibition of transforming growth factor β (TGF-β)/Smad3 signaling. Med. Sci. Monit. Int. 2018, 24, 1633–1641. [Google Scholar] [CrossRef] [PubMed]
- Ganai, A.A.; Husain, M. Genistein attenuates D-GalN induced liver fibrosis/chronic liver damage in rats by blocking the TGF-β/Smad signaling pathways. Chem. Biol. Interact. 2017, 261, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kong, D.; Qiu, J.; Xie, Y.; Lu, Z.; Zhou, C.; Liu, X.; Zhang, R.; Wang, Y. Praziquantel Ameliorates ccl4 -induced liver fibrosis in mice by inhibiting TGF-β/smad signalling via upregulating smad7 in hepatic stellate cells. Br. J. Pharm. 2019. [Google Scholar] [CrossRef] [PubMed]
- Delire, B.; Stärkel, P.; Leclercq, I. Animal models for fibrotic liver diseases: What we have, what we need, and what is under development. J. Clin. Transl. Hepatol. 2015, 3, 53–66. [Google Scholar] [PubMed]
- Hayashi, H.; Sakai, T. Animal models for the study of liver fibrosis: New insights from knockout mouse models. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G729–G738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, M.; Matsuura, T.; Kojima, S. TGF-β LAP Degradation Products, a Novel Biomarker and Promising Therapeutic Target for Liver Fibrogenesis. In Innovative Medicine: Basic Research and Development; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015. [Google Scholar]
- Qiao, X.; Rao, P.; Zhang, Y.; Liu, L.; Pang, M.; Wang, H.; Hu, M.; Tian, X.; Zhang, J.; Zhao, Y.; et al. Redirecting TGF-β signaling through the β-catenin/foxo complex prevents kidney fibrosis. J. Am. Soc. Nephrol. 2018, 29, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Akcora, B.Ö.; Storm, G.; Bansal, R. Inhibition of canonical WNT signaling pathway by β-catenin/CBP inhibitor ICG-001 ameliorates liver fibrosis in vivo through suppression of stromal CXCL12. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Ikoma, A.; Shibakawa, M.; Shimoda, S.; Harada, K.; Saio, M.; Imamura, J.; Osawa, Y.; Kimura, M.; Nishikawa, K.; et al. Safety, tolerability, and preliminary efficacy of the anti-fibrotic small molecule pri-724, a cbp/β-catenin inhibitor, in patients with hepatitis c virus-related cirrhosis: A single-center, open-label, dose escalation phase 1 trial. EBioMedicine 2017, 23, 79–87. [Google Scholar] [CrossRef] [PubMed]
- El-Wakeel, S.A.; Rahmo, R.M.; El-Abhar, H.S. Anti-fibrotic impact of Carvedilol in a CCl-4 model of liver fibrosis via serum microRNA-200a/SMAD7 enhancement to bridle TGF-β1/EMT track. Sci. Rep. 2018, 8, 14327. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.; Park, S.Y.; Le, C.T.; Park, W.S.; Choi, D.H.; Cho, E.-H. Metformin ameliorates activation of hepatic stellate cells and hepatic fibrosis by succinate and GPR91 inhibition. Biochem. Biophys. Res. Commun. 2018, 495, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Wu, K.; Lin, L.; Ge, P.; Dai, J.; He, X.; Hu, K.; Zhang, L. Metformin mitigates carbon tetrachloride-induced TGF-β1/Smad3 signaling and liver fibrosis in mice. Biomed. Pharm. 2017, 90, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massagué, J. Transforming growth factor-β signaling in immunity and cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, a.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells 2019, 8, 1419. https://doi.org/10.3390/cells8111419
Dewidar B, Meyer C, Dooley S, Meindl-Beinker aN. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells. 2019; 8(11):1419. https://doi.org/10.3390/cells8111419
Chicago/Turabian StyleDewidar, Bedair, Christoph Meyer, Steven Dooley, and and Nadja Meindl-Beinker. 2019. "TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019" Cells 8, no. 11: 1419. https://doi.org/10.3390/cells8111419
APA StyleDewidar, B., Meyer, C., Dooley, S., & Meindl-Beinker, a. N. (2019). TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells, 8(11), 1419. https://doi.org/10.3390/cells8111419