JNK-Dependent cJun Phosphorylation Mitigates TGFβ- and EGF-Induced Pre-Malignant Breast Cancer Cell Invasion by Suppressing AP-1-Mediated Transcriptional Responses

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents and Antibodies
2.3. 3D Spheroid Invasion Assays
2.4. In Vitro Wound-Healing Assay
2.5. Immunoblot Analysis
2.6. RNA Isolation, cDNA Synthesis, and Quantitative Real-Time-PCR
2.7. DNA Transfer and Constructs
2.8. Statistical Analysis
3. Results
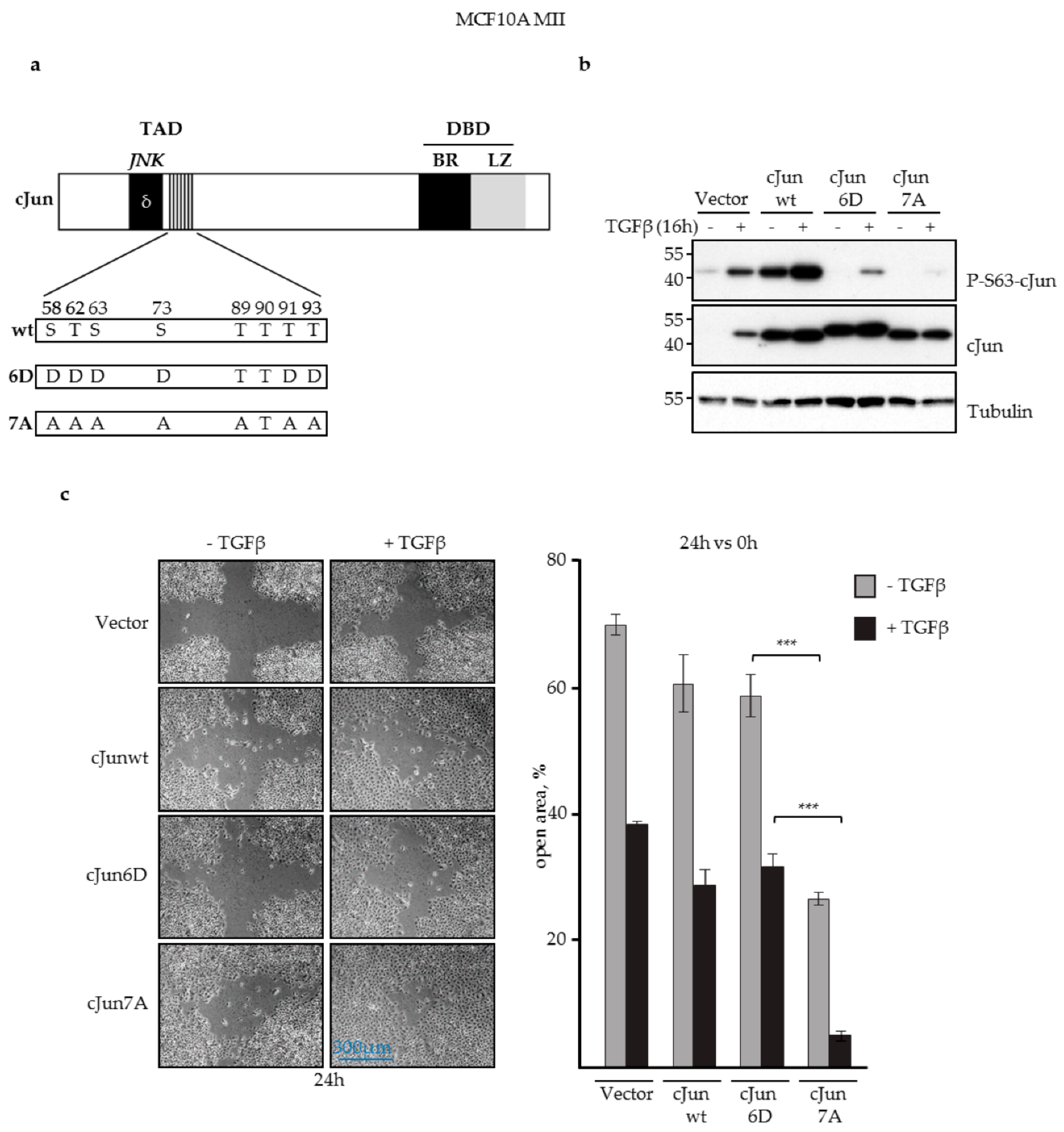
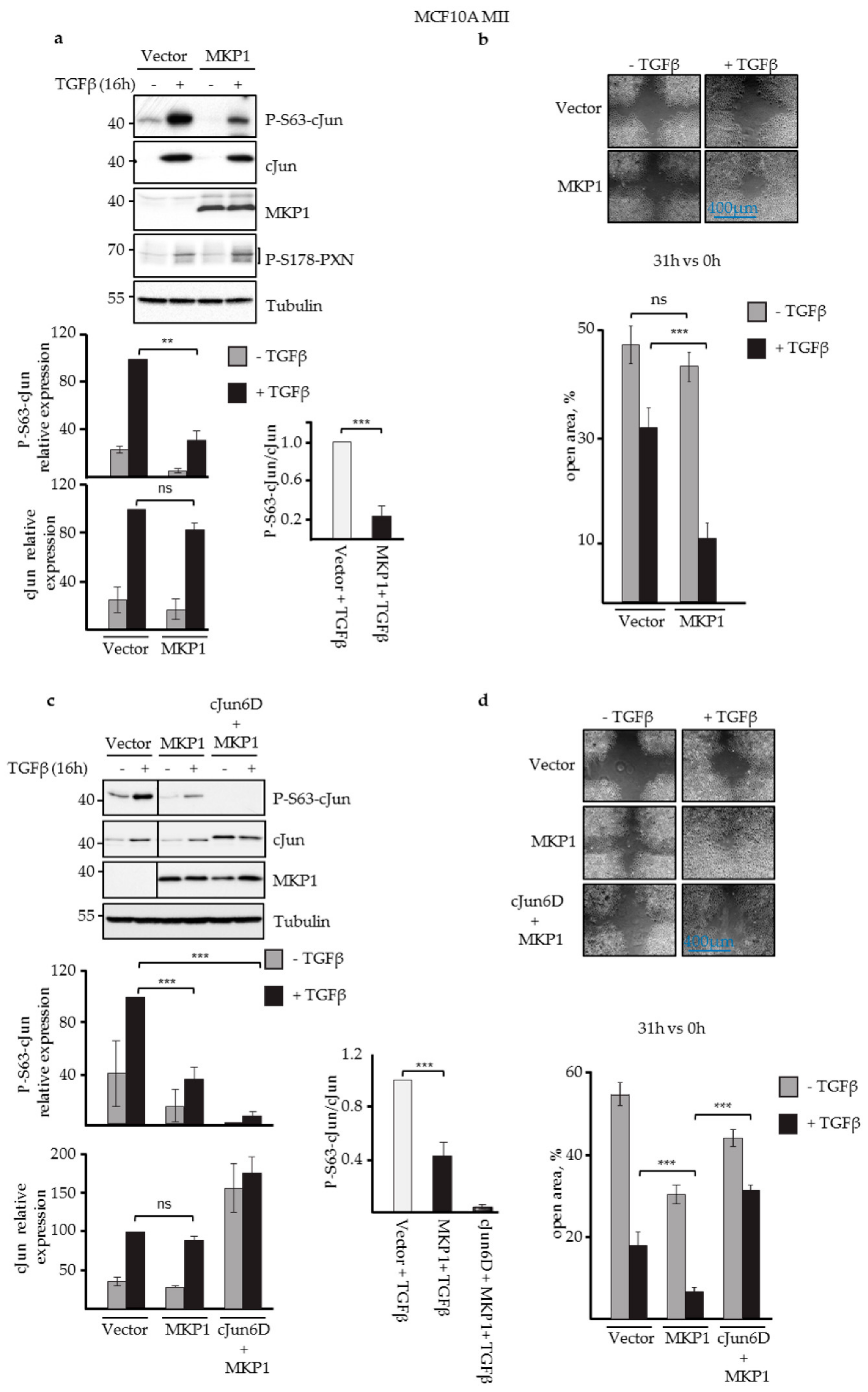
3.1. JNK-Dependent Phosphorylation of cJun Negatively Affected MCF10A MII Cell Migration
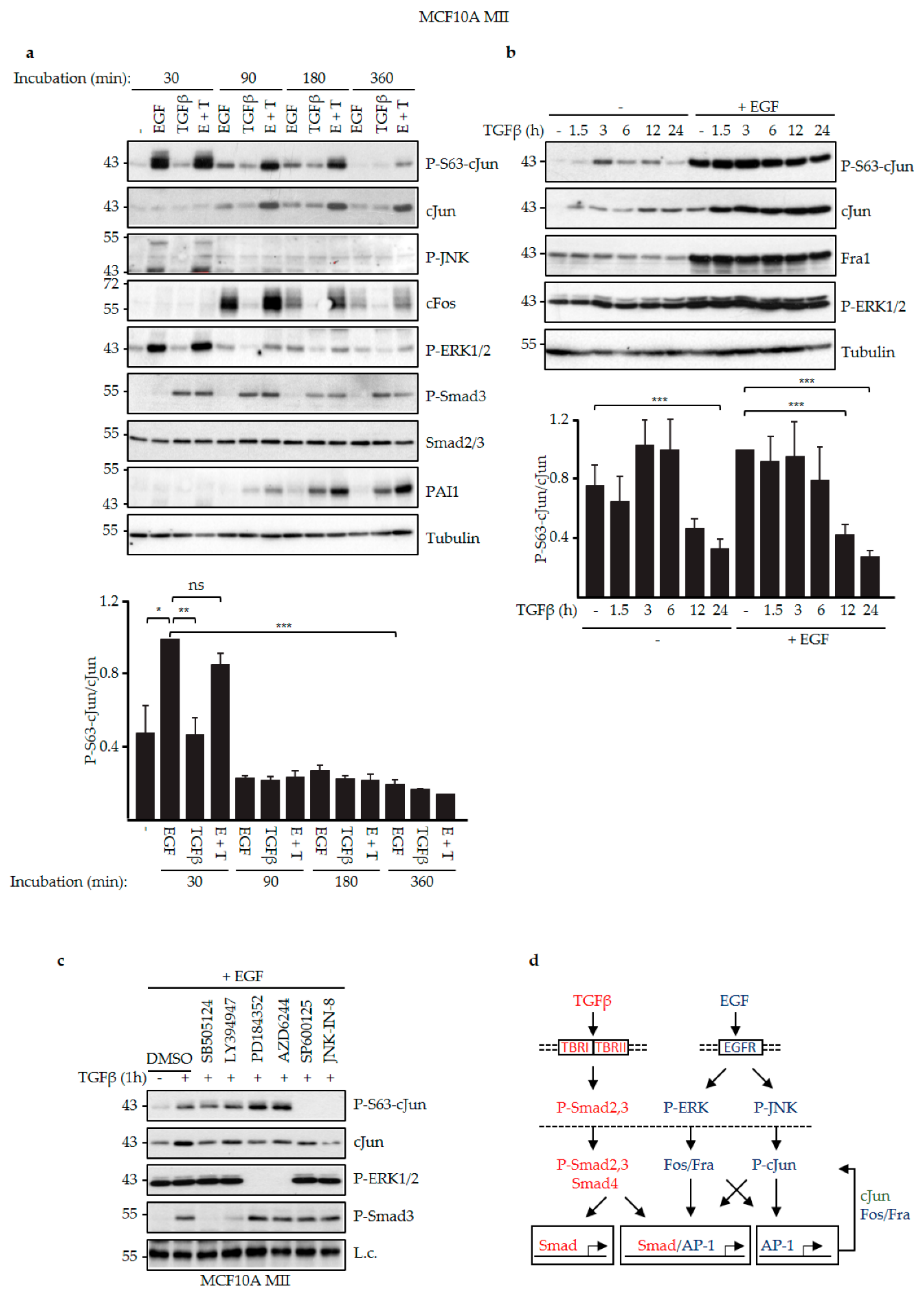
3.2. EGF and TGFβ Signaling Had Different Effects on JNK-Dependent cJun Phosphorylation
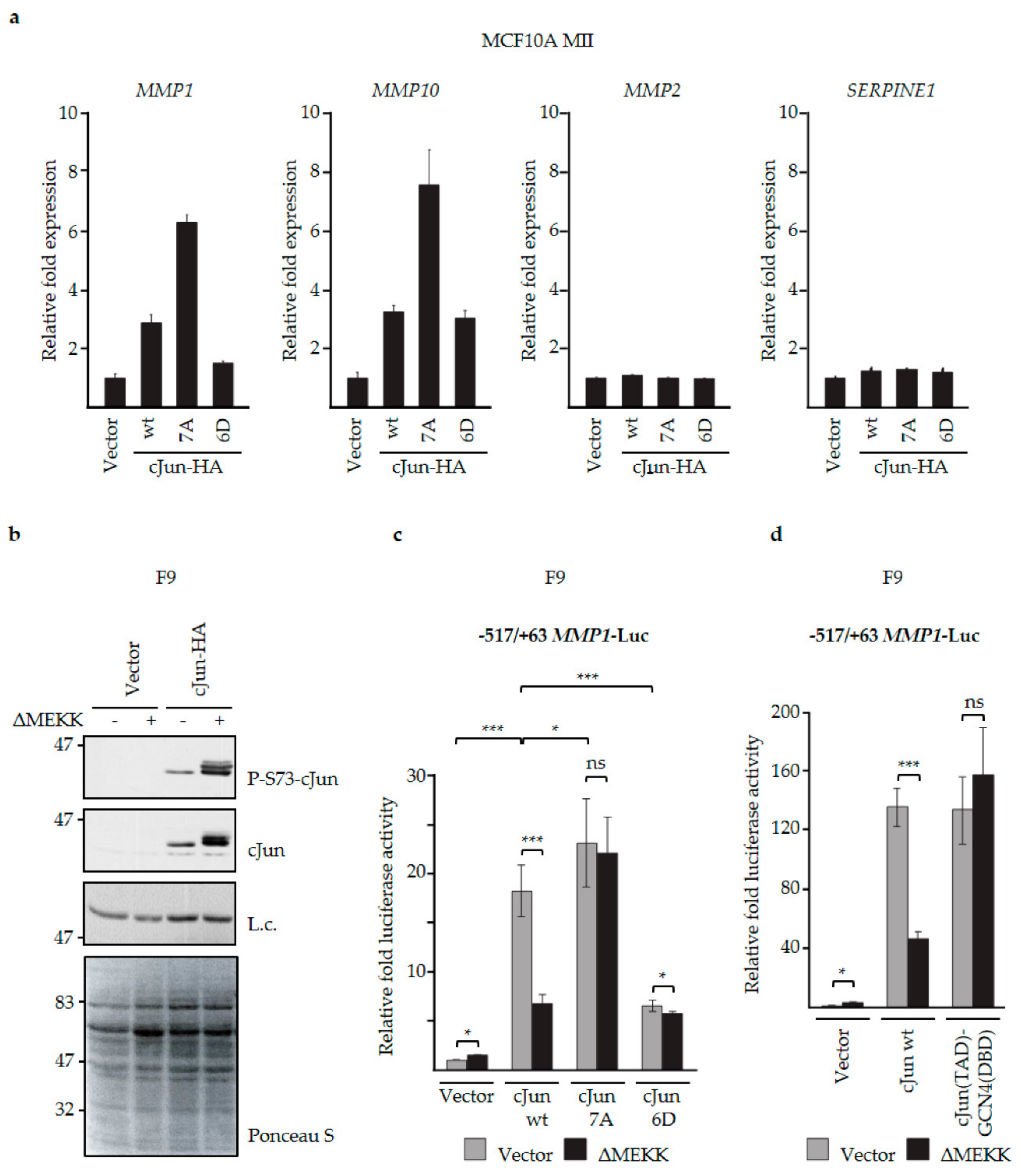
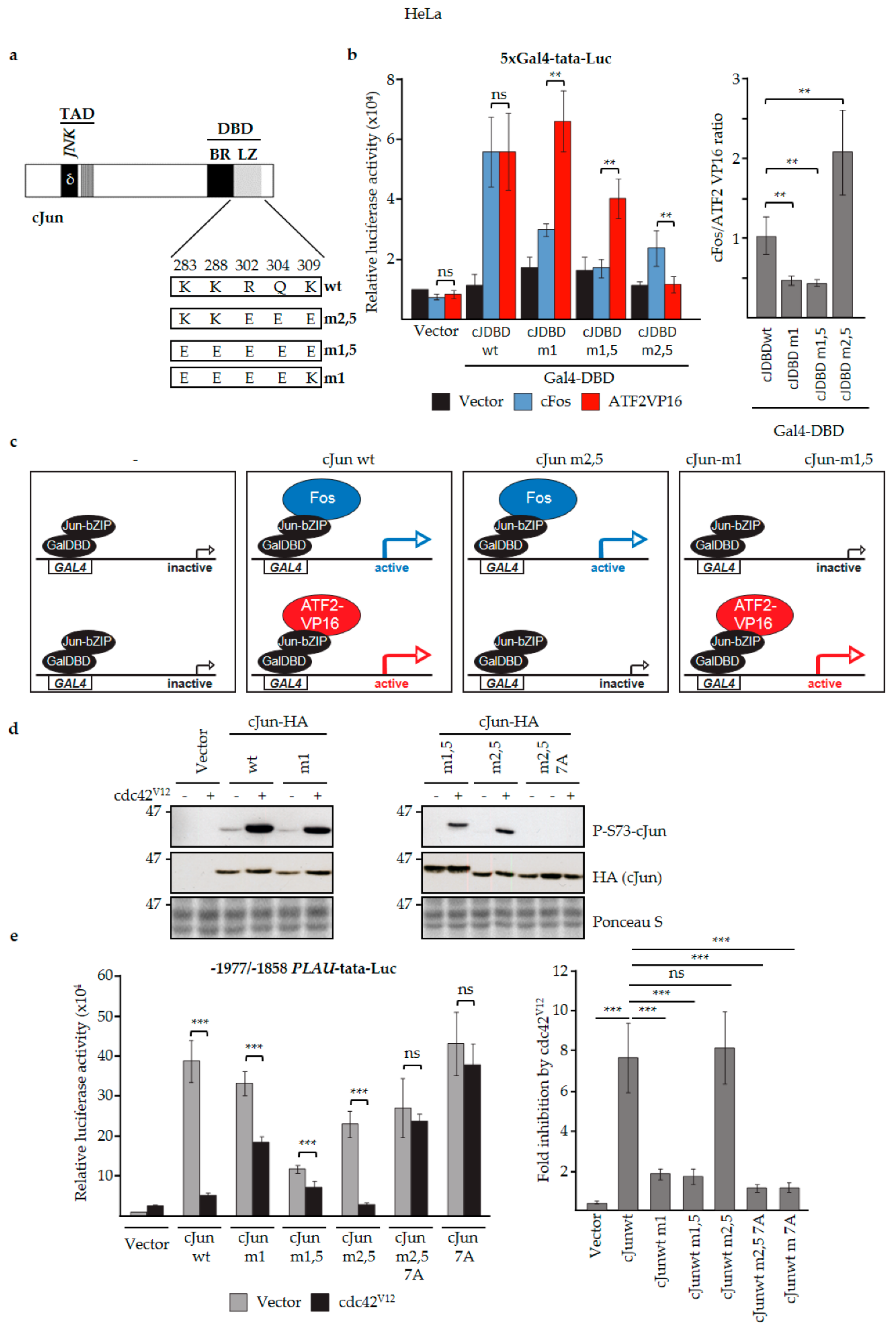
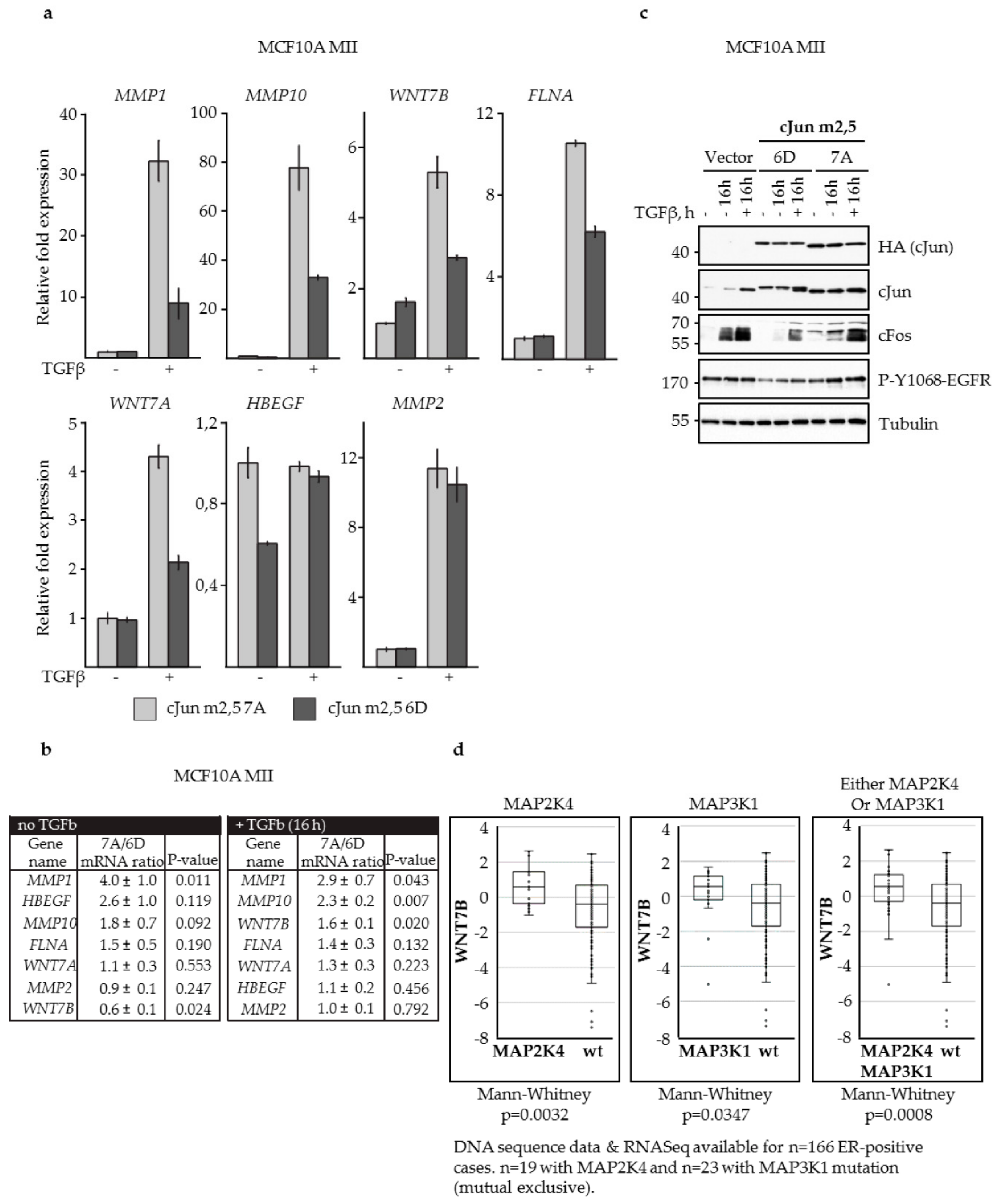
3.3. N-Terminal Phosphorylation of cJun Negatively Affected the Activation of MMP1 and MMP10
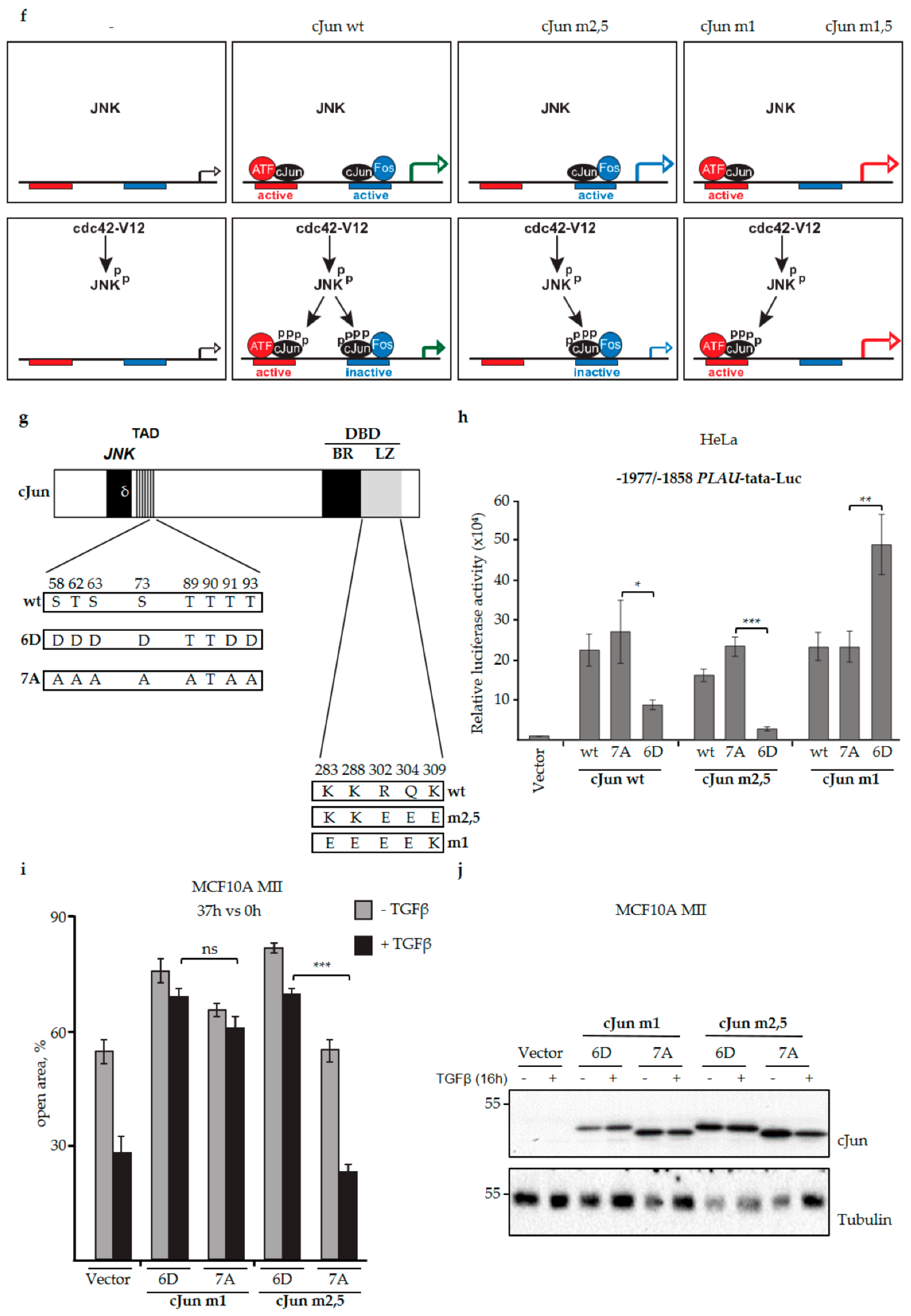
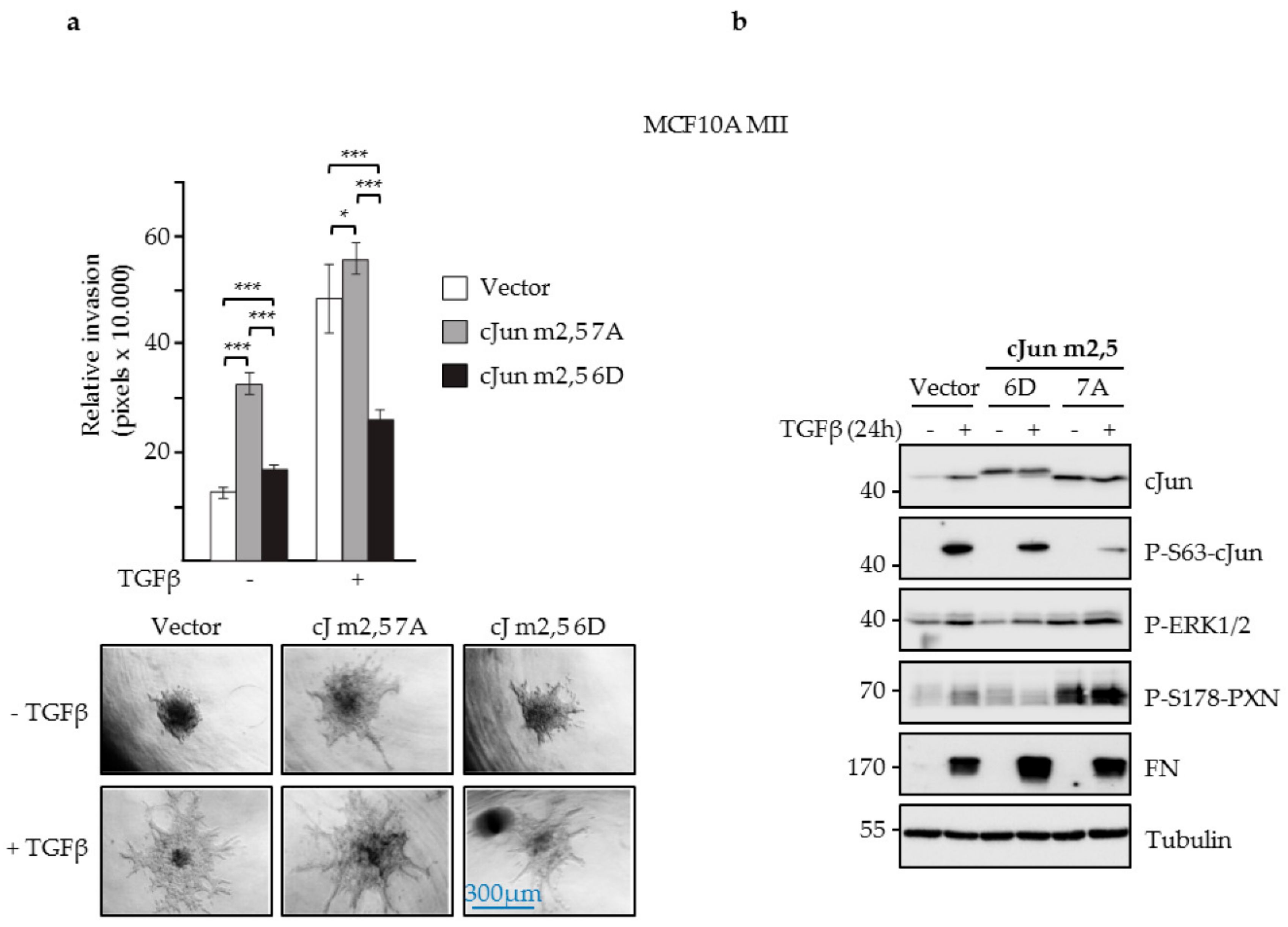
3.4. JNK-Dependent Phosphorylation of cJun Specifically Inhibited Gene Activation and Migration by Jun/Fos Dimers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- David, C.J.; Massague, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell. Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Padgett, R.W. Matters of context guide future research in TGFβ superfamily signaling. Sci. Signal. 2015, 8, re10. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, X.H.; Derynck, R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature 1998, 394, 909–913. [Google Scholar] [CrossRef]
- Koinuma, D.; Tsutsumi, S.; Kamimura, N.; Taniguchi, H.; Miyazawa, K.; Sunamura, M.; Imamura, T.; Miyazono, K.; Aburatani, H. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor β signaling. Mol. Cell. Biol. 2009, 29, 172–186. [Google Scholar] [CrossRef]
- Ikushima, H.; Miyazono, K. TGF-β signal transduction spreading to a wider field: A broad variety of mechanisms for context-dependent effects of TGF-β. Cell Tissue Res. 2012, 347, 37–49. [Google Scholar] [CrossRef]
- Massague, J. TGF-β signaling in context. Nat. Rev. Mol. Cell. Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef]
- Zhang, Y.E. Mechanistic insight into contextual TGF-β signaling. Curr. Opin. Cell Biol. 2018, 51, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, A.; Ten Dijke, P.; van Dam, H. Key signaling nodes in mammary gland development and cancer: Smad signal integration in epithelial cell plasticity. Breast Cancer Res. 2012, 14, 204. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Bitzer, M.; Liang, D.; Yang, Y.C.; Massimi, A.; Kneitz, S.; Piek, E.; Bottinger, E.P. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc. Natl. Acad. Sci. USA 2001, 98, 6686–6691. [Google Scholar] [CrossRef] [PubMed]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef]
- Thuault, S.; Tan, E.J.; Peinado, H.; Cano, A.; Heldin, C.H.; Moustakas, A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 2008, 283, 33437–33446. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- ten Dijke, P.; Arthur, H.M. Extracellular control of TGFβ signalling in vascular development and disease. Nat. Rev. Mol. Cell. Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef]
- Drabsch, Y.; ten Dijke, P. TGF-β signaling in breast cancer cell invasion and bone metastasis. J. Mammary Gland. Biol. Neoplasia 2011, 16, 97–108. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-β signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef] [PubMed]
- Ozanne, B.W.; Spence, H.J.; McGarry, L.C.; Hennigan, R.F. Transcription factors control invasion: AP-1 the first among equals. Oncogene 2007, 26, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E. AP-1—The Jun proteins: Oncogenes or tumor suppressors in disguise? Cell. Signal. 2010, 22, 894–899. [Google Scholar] [CrossRef]
- Lopez-Bergami, P.; Lau, E.; Ronai, Z. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat. Rev. Cancer 2010, 10, 65–76. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ling, E.; Cowley, C.J.; Couch, C.H.; Wang, X.; Harmin, D.A.; Roberts, C.W.M.; Greenberg, M.E. AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol. Cell 2017, 68, 1067–1082. [Google Scholar] [CrossRef]
- Madrigal, P.; Alasoo, K. AP-1 Takes Centre Stage in Enhancer Chromatin Dynamics. Trends Cell Biol. 2018, 28, 509–511. [Google Scholar] [CrossRef]
- Belguise, K.; Kersual, N.; Galtier, F.; Chalbos, D. FRA-1 expression level regulates proliferation and invasiveness of breast cancer cells. Oncogene 2005, 24, 1434–1444. [Google Scholar] [CrossRef]
- Trop-Steinberg, S.; Azar, Y. AP-1 Expression and its Clinical Relevance in Immune Disorders and Cancer. Am. J. Med. Sci 2017, 353, 474–483. [Google Scholar] [CrossRef]
- Desmet, C.J.; Gallenne, T.; Prieur, A.; Reyal, F.; Visser, N.L.; Wittner, B.S.; Smit, M.A.; Geiger, T.R.; Laoukili, J.; Iskit, S.; et al. Identification of a pharmacologically tractable Fra-1/ADORA2B axis promoting breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 5139–5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein kinase C alpha is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 2013, 24, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Tulchinsky, E. FRA-1 as a driver of tumour heterogeneity: A nexus between oncogenes and embryonic signalling pathways in cancer. Oncogene 2015, 34, 4421–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakiri, L.; Macho-Maschler, S.; Custic, I.; Niemiec, J.; Guio-Carrion, A.; Hasenfuss, S.C.; Eger, A.; Muller, M.; Beug, H.; Wagner, E.F. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFβ expression. Cell Death Differ. 2015, 22, 336–350. [Google Scholar] [CrossRef] [Green Version]
- Hefetz-Sela, S.; Stein, I.; Klieger, Y.; Porat, R.; Sade-Feldman, M.; Zreik, F.; Nagler, A.; Pappo, O.; Quagliata, L.; Dazert, E.; et al. Acquisition of an immunosuppressive protumorigenic macrophage phenotype depending on c-Jun phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, 17582–17587. [Google Scholar] [CrossRef] [Green Version]
- Trierweiler, C.; Hockenjos, B.; Zatloukal, K.; Thimme, R.; Blum, H.E.; Wagner, E.F.; Hasselblatt, P. The transcription factor c-JUN/AP-1 promotes HBV-related liver tumorigenesis in mice. Cell Death Differ. 2016, 23, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Sundqvist, A.; Zieba, A.; Vasilaki, E.; Herrera Hidalgo, C.; Soderberg, O.; Koinuma, D.; Miyazono, K.; Heldin, C.H.; Landegren, U.; Ten Dijke, P.; et al. Specific interactions between Smad proteins and AP-1 components determine TGFβ-induced breast cancer cell invasion. Oncogene 2013, 32, 3606–3615. [Google Scholar] [CrossRef] [Green Version]
- Sundqvist, A.; Morikawa, M.; Ren, J.; Vasilaki, E.; Kawasaki, N.; Kobayashi, M.; Koinuma, D.; Aburatani, H.; Miyazono, K.; Heldin, C.H.; et al. JUNB governs a feed-forward network of TGFβ signaling that aggravates breast cancer invasion. Nucleic Acids Res. 2018, 46, 1180–1195. [Google Scholar] [CrossRef] [Green Version]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D., Jr.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar]
- Dawson, P.J.; Wolman, S.R.; Tait, L.; Heppner, G.H.; Miller, F.R. MCF10AT: A model for the evolution of cancer from proliferative breast disease. Am. J. Pathol. 1996, 148, 313–319. [Google Scholar]
- Sundqvist, A.; Vasilaki, E.; Voytyuk, O.; Morikawa, M.; Moustakas, A.; Miyazono, K.; Heldin, C.-H.; ten Dijke, P.; van Dam, H. TGFβ-SMAD and EGF-MEK synergize to induce AP-1- and p63-dependent genes enhancing invasion of breast cancer cells. Submitted.
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Ding, L.; Shen, D.; Luo, J.; Suman, V.J.; Wallis, J.W.; Van Tine, B.A.; Hoog, J.; Goiffon, R.J.; Goldstein, T.C.; et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012, 486, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Network, C.G.A.; Network, T.C.G.A.; Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Z.; Vis, D.J.; Bruna, A.; Sustic, T.; van Wageningen, S.; Batra, A.S.; Rueda, O.M.; Bosdriesz, E.; Caldas, C.; Wessels, L.F.A.; et al. MAP3K1 and MAP2K4 mutations are associated with sensitivity to MEK inhibitors in multiple cancer models. Cell Res. 2018, 28, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Cellurale, C.; Girnius, N.; Jiang, F.; Cavanagh-Kyros, J.; Lu, S.; Garlick, D.S.; Mercurio, A.M.; Davis, R.J. Role of JNK in mammary gland development and breast cancer. Cancer Res. 2012, 72, 472–481. [Google Scholar] [CrossRef] [Green Version]
- Angel, P.; Hattori, K.; Smeal, T.; Karin, M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell 1988, 55, 875–885. [Google Scholar] [CrossRef]
- Radler-Pohl, A.; Sachsenmaier, C.; Gebel, S.; Auer, H.P.; Bruder, J.T.; Rapp, U.; Angel, P.; Rahmsdorf, H.J.; Herrlich, P. UV-induced activation of AP-1 involves obligatory extranuclear steps including Raf-1 kinase. EMBO J. 1993, 12, 1005–1012. [Google Scholar] [CrossRef]
- Van Dam, H.; Duyndam, M.; Rottier, R.; Bosch, A.; de Vries-Smits, L.; Herrlich, P.; Zantema, A.; Angel, P.; van der Eb, A.J. Heterodimer formation of cJun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus E1A protein. EMBO J. 1993, 12, 479–487. [Google Scholar] [CrossRef]
- Van Dam, H.; Wilhelm, D.; Herr, I.; Steffen, A.; Herrlich, P.; Angel, P. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 1995, 14, 1798–1811. [Google Scholar] [CrossRef]
- Wiercinska, E.; Naber, H.P.; Pardali, E.; van der Pluijm, G.; van Dam, H.; ten Dijke, P. The TGF-β/Smad pathway induces breast cancer cell invasion through the up-regulation of matrix metalloproteinase 2 and 9 in a spheroid invasion model system. Breast Cancer Res. Treat. 2011, 128, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdi, M.; Kool, J.; Cornelissen-Steijger, P.; Carlotti, F.; Popeijus, H.E.; van der Burgt, C.; Janssen, J.M.; Yasui, A.; Hoeben, R.C.; Terleth, C.; et al. DNA damage in transcribed genes induces apoptosis via the JNK pathway and the JNK-phosphatase MKP-1. Oncogene 2005, 24, 7135–7144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duyndam, M.C.; van Dam, H.; Smits, P.H.; Verlaan, M.; van der Eb, A.J.; Zantema, A. The N-terminal transactivation domain of ATF2 is a target for the co-operative activation of the c-jun promoter by p300 and 12S E1A. Oncogene 1999, 18, 2311–2321. [Google Scholar] [CrossRef] [PubMed]
- De Cesare, D.; Vallone, D.; Caracciolo, A.; Sassone-Corsi, P.; Nerlov, C.; Verde, P. Heterodimerization of c-Jun with ATF-2 and c-Fos is required for positive and negative regulation of the human urokinase enhancer. Oncogene 1995, 11, 365–376. [Google Scholar]
- Musti, A.M.; Treier, M.; Bohmann, D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science 1997, 275, 400–402. [Google Scholar] [CrossRef]
- Papavassiliou, A.G.; Treier, M.; Bohmann, D. Intramolecular signal transduction in c-Jun. EMBO J. 1995, 14, 2014–2019. [Google Scholar] [CrossRef]
- Treier, M.; Bohmann, D.; Mlodzik, M. JUN cooperates with the ETS domain protein pointed to induce photoreceptor R7 fate in the Drosophila eye. Cell 1995, 83, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Oliviero, S.; Robinson, G.S.; Struhl, K.; Spiegelman, B.M. Yeast GCN4 as a probe for oncogenesis by AP-1 transcription factors: Transcriptional activation through AP-1 sites is not sufficient for cellular transformation. Genes Dev. 1992, 6, 1799–1809. [Google Scholar] [CrossRef] [Green Version]
- van Dam, H.; Huguier, S.; Kooistra, K.; Baguet, J.; Vial, E.; van der Eb, A.J.; Herrlich, P.; Angel, P.; Castellazzi, M. Autocrine growth and anchorage independence: Two complementing Jun-controlled genetic programs of cellular transformation. Genes Dev. 1998, 12, 1227–1239. [Google Scholar] [CrossRef] [Green Version]
- Carlotti, F.; Bazuine, M.; Kekarainen, T.; Seppen, J.; Pognonec, P.; Maassen, J.A.; Hoeben, R.C. Lentiviral vectors efficiently transduce quiescent mature 3T3-L1 adipocytes. Mol. Ther. 2004, 9, 209–217. [Google Scholar] [CrossRef]
- Watson, A.; Eilers, A.; Lallemand, D.; Kyriakis, J.; Rubin, L.L.; Ham, J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J. Neurosci. 1998, 18, 751–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slack, D.N.; Seternes, O.M.; Gabrielsen, M.; Keyse, S.M. Distinct binding determinants for ERK2/p38alpha and JNK map kinases mediate catalytic activation and substrate selectivity of map kinase phosphatase-1. J. Biol. Chem. 2001, 276, 16491–16500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeke, A.; Misheva, M.; Remenyi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Smeal, T.; Binetruy, B.; Mercola, D.A.; Birrer, M.; Karin, M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature 1991, 354, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Pulverer, B.J.; Kyriakis, J.M.; Avruch, J.; Nikolakaki, E.; Woodgett, J.R. Phosphorylation of c-jun mediated by MAP kinases. Nature 1991, 353, 670–674. [Google Scholar] [CrossRef]
- Binetruy, B.; Smeal, T.; Karin, M. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature 1991, 351, 122–127. [Google Scholar] [CrossRef]
- Angel, P.; Allegretto, E.A.; Okino, S.T.; Hattori, K.; Boyle, W.J.; Hunter, T.; Karin, M. Oncogene jun encodes a sequence-specific trans-activator similar to AP-1. Nature 1988, 332, 166–171. [Google Scholar] [CrossRef]
- Behrens, A.; Sibilia, M.; Wagner, E.F. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 1999, 21, 326–329. [Google Scholar] [CrossRef]
- Nateri, A.S.; Spencer-Dene, B.; Behrens, A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 2005, 437, 281–285. [Google Scholar] [CrossRef]
- Weiss, C.; Schneider, S.; Wagner, E.F.; Zhang, X.; Seto, E.; Bohmann, D. JNK phosphorylation relieves HDAC3-dependent suppression of the transcriptional activity of c-Jun. EMBO J. 2003, 22, 3686–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huguier, S.; Baguet, J.; Perez, S.; van Dam, H.; Castellazzi, M. Transcription factor ATF2 cooperates with v-Jun to promote growth factor-independent proliferation in vitro and tumor formation in vivo. Mol. Cell. Biol. 1998, 18, 7020–7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, E.K.; Rutkowski, R.; Kim, P.S. Mechanism of specificity in the Fos-Jun oncoprotein heterodimer. Cell 1992, 68, 699–708. [Google Scholar] [CrossRef]
- Vasilaki, E.; Morikawa, M.; Koinuma, D.; Mizutani, A.; Hirano, Y.; Ehata, S.; Sundqvist, A.; Kawasaki, N.; Cedervall, J.; Olsson, A.K.; et al. Ras and TGF-β signaling enhance cancer progression by promoting the DeltaNp63 transcriptional program. Sci. Signal. 2016, 9, ra84. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J. Targeting TGF-β Signaling for Therapeutic Gain. Cold Spring Harb. Perspect. Biol. 2017, 9, a022301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Wong, K.K. Recent developments in anti-cancer agents targeting the Ras/Raf/MEK/ERK pathway. Recent Pat. Anti-Cancer Drug Discov. 2009, 4, 28–35. [Google Scholar] [CrossRef]
- Chappell, W.H.; Steelman, L.S.; Long, J.M.; Kempf, R.C.; Abrams, S.L.; Franklin, R.A.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: Rationale and importance to inhibiting these pathways in human health. Oncotarget 2011, 2, 135–164. [Google Scholar] [CrossRef]
- Das, M.; Garlick, D.S.; Greiner, D.L.; Davis, R.J. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011, 25, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Gozdecka, M.; Lyons, S.; Kondo, S.; Taylor, J.; Li, Y.; Walczynski, J.; Thiel, G.; Breitwieser, W.; Jones, N. JNK suppresses tumor formation via a gene-expression program mediated by ATF2. Cell Rep. 2014, 9, 1361–1374. [Google Scholar] [CrossRef]
- Abell, A.N.; Granger, D.A.; Johnson, N.L.; Vincent-Jordan, N.; Dibble, C.F.; Johnson, G.L. Trophoblast stem cell maintenance by fibroblast growth factor 4 requires MEKK4 activation of Jun N-terminal kinase. Mol. Cell. Biol. 2009, 29, 2748–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Diefenbacher, M.; Sekula, S.; Heilbock, C.; Maier, J.V.; Litfin, M.; van Dam, H.; Castellazzi, M.; Herrlich, P.; Kassel, O. Restriction to Fos family members of Trip6-dependent coactivation and glucocorticoid receptor-dependent trans-repression of activator protein-1. Mol. Endocrinol. 2008, 22, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sanchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef]
- Huang, S.; Holzel, M.; Knijnenburg, T.; Schlicker, A.; Roepman, P.; McDermott, U.; Garnett, M.; Grernrum, W.; Sun, C.; Prahallad, A.; et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-β receptor signaling. Cell 2012, 151, 937–950. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Chen, S.; Tan, X.; Li, N.; Liu, C.; Li, Z.; Liu, Z.; Stupack, D.G.; Reisfeld, R.A.; Xiang, R. Fra-1 promotes breast cancer chemosensitivity by driving cancer stem cells from dormancy. Cancer Res. 2012, 72, 3451–3456. [Google Scholar] [CrossRef] [Green Version]
- Van Staalduinen, J.; Baker, D.; Ten Dijke, P.; van Dam, H. Epithelial-mesenchymal-transition-inducing transcription factors: New targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sundqvist, A.; Voytyuk, O.; Hamdi, M.; Popeijus, H.E.; Bijlsma-van der Burgt, C.; Janssen, J.; Martens, J.W.M.; Moustakas, A.; Heldin, C.-H.; ten Dijke, P.; et al. JNK-Dependent cJun Phosphorylation Mitigates TGFβ- and EGF-Induced Pre-Malignant Breast Cancer Cell Invasion by Suppressing AP-1-Mediated Transcriptional Responses. Cells 2019, 8, 1481. https://doi.org/10.3390/cells8121481
Sundqvist A, Voytyuk O, Hamdi M, Popeijus HE, Bijlsma-van der Burgt C, Janssen J, Martens JWM, Moustakas A, Heldin C-H, ten Dijke P, et al. JNK-Dependent cJun Phosphorylation Mitigates TGFβ- and EGF-Induced Pre-Malignant Breast Cancer Cell Invasion by Suppressing AP-1-Mediated Transcriptional Responses. Cells. 2019; 8(12):1481. https://doi.org/10.3390/cells8121481
Chicago/Turabian StyleSundqvist, Anders, Oleksandr Voytyuk, Mohamed Hamdi, Herman E. Popeijus, Corina Bijlsma-van der Burgt, Josephine Janssen, John W.M. Martens, Aristidis Moustakas, Carl-Henrik Heldin, Peter ten Dijke, and et al. 2019. "JNK-Dependent cJun Phosphorylation Mitigates TGFβ- and EGF-Induced Pre-Malignant Breast Cancer Cell Invasion by Suppressing AP-1-Mediated Transcriptional Responses" Cells 8, no. 12: 1481. https://doi.org/10.3390/cells8121481
APA StyleSundqvist, A., Voytyuk, O., Hamdi, M., Popeijus, H. E., Bijlsma-van der Burgt, C., Janssen, J., Martens, J. W. M., Moustakas, A., Heldin, C. -H., ten Dijke, P., & van Dam, H. (2019). JNK-Dependent cJun Phosphorylation Mitigates TGFβ- and EGF-Induced Pre-Malignant Breast Cancer Cell Invasion by Suppressing AP-1-Mediated Transcriptional Responses. Cells, 8(12), 1481. https://doi.org/10.3390/cells8121481