TGFβ Family Signaling Pathways in Pluripotent and Teratocarcinoma Stem Cells’ Fate Decisions: Balancing Between Self-Renewal, Differentiation, and Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
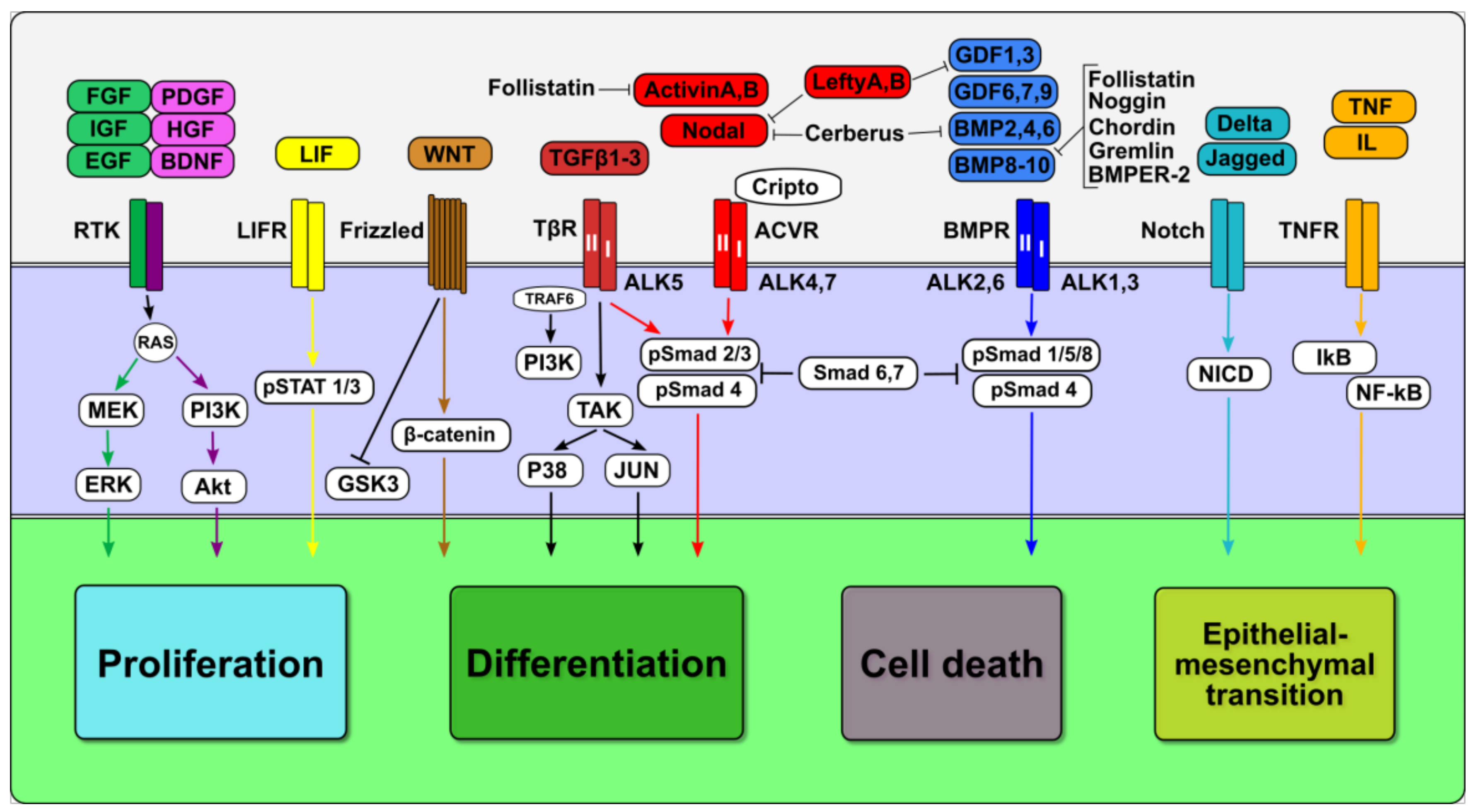
2. Overview of TGFβ Family Signaling
2.1. Canonical and Non-Canonical TGFβ Family Signaling
2.2. Crosstalk Between TGFβ Family and Other Signaling Pathways
2.3. Context-Dependent Activity and Roles of TGFβ Family Signaling
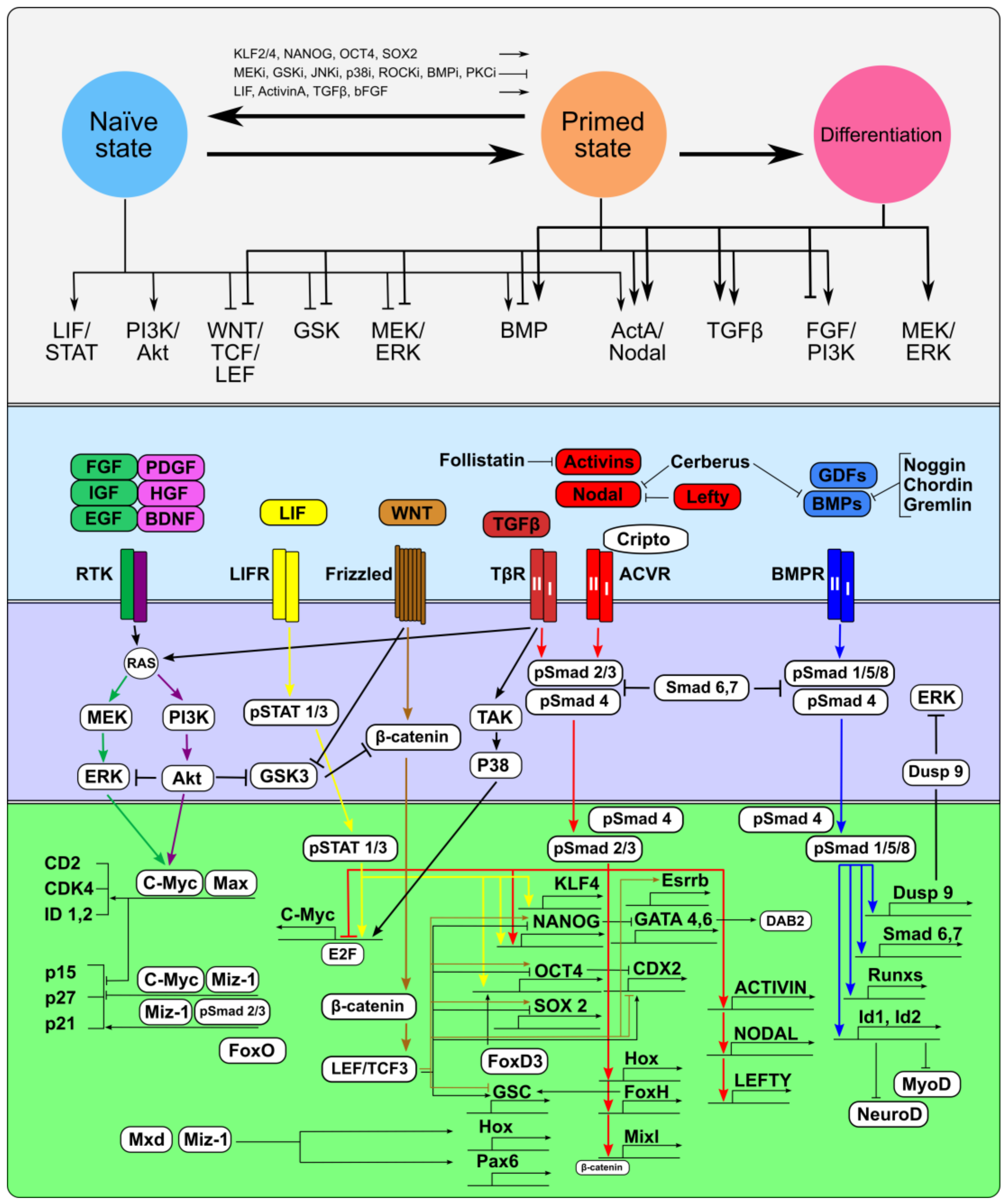
3. TGFβ Family Signaling Pathways in Regulation of Pluripotency, Self-Renewal, and Differentiation
3.1. TGFβ Family in Signaling Networks of Naїve and Primed Pluripotent Stem Cells
3.2. Signaling Pathway Rearrangements during Interconversion Between Naїve and Primed Pluripotent States
3.3. TGFβ Family Signaling during the Onset of Pluripotent Stem Cell Differentiation
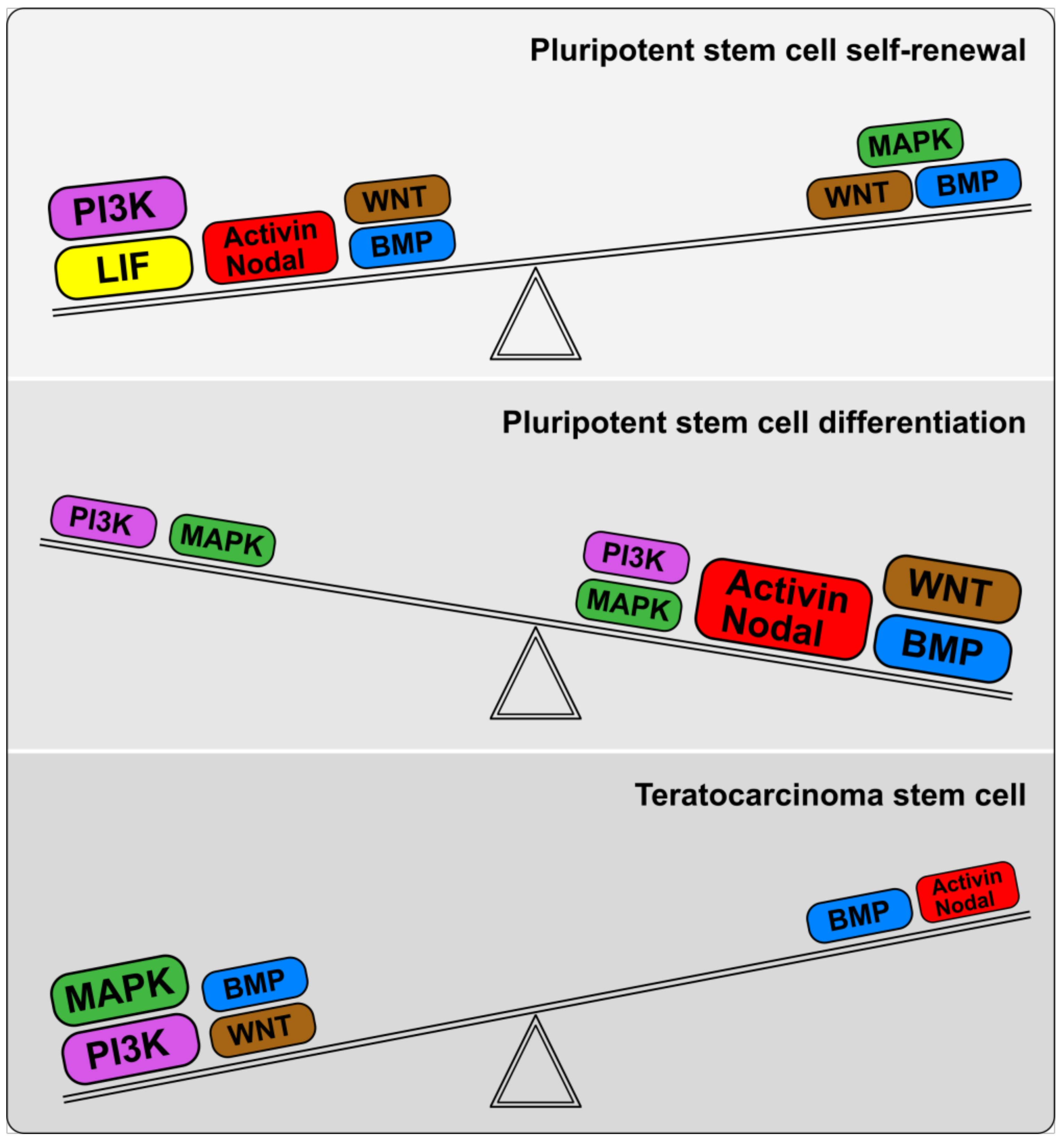
4. Imbalance of TGFβ/BMP Signaling Pathways in Teratocarcinoma Stem Cells
4.1. Aberrant Characteristics and Cell States of Malignant Embryonal Carcinoma (Teratocarcinoma) Cell Lines
4.2. TGFβ Family Signaling Pathways Contribute to the Imbalance of Self-Renewal and Differentiation in Embryonal Carcinoma Cells
5. TGFβ Family Signaling Pathways in Regulation of Tumorigenicity of Pluripotent and Teratocarcinoma Stem Cells
6. Conclusions
Funding
Conflicts of Interest
References
- Derynck, R.; Miyazono, K. TGF-β and the TGF-β family. In The TGF-β family; Derynck, R., Miyazono, K., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2008; pp. 29–43. [Google Scholar]
- Schilling, S.H.; Hjelmeland, A.B.; Rich, J.N.; Wang, X. TGF-β: A multipotential cytokine. In The TGF-β Family; Derynck, R., Miyazono, K., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2008; pp. 45–77. [Google Scholar]
- Mullen, A.C.; Wrana, J.L. TGF-β Family Signaling in Embryonic and Somatic Stem-Cell Renewal and Differentiation. Cold Spring Harb. Perspect. Boil. 2017, 9, a022186. [Google Scholar] [CrossRef]
- Oshimori, N.; Fuchs, E. The harmonies played by TGF-β in stem cell biology. Cell Stem Cell 2012, 11, 751–764. [Google Scholar] [CrossRef]
- Caisander, G.; Park, H.; Frej, K.; Lindqvist, J.; Bergh, C.; Lundin, K.; Hanson, C. Chromosomal integrity maintained in five human embryonic stem cell lines after prolonged in vitro culture. Chromosome Res. 2006, 14, 131–137. [Google Scholar] [CrossRef]
- Urbach, A.; Benvenisty, N. Studying early lethality of 45, XO (Turner’s syndrome) embryos using human embryonic stem cells. PLoS ONE 2009, 4, e4175. [Google Scholar] [CrossRef]
- Hovatta, O.; Jaconi, M.; Töhönen, V.; Béna, F.; Gimelli, S.; Bosman, A.; Holm, F.; Wyder, S.; Zdobnov, E.M.; Irion, O.; et al. A teratocarcinoma-like human embryonic stem cell (hESC) line and four hESC lines reveal potentially oncogenic genomic changes. PLoS ONE 2010, 5, e10263. [Google Scholar] [CrossRef]
- Gore, A.; Li, Z.; Fung, H.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W. From teratocarcinomas to embryonic stem cells. Philos. Trans. R. Soc. Lond. B: Biol. Sci. 2002, 357, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; Hill, C.S. How the Smads regulate transcription. Int. J. Biochem. Cell Biol. 2008, 40, 383–408. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Boil. 2016, 8, a022079. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Chen, Y.; Feng, X. Transcriptional control via Smads. In The TGF-β Family; Derynck, R., Miyazono, K., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2008; pp. 287–332. [Google Scholar]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, X. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gudey, S.K.; Landström, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Boil. 2017, 9, a022137. [Google Scholar] [CrossRef] [PubMed]
- Onichtchouk, D.; Chen, Y.G.; Dosch, R.; Gawantka, V.; Delius, H.; Massagué, J.; Niehrs, C. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 1999, 401, 480–485. [Google Scholar] [CrossRef]
- Samarakoon, R.; Chitnis, S.S.; Higgins, S.P.; Higgins, C.E.; Krepinsky, J.C.; Higgins, P.J. Redox-induced Src kinase and caveolin-1 signaling in TGF-β1-initiated SMAD2/3 activation and PAI-1 expression. PLoS ONE 2011, 6, e22896. [Google Scholar] [CrossRef]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef]
- Chang, C. Agonist and antagonist of the TGF-β family ligands. In The TGF-β Family; Derynck, R., Miyazono, K., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2008; pp. 203–257. [Google Scholar]
- Thompson, T.B.; Lerch, T.F.; Cook, R.W.; Woodruff, T.K.; Jardetzky, T.S. The structure of the follistatin: Activin complex reveals antagonism of both type I and type II receptor binding. Dev. Cell 2005, 9, 535–543. [Google Scholar] [CrossRef]
- Fainsod, A.; Deissler, K.; Yelin, R.; Marom, K.; Epstein, M.; Pillemer, G.; Steinbeisser, H.; Blum, M. The dorsalizing and neural inducing gene follistatin is an antagonist of BMP-4. Mech. Dev. 1997, 63, 39–50. [Google Scholar] [CrossRef]
- Iemura, S.; Yamamoto, T.S.; Takagi, C.; Uchiyama, H.; Natsume, T.; Shimasaki, S.; Sugino, H.; Ueno, N. Direct binding of follistatin to a complex of bone-morphogenetic protein and its receptor inhibits ventral and epidermal cell fates in early Xenopus embryo. Proc. Natl. Acad. Sci. USA 1998, 95, 9337–9342. [Google Scholar] [CrossRef] [PubMed]
- Amthor, H.; Christ, B.; Rashid-Doubell, F.; Kemp, C.F.; Lang, E.; Patel, K. Follistatin regulates bone morphogenetic protein-7 (BMP-7) activity to stimulate embryonic muscle growth. Dev. Biol. 2002, 243, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Amthor, H.; Nicholas, G.; McKinnell, I.; Kemp, C.F.; Sharma, M.; Kambadur, R.; Patel, K. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev. Biol. 2004, 270, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.C.; Harland, R.M. Expression cloning of noggin, a new dorsalizing factor localized to the Spemann organizer in Xenopus embryos. Cell 1992, 70, 829–840. [Google Scholar] [CrossRef]
- Piccolo, S.; Sasai, Y.; Lu, B.; De Robertis, E.M. Dorsoventral patterning in Xenopus: Inhibition of ventral signals by direct binding of chordin to BMP-4. Cell 1996, 86, 589–598. [Google Scholar] [CrossRef]
- Blitz, I.L.; Cho, K.W.Y.; Chang, C. Twisted gastrulation loss-of-function analyses support its role as a BMP inhibitor during early Xenopus embryogenesis. Development 2003, 130, 4975–4988. [Google Scholar] [CrossRef]
- Bouwmeester, T.; Kim, S.; Sasai, Y.; Lu, B.; De Robertis, E.M. Cerberus is a head-inducing secreted factor expressed in the anterior endoderm of Spemann’s organizer. Nature 1996, 382, 595–601. [Google Scholar] [CrossRef]
- Hsu, D.R.; Economides, A.N.; Wang, X.; Eimon, P.M.; Harland, R.M. The Xenopus dorsalizing factor Gremlin identifies a novel family of secreted proteins that antagonize BMP activities. Mol. Cell 1998, 1, 673–683. [Google Scholar] [CrossRef]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef]
- Coffinier, C.; Ketpura, N.; Tran, U.; Geissert, D.; De Robertis, E.M. Mouse Crossveinless-2 is the vertebrate homolog of a Drosophila extracellular regulator of BMP signaling. Mech. Dev. 2002, 119, S179–S184. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.; Binder, O.; Wu, Y.; Aitsebaomo, J.; Ren, R.; Bode, C.; Bautch, V.L.; Conlon, F.L.; Patterson, C. BMPER, a novel endothelial cell precursor-derived protein, antagonizes bone morphogenetic protein signaling and endothelial cell differentiation. Mol. Cell. Biol. 2003, 23, 5664–5679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coles, E.; Christiansen, J.; Economou, A.; Bronner-Fraser, M.; Wilkinson, D.G. A vertebrate crossveinless 2 homologue modulates BMP activity and neural crest cell migration. Development 2004, 131, 5309–5317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrand, A.; Romarís, M.; Rasmussen, L.M.; Heinegård, D.; Twardzik, D.R.; Border, W.A.; Ruoslahti, E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem. J. 1994, 302, 527–534. [Google Scholar] [CrossRef]
- Doliana, R.; Mongiat, M.; Bucciotti, F.; Giacomello, E.; Deutzmann, R.; Volpin, D.; Bressan, G.M.; Colombatti, A. EMILIN, a component of the elastic fiber and a new member of the C1q/tumor necrosis factor superfamily of proteins. J. Biol. Chem. 1999, 274, 16773–16781. [Google Scholar] [CrossRef] [Green Version]
- Viviano, B.L.; Paine-Saunders, S.; Gasiunas, N.; Gallagher, J.; Saunders, S. Domain-specific modification of heparan sulfate by Qsulf1 modulates the binding of the bone morphogenetic protein antagonist Noggin. J. Biol. Chem. 2004, 279, 5604–5611. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.S.; Alliston, T.; Delston, R.; Derynck, R. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 2005, 24, 2543–2555. [Google Scholar] [CrossRef] [Green Version]
- Hannon, G.J.; Beach, D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 1994, 371, 257–261. [Google Scholar] [CrossRef]
- Datto, M.B.; Yu, Y.; Wang, X.F. Functional analysis of the transforming growth factor beta responsive elements in the WAF1/Cip1/p21 promoter. J. Biol. Chem. 1995, 270, 28623–28628. [Google Scholar] [CrossRef] [Green Version]
- Staller, P.; Peukert, K.; Kiermaier, A.; Seoane, J.; Lukas, J.; Karsunky, H.; Möröy, T.; Bartek, J.; Massagué, J.; Hänel, F.; et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001, 3, 392–399. [Google Scholar] [CrossRef]
- Lasorella, A.; Noseda, M.; Beyna, M.; Yokota, Y.; Iavarone, A. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature 2000, 407, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massagué, J. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Eivers, E.; Fuentealba, L.C.; De Robertis, E.M. Integrating positional information at the level of Smad1/5/8. Curr. Opin. Genet. Dev. 2008, 18, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, D.; Datta, P.K.; Beauchamp, R.D. Oncogenic ras represses transforming growth factor-beta /Smad signaling by degrading tumor suppressor Smad4. J. Biol. Chem. 2001, 276, 29531–29537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.A.; Rahnama, M.; Wang, S.; Sosu-Sedzorme, W.; Verheyen, E.M. Drosophila Nemo antagonizes BMP signaling by phosphorylation of Mad and inhibition of its nuclear accumulation. Development 2007, 134, 2061–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, K.; Suzuki, H.; Ohashi, T.; Nitta, K.; Yumura, W.; Nihei, H. Involvement of MAP kinase cascades in Smad7 transcriptional regulation. Biochem. Biophys. Res. Commun. 2001, 289, 376–381. [Google Scholar] [CrossRef]
- Conery, A.R.; Cao, Y.; Thompson, E.A.; Townsend, C.M.J.; Ko, T.C.; Luo, K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat. Cell Biol. 2004, 6, 366–372. [Google Scholar] [CrossRef]
- Remy, I.; Montmarquette, A.; Michnick, S.W. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat. Cell Biol. 2004, 6, 358–365. [Google Scholar] [CrossRef]
- Song, K.; Wang, H.; Krebs, T.L.; Danielpour, D. Novel roles of Akt and mTOR in suppressing TGF-beta/ALK5-mediated Smad3 activation. EMBO J. 2006, 25, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Valderrama-Carvajal, H.; Cocolakis, E.; Lacerte, A.; Lee, E.; Krystal, G.; Ali, S.; Lebrun, J. Activin/TGF-beta induce apoptosis through Smad-dependent expression of the lipid phosphatase SHIP. Nat. Cell Biol. 2002, 4, 963–969. [Google Scholar] [CrossRef]
- Seoane, J.; Le, H.; Shen, L.; Anderson, S.A.; Massagué, J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Wilkes, M.C.; Mitchell, H.; Penheiter, S.G.; Doré, J.J.; Suzuki, K.; Edens, M.; Sharma, D.K.; Pagano, R.E.; Leof, E.B. Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. 2005, 65, 10431–10440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, J.C.; Lee, D.Y.; Waghray, M.; Keshamouni, V.G.; Thomas, P.E.; Zhang, H.; Cui, Z.; Thannickal, V.J. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J. Biol. Chem. 2004, 279, 1359–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Culmsee, C.; Klumpp, S.; Krieglstein, J. Neuroprotection by transforming growth factor-beta1 involves activation of nuclear factor-kappaB through phosphatidylinositol-3-OH kinase/Akt and mitogen-activated protein kinase-extracellular-signal regulated kinase1,2 signaling pathways. Neuroscience 2004, 123, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.; Landström, M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, A.; Song, J.; Thakur, N.; Itoh, S.; Marcusson, A.; Bergh, A.; Heldin, C.; Landström, M. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci. Signal. 2017, 10, eaal4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willert, J.; Epping, M.; Pollack, J.R.; Brown, P.O.; Nusse, R. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev. Biol. 2002, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Theil, T.; Aydin, S.; Koch, S.; Grotewold, L.; Rüther, U. Wnt and Bmp signalling cooperatively regulate graded Emx2 expression in the dorsal telencephalon. Development 2002, 129, 3045–3054. [Google Scholar]
- Lei, S.; Dubeykovskiy, A.; Chakladar, A.; Wojtukiewicz, L.; Wang, T.C. The murine gastrin promoter is synergistically activated by transforming growth factor-beta/Smad and Wnt signaling pathways. J. Biol. Chem. 2004, 279, 42492–42502. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, A.; Katagiri, T.; Tamura, M. Cross-talk between Wnt and bone morphogenetic protein 2 (BMP-2) signaling in differentiation pathway of C2C12 myoblasts. J. Biol. Chem. 2005, 280, 37660–37668. [Google Scholar] [CrossRef] [Green Version]
- Kosinski, C.; Li, V.S.W.; Chan, A.S.Y.; Zhang, J.; Ho, C.; Tsui, W.Y.; Chan, T.L.; Mifflin, R.C.; Powell, D.W.; Yuen, S.T.; et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc. Natl. Acad. Sci. USA 2007, 104, 15418–15423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Esteban, C.; Capdevila, J.; Kawakami, Y.; Izpisúa Belmonte, J.C. Wnt signaling and PKA control Nodal expression and left-right determination in the chick embryo. Development 2001, 128, 3189–3195. [Google Scholar] [PubMed]
- Jin, E.; Lee, S.; Choi, Y.; Jung, J.; Bang, O.; Kang, S. BMP-2-enhanced chondrogenesis involves p38 MAPK-mediated down-regulation of Wnt-7a pathway. Mol. Cells 2006, 22, 353–359. [Google Scholar] [PubMed]
- Morkel, M.; Huelsken, J.; Wakamiya, M.; Ding, J.; van de Wetering, M.; Clevers, H.; Taketo, M.M.; Behringer, R.R.; Shen, M.M.; Birchmeier, W. Beta-catenin regulates Cripto- and Wnt3-dependent gene expression programs in mouse axis and mesoderm formation. Development 2003, 130, 6283–6294. [Google Scholar] [CrossRef] [Green Version]
- Fuentealba, L.C.; Eivers, E.; Ikeda, A.; Hurtado, C.; Kuroda, H.; Pera, E.M.; De Robertis, E.M. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 2007, 131, 980–993. [Google Scholar] [CrossRef] [Green Version]
- Aragón, E.; Goerner, N.; Zaromytidou, A.; Xi, Q.; Escobedo, A.; Massagué, J.; Macias, M.J. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 2011, 25, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M.; Yagi, K.; Yamamoto, H.; Furukawa, Y.; Shimada, S.; Nakamura, Y.; Kikuchi, A.; Miyazono, K.; Kato, M. Axin facilitates Smad3 activation in the transforming growth factor beta signaling pathway. Mol. Cell. Biol. 2001, 21, 5132–5141. [Google Scholar] [CrossRef] [Green Version]
- Jian, H.; Shen, X.; Liu, I.; Semenov, M.; He, X.; Wang, X. Smad3-dependent nuclear translocation of beta-catenin is required for TGF-beta1-induced proliferation of bone marrow-derived adult human mesenchymal stem cells. Genes Dev. 2006, 20, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Klüppel, M.; Wrana, J.L. Turning it up a Notch: Cross-talk between TGF beta and Notch signaling. Bioessays 2005, 27, 115–118. [Google Scholar] [CrossRef]
- Zavadil, J.; Cermak, L.; Soto-Nieves, N.; Böttinger, E.P. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004, 23, 1155–1165. [Google Scholar] [CrossRef] [Green Version]
- Nyhan, K.C.; Faherty, N.; Murray, G.; Cooey, L.B.; Godson, C.; Crean, J.K.; Brazil, D.P. Jagged/Notch signalling is required for a subset of TGFβ1 responses in human kidney epithelial cells. Biochim. Biophys. Acta 2010, 1803, 1386–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlqvist, C.; Blokzijl, A.; Chapman, G.; Falk, A.; Dannaeus, K.; Ibâñez, C.F.; Lendahl, U. Functional Notch signaling is required for BMP4-induced inhibition of myogenic differentiation. Development 2003, 130, 6089–6099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamurovic, N.; Cappellen, D.; Rohner, D.; Susa, M. Coordinated activation of notch, Wnt, and transforming growth factor-beta signaling pathways in bone morphogenic protein 2-induced osteogenesis. Notch target gene Hey1 inhibits mineralization and Runx2 transcriptional activity. J. Biol. Chem. 2004, 279, 37704–37715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takizawa, T.; Ochiai, W.; Nakashima, K.; Taga, T. Enhanced gene activation by Notch and BMP signaling cross-talk. Nucleic Acids Res. 2003, 31, 5723–5731. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, A.; Dahlqvist, C.; Reissmann, E.; Falk, A.; Moliner, A.; Lendahl, U.; Ibáñez, C.F. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J. Cell Biol. 2003, 163, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Kumano, K.; Shimizu, K.; Imai, Y.; Kurokawa, M.; Ogawa, S.; Miyagishi, M.; Taira, K.; Hirai, H.; Chiba, S. Notch1 oncoprotein antagonizes TGF-beta/Smad-mediated cell growth suppression via sequestration of coactivator p300. Cancer Sci. 2005, 96, 274–282. [Google Scholar] [CrossRef]
- Sun, Y.; Lowther, W.; Kato, K.; Bianco, C.; Kenney, N.; Strizzi, L.; Raafat, D.; Hirota, M.; Khan, N.I.; Bargo, S.; et al. Notch4 intracellular domain binding to Smad3 and inhibition of the TGF-beta signaling. Oncogene 2005, 24, 5365–5374. [Google Scholar] [CrossRef] [Green Version]
- Valdez, J.M.; Zhang, L.; Su, Q.; Dakhova, O.; Zhang, Y.; Shahi, P.; Spencer, D.M.; Creighton, C.J.; Ittmann, M.M.; Xin, L. Notch and TGFβ form a reciprocal positive regulatory loop that suppresses murine prostate basal stem/progenitor cell activity. Cell Stem Cell 2012, 11, 676–688. [Google Scholar] [CrossRef] [Green Version]
- López-Rovira, T.; Chalaux, E.; Rosa, J.L.; Bartrons, R.; Ventura, F. Interaction and functional cooperation of NF-kappa B with Smads. Transcriptional regulation of the junB promoter. J. Biol. Chem. 2000, 275, 28937–28946. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.; Chiao, C.; Kuo, W.; Hsiao, Y.; Chen, Y.; Wei, Y.; Lai, T.; Fong, Y.; Tang, C. TGF-beta1 increases motility and alphavbeta3 integrin up-regulation via PI3K, Akt and NF-kappaB-dependent pathway in human chondrosarcoma cells. Biochem. Pharmacol. 2008, 75, 1292–1301. [Google Scholar] [CrossRef]
- Descargues, P.; Sil, A.K.; Sano, Y.; Korchynskyi, O.; Han, G.; Owens, P.; Wang, X.; Karin, M. IKKalpha is a critical coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 2487–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, K.A.; Ravindran, A.; Podolsky, M.A.; Glick, A.B. The TGFβ1 pathway is required for NFκB dependent gene expression in mouse keratinocytes. Cytokine 2013, 64, 652–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Yu, Y.; Sun, C.; Liu, T.; Liang, T.; Zhan, L.; Lin, X.; Feng, X. STAT3 selectively interacts with Smad3 to antagonize TGF-β signalling. Oncogene 2016, 35, 4388–4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulloa, L.; Doody, J.; Massagué, J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature 1999, 397, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Massagué, J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, A.; Massagué, J. E2F and histone deacetylase mediate transforming growth factor beta repression of cdc25A during keratinocyte cell cycle arrest. Mol. Cell. Biol. 1999, 19, 916–922. [Google Scholar] [CrossRef] [Green Version]
- Lebrin, F.; Goumans, M.; Jonker, L.; Carvalho, R.L.C.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Gifford, C.C.; Samarakoon, R.; Higgins, P.J. Deregulation of Negative Controls on TGF-β1 Signaling in Tumor Progression. Cancers 2018, 10, 159. [Google Scholar] [CrossRef] [Green Version]
- Prehn, J.H.; Bindokas, V.P.; Marcuccilli, C.J.; Krajewski, S.; Reed, J.C.; Miller, R.J. Regulation of neuronal Bcl2 protein expression and calcium homeostasis by transforming growth factor type beta confers wide-ranging protection on rat hippocampal neurons. Proc. Natl. Acad. Sci. USA 1994, 91, 12599–12603. [Google Scholar] [CrossRef] [Green Version]
- Hishikawa, K.; Oemar, B.S.; Tanner, F.C.; Nakaki, T.; Lüscher, T.F.; Fujii, T. Connective tissue growth factor induces apoptosis in human breast cancer cell line MCF-7. J. Biol. Chem. 1999, 274, 37461–37466. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Bhat, M.; Hsing, A.Y.; Ma, J.; Danielpour, D. Bcl-xL blocks transforming growth factor-beta 1-induced apoptosis by inhibiting cytochrome c release and not by directly antagonizing Apaf-1-dependent caspase activation in prostate epithelial cells. J. Biol. Chem. 2001, 276, 26614–26621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Park, B.J.; Park, J.H.; Yang, M.H.; Chi, S.G. TGF-beta1 inhibition of apoptosis through the transcriptional up-regulation of Bcl-X(L) in human monocytic leukemia U937 cells. Exp. Mol. Med. 1999, 31, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Yang, G.; Ahlemeyer, B.; Pang, L.; Che, X.; Culmsee, C.; Klumpp, S.; Krieglstein, J. Transforming growth factor-beta 1 increases bad phosphorylation and protects neurons against damage. J. Neurosci. 2002, 22, 3898–3909. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, T.; Kiss, A.; Thorgeirsson, S.S. Induction of p53 and Bax during TGF-beta 1 initiated apoptosis in rat liver epithelial cells. Biochem. Biophys. Res. Commun. 1998, 251, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Chen, C.; Chen, C.; Chen, J.; Su, Y.; Chen, R. TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Ghiassi, M.; Jirmanova, L.; Balliet, A.G.; Hoffman, B.; Fornace, A.J.J.; Liebermann, D.A.; Bottinger, E.P.; Roberts, A.B. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J. Biol. Chem. 2003, 278, 43001–43007. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ozaki, I.; Mizuta, T.; Hamajima, H.; Yasutake, T.; Eguchi, Y.; Ideguchi, H.; Yamamoto, K.; Matsuhashi, S. Involvement of programmed cell death 4 in transforming growth factor-beta1-induced apoptosis in human hepatocellular carcinoma. Oncogene 2006, 25, 6101–6112. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J.; Xi, Q. TGF-β control of stem cell differentiation genes. FEBS Lett. 2012, 586, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.C.; Orlando, D.A.; Newman, J.J.; Lovén, J.; Kumar, R.M.; Bilodeau, S.; Reddy, J.; Guenther, M.G.; DeKoter, R.P.; Young, R.A. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 2011, 147, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Robertson, E.J.; Norris, D.P.; Brennan, J.; Bikoff, E.K. Control of early anterior-posterior patterning in the mouse embryo by TGF-beta signalling. Philos. Trans. R. Soc. Lond. B: Biol. Sci. 2003, 358, 1351–1357. [Google Scholar] [CrossRef] [Green Version]
- Tam, P.P.L.; Loebel, D.A.F. Gene function in mouse embryogenesis: Get set for gastrulation. Nat. Rev. Genet. 2007, 8, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Keller, G. Differentiation of embryonic stem cells to clinically relevant populations: Lessons from embryonic development. Cell 2008, 132, 661–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, T.A.; Narimatsu, M.; Weiss, A.; David, L.; Wrana, J.L. The TGFβ superfamily in stem cell biology and early mammalian embryonic development. Biochim. Biophys. Acta 2013, 1830, 2268–2279. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Gaunt, S.J.; Cho, K.W.; Steinbeisser, H.; Blumberg, B.; Bittner, D.; De Robertis, E.M. Gastrulation in the mouse: The role of the homeobox gene goosecoid. Cell 1992, 69, 1097–1106. [Google Scholar] [CrossRef]
- Hart, A.H.; Hartley, L.; Sourris, K.; Stadler, E.S.; Li, R.; Stanley, E.G.; Tam, P.P.L.; Elefanty, A.G.; Robb, L. Mixl1 is required for axial mesendoderm morphogenesis and patterning in the murine embryo. Development 2002, 129, 3597–3608. [Google Scholar] [PubMed]
- Chen, X.; Weisberg, E.; Fridmacher, V.; Watanabe, M.; Naco, G.; Whitman, M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature 1997, 389, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Vallier, L.; Touboul, T.; Chng, Z.; Brimpari, M.; Hannan, N.; Millan, E.; Smithers, L.E.; Trotter, M.; Rugg-Gunn, P.; Weber, A.; et al. Early cell fate decisions of human embryonic stem cells and mouse epiblast stem cells are controlled by the same signalling pathways. PLoS ONE 2009, 4, e6082. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Black, B.L.; Derynck, R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 2001, 15, 2950–2966. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Kang, J.S.; Derynck, R. TGF-beta-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. EMBO J. 2004, 23, 1557–1566. [Google Scholar] [CrossRef] [Green Version]
- Padua, D.; Massagué, J. Roles of TGFbeta in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowanetz, M.; Valcourt, U.; Bergström, R.; Heldin, C.; Moustakas, A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol. Cell. Biol. 2004, 24, 4241–4254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, M.; Cubillo, E.; Tobiume, K.; Shirakihara, T.; Fukuda, N.; Suzuki, H.; Shimizu, K.; Takehara, K.; Cano, A.; Saitoh, M.; et al. A role for Id in the regulation of TGF-beta-induced epithelial-mesenchymal transdifferentiation. Cell Death Differ. 2004, 11, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Ellenrieder, V.; Hendler, S.F.; Boeck, W.; Seufferlein, T.; Menke, A.; Ruhland, C.; Adler, G.; Gress, T.M. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 2001, 61, 4222–4228. [Google Scholar]
- Yu, L.; Hébert, M.C.; Zhang, Y.E. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Invest. 2004, 114, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Oft, M.; Peli, J.; Rudaz, C.; Schwarz, H.; Beug, H.; Reichmann, E. TGF-beta1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996, 10, 2462–2477. [Google Scholar] [CrossRef] [Green Version]
- Ekman, M.; Mu, Y.; Lee, S.Y.; Edlund, S.; Kozakai, T.; Thakur, N.; Tran, H.; Qian, J.; Groeden, J.; Heldin, C.; et al. APC and Smad7 link TGFβ type I receptors to the microtubule system to promote cell migration. Mol. Biol. Cell 2012, 23, 2109–2121. [Google Scholar] [CrossRef]
- Thakur, N.; Gudey, S.K.; Marcusson, A.; Fu, J.Y.; Bergh, A.; Heldin, C.; Landström, M. TGFβ-induced invasion of prostate cancer cells is promoted by c-Jun-dependent transcriptional activation of Snail1. Cell Cycle 2014, 13, 2400–2414. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.; Landström, M.; Moustakas, A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Sundar, R.; Thakur, N.; Ekman, M.; Gudey, S.K.; Yakymovych, M.; Hermansson, A.; Dimitriou, H.; Bengoechea-Alonso, M.T.; Ericsson, J.; et al. TRAF6 ubiquitinates TGFβ type I receptor to promote its cleavage and nuclear translocation in cancer. Nat. Commun. 2011, 2, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreesen, O.; Brivanlou, A.H. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev. 2007, 3, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.M.; Reynolds, D.; Cliff, T.; Ohtsuka, S.; Mattheyses, A.L.; Sun, Y.; Menendez, L.; Kulik, M.; Dalton, S. Signaling network crosstalk in human pluripotent cells: A Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell 2012, 10, 312–326. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Wu, C.; Chu, G.C.; Xiao, Y.; Ho, D.; Zhang, J.; Perry, S.R.; Labrot, E.S.; Wu, X.; Lis, R.; et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 2011, 470, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Xiong, S.; Cheng, J.; Klausen, C.; Zhao, J.; Leung, P.C.K. TGF-β1 stimulates migration of type II endometrial cancer cells by down-regulating PTEN via activation of SMAD and ERK1/2 signaling pathways. Oncotarget 2016, 7, 61262–61272. [Google Scholar]
- Buijs, J.T.; Rentsch, C.A.; van der Horst, G.; van Overveld, P.G.M.; Wetterwald, A.; Schwaninger, R.; Henriquez, N.V.; Ten Dijke, P.; Borovecki, F.; Markwalder, R.; et al. BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am. J. Pathol. 2007, 171, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Ying, X.; Sun, Y.; He, P. MicroRNA-137 inhibits BMP7 to enhance the epithelial-mesenchymal transition of breast cancer cells. Oncotarget 2017, 8, 18348–18358. [Google Scholar] [CrossRef]
- Stolfi, C.; Marafini, I.; De Simone, V.; Pallone, F.; Monteleone, G. The dual role of Smad7 in the control of cancer growth and metastasis. Int J Mol Sci 2013, 14, 23774–23790. [Google Scholar] [CrossRef] [Green Version]
- Khin, S.S.; Kitazawa, R.; Win, N.; Aye, T.T.; Mori, K.; Kondo, T.; Kitazawa, S. BAMBI gene is epigenetically silenced in subset of high-grade bladder cancer. Int. J. Cancer 2009, 125, 328–338. [Google Scholar] [CrossRef]
- Yu, W.; Chai, H. Inhibition of BAMBI reduces the viability and motility of colon cancer via activating TGF-β/Smad pathway. Oncol. Lett. 2017, 14, 4793–4799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deheuninck, J.; Luo, K. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res. 2009, 19, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.; Smith, A. Capturing pluripotency. Cell 2008, 132, 532–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackett, J.A.; Surani, M.A. Regulatory principles of pluripotency: From the ground state up. Cell Stem Cell 2014, 15, 416–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberger, L.; Ayyash, M.; Novershtern, N.; Hanna, J.H. Dynamic stem cell states: Naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 2016, 17, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.L.; Zhang, W.; Jiang, J.; et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Pfeuty, B.; Kress, C.; Pain, B. Network Features and Dynamical Landscape of Naive and Primed Pluripotency. Biophys. J. 2018, 114, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.L.; Hilton, D.J.; Pease, S.; Willson, T.A.; Stewart, C.L.; Gearing, D.P.; Wagner, E.F.; Metcalf, D.; Nicola, N.A.; Gough, N.M. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 1988, 336, 684–687. [Google Scholar] [CrossRef]
- Nichols, J.; Davidson, D.; Taga, T.; Yoshida, K.; Chambers, I.; Smith, A. Complementary tissue-specific expression of LIF and LIF-receptor mRNAs in early mouse embryogenesis. Mech. Dev. 1996, 57, 123–131. [Google Scholar] [CrossRef]
- Boeuf, H.; Hauss, C.; Graeve, F.D.; Baran, N.; Kedinger, C. Leukemia inhibitory factor-dependent transcriptional activation in embryonic stem cells. J. Cell Biol. 1997, 138, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H.; Burdon, T.; Chambers, I.; Smith, A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998, 12, 2048–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdon, T.; Smith, A.; Savatier, P. Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol. 2002, 12, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, P.; McLean, C.; Sheppard, A.; Rivett, D.; Jones, K.; Dalton, S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 2005, 132, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Brons, I.G.M.; Smithers, L.E.; Trotter, M.W.B.; Rugg-Gunn, P.; Sun, B.; Chuva de Sousa Lopes, S.M.; Howlett, S.K.; Clarkson, A.; Ahrlund-Richter, L.; Pedersen, R.A.; et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007, 448, 191–195. [Google Scholar] [CrossRef]
- Tesar, P.J.; Chenoweth, J.G.; Brook, F.A.; Davies, T.J.; Evans, E.P.; Mack, D.L.; Gardner, R.L.; McKay, R.D.G. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 2007, 448, 196–199. [Google Scholar] [CrossRef]
- Niwa, H.; Ogawa, K.; Shimosato, D.; Adachi, K. A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature 2009, 460, 118–122. [Google Scholar] [CrossRef]
- Kunath, T.; Saba-El-Leil, M.K.; Almousailleakh, M.; Wray, J.; Meloche, S.; Smith, A. FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development 2007, 134, 2895–2902. [Google Scholar] [CrossRef] [Green Version]
- Besser, D. Expression of nodal, lefty-a, and lefty-B in undifferentiated human embryonic stem cells requires activation of Smad2/3. J. Biol. Chem. 2004, 279, 45076–45084. [Google Scholar] [CrossRef] [Green Version]
- Vallier, L.; Alexander, M.; Pedersen, R.A. Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J. Cell. Sci. 2005, 118, 4495–4509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahéron, L.; Opitz, S.L.; Zaehres, H.; Lensch, M.W.; Andrews, P.W.; Itskovitz-Eldor, J.; Daley, G.Q. LIF/STAT3 signaling fails to maintain self-renewal of human embryonic stem cells. Stem Cells 2004, 22, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Pébay, A.; Wong, R.C.B.; Pitson, S.M.; Wolvetang, E.J.; Peh, G.S.; Filipczyk, A.; Koh, K.L.L.; Tellis, I.; Nguyen, L.T.V.; Pera, M.F. Essential roles of sphingosine-1-phosphate and platelet-derived growth factor in the maintenance of human embryonic stem cells. Stem Cells 2005, 23, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Schulz, T.C.; Sherrer, E.S.; Dauphin, D.S.; Shin, S.; Nelson, A.M.; Ware, C.B.; Zhan, M.; Song, C.; Chen, X.; et al. Self-renewal of human embryonic stem cells requires insulin-like growth factor-1 receptor and ERBB2 receptor signaling. Blood 2007, 110, 4111–4119. [Google Scholar] [CrossRef] [Green Version]
- Eiselleova, L.; Matulka, K.; Kriz, V.; Kunova, M.; Schmidtova, Z.; Neradil, J.; Tichy, B.; Dvorakova, D.; Pospisilova, S.; Hampl, A.; et al. A complex role for FGF-2 in self-renewal, survival, and adhesion of human embryonic stem cells. Stem Cells 2009, 27, 1847–1857. [Google Scholar] [CrossRef] [Green Version]
- Greber, B.; Lehrach, H.; Adjaye, J. Fibroblast growth factor 2 modulates transforming growth factor beta signaling in mouse embryonic fibroblasts and human ESCs (hESCs) to support hESC self-renewal. Stem Cells 2007, 25, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Na, J.; Furue, M.K.; Andrews, P.W. Inhibition of ERK1/2 prevents neural and mesendodermal differentiation and promotes human embryonic stem cell self-renewal. Stem Cell Res. 2010, 5, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Burdon, T.; Stracey, C.; Chambers, I.; Nichols, J.; Smith, A. Suppression of SHP-2 and ERK signalling promotes self-renewal of mouse embryonic stem cells. Dev. Biol. 1999, 210, 30–43. [Google Scholar] [CrossRef] [Green Version]
- Ying, Q.; Wray, J.; Nichols, J.; Batlle-Morera, L.; Doble, B.; Woodgett, J.; Cohen, P.; Smith, A. The ground state of embryonic stem cell self-renewal. Nature 2008, 453, 519–523. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Guo, R.; Zhang, Q.; Guo, H.; Yang, M.; Wu, Z.; Gao, S.; Liu, L.; Chen, L. Erk signaling is indispensable for genomic stability and self-renewal of mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E5936–E5943. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, L.; Hughes, O.; Yung, S.; Hyslop, L.; Stewart, R.; Wappler, I.; Peters, H.; Walter, T.; Stojkovic, P.; Evans, J.; et al. The role of PI3K/AKT, MAPK/ERK and NFkappabeta signalling in the maintenance of human embryonic stem cell pluripotency and viability highlighted by transcriptional profiling and functional analysis. Hum. Mol. Genet. 2006, 15, 1894–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, G.; Wang, C.; Zhao, Y.; Zhang, H.; Tan, Z.; Song, Z.; Ding, M.; Deng, H. MEK/ERK signaling contributes to the maintenance of human embryonic stem cell self-renewal. Differentiation 2007, 75, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Meijer, L.; Skaltsounis, L.; Greengard, P.; Brivanlou, A.H. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 2004, 10, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.M.; Bechard, M.; Smith, K.; Dalton, S. Reconciling the different roles of Gsk3β in “naïve” and “primed” pluripotent stem cells. Cell Cycle 2012, 11, 2991–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Robitaille, A.M.; Berndt, J.D.; Davidson, K.C.; Fischer, K.A.; Mathieu, J.; Potter, J.C.; Ruohola-Baker, H.; Moon, R.T. Wnt/β-catenin signaling promotes self-renewal and inhibits the primed state transition in naïve human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6382–E6390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Sampsell-Barron, T.L.; Gu, F.; Root, S.; Peck, R.M.; Pan, G.; Yu, J.; Antosiewicz-Bourget, J.; Tian, S.; Stewart, R.; et al. NANOG is a direct target of TGFbeta/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell 2008, 3, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Vallier, L.; Mendjan, S.; Brown, S.; Chng, Z.; Teo, A.; Smithers, L.E.; Trotter, M.W.B.; Cho, C.H.; Martinez, A.; Rugg-Gunn, P.; et al. Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development 2009, 136, 1339–1349. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.; Teo, A.; Pauklin, S.; Hannan, N.; Cho, C.H.; Lim, B.; Vardy, L.; Dunn, N.R.; Trotter, M.; Pedersen, R.; et al. Activin/Nodal signaling controls divergent transcriptional networks in human embryonic stem cells and in endoderm progenitors. Stem Cells 2011, 29, 1176–1185. [Google Scholar] [CrossRef]
- James, D.; Levine, A.J.; Besser, D.; Hemmati-Brivanlou, A. TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 2005, 132, 1273–1282. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, K.; Saito, A.; Matsui, H.; Suzuki, H.; Ohtsuka, S.; Shimosato, D.; Morishita, Y.; Watabe, T.; Niwa, H.; Miyazono, K. Activin-Nodal signaling is involved in propagation of mouse embryonic stem cells. J. Cell. Sci. 2007, 120, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Greber, B.; Lehrach, H.; Adjaye, J. Control of early fate decisions in human ES cells by distinct states of TGFbeta pathway activity. Stem Cells Dev. 2008, 17, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Ying, Q.L.; Nichols, J.; Chambers, I.; Smith, A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 2003, 115, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Raya, Á.; Kawakami, Y.; Morita, M.; Matsui, T.; Nakashima, K.; Gage, F.H.; Rodríguez-Esteban, C.; Izpisúa Belmonte, J.C. Nanog binds to Smad1 and blocks bone morphogenetic protein-induced differentiation of embryonic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 10294–10299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Fei, T.; Zhang, J.; Zhu, G.; Wang, L.; Lu, D.; Chi, X.; Teng, Y.; Hou, N.; Yang, X.; et al. BMP4 Signaling Acts via dual-specificity phosphatase 9 to control ERK activity in mouse embryonic stem cells. Cell Stem Cell 2012, 10, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Chen, X.; Li, D.S.; Li, R.; Addicks, G.C.; Glennon, C.; Zwaka, T.P.; Thomson, J.A. BMP4 initiates human embryonic stem cell differentiation to trophoblast. Nat. Biotechnol. 2002, 20, 1261–1264. [Google Scholar] [CrossRef]
- Xu, R.; Peck, R.M.; Li, D.S.; Feng, X.; Ludwig, T.; Thomson, J.A. Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat. Methods 2005, 2, 185–190. [Google Scholar] [CrossRef]
- Zhang, P.; Li, J.; Tan, Z.; Wang, C.; Liu, T.; Chen, L.; Yong, J.; Jiang, W.; Sun, X.; Du, L.; et al. Short-term BMP-4 treatment initiates mesoderm induction in human embryonic stem cells. Blood 2008, 111, 1933–1941. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, A.S.; Faial, T.; Gardner, L.; Niakan, K.K.; Ortmann, D.; Senner, C.E.; Callery, E.M.; Trotter, M.W.; Hemberger, M.; Smith, J.C.; et al. BRACHYURY and CDX2 mediate BMP-induced differentiation of human and mouse pluripotent stem cells into embryonic and extraembryonic lineages. Cell Stem Cell 2011, 9, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Lifantseva, N.V.; Kol’tsova, A.M.; Polianskaia, G.G.; Gordeeva, O.F. Expression of TGFbeta family factors and FGF2 in mouse and human embryonic stem cells maintained in different culture systems. Ontogenez 2013, 44, 10–23. [Google Scholar]
- Toyooka, Y.; Shimosato, D.; Murakami, K.; Takahashi, K.; Niwa, H. Identification and characterization of subpopulations in undifferentiated ES cell culture. Development 2008, 135, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Barbacioru, C.; Bao, S.; Lee, C.; Nordman, E.; Wang, X.; Lao, K.; Surani, M.A. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell 2010, 6, 468–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, K.M.; Lim, B. A precarious balance: Pluripotency factors as lineage specifiers. Cell Stem Cell 2011, 8, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, C.; Ying, Q. Gbx2, a LIF/Stat3 target, promotes reprogramming to and retention of the pluripotent ground state. J. Cell. Sci. 2013, 126, 1093–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, S.; Li, P.; Tong, C.; Ying, Q. Embryonic stem cell self-renewal pathways converge on the transcription factor Tfcp2l1. EMBO J. 2013, 32, 2548–2560. [Google Scholar] [CrossRef] [Green Version]
- Yeo, J.; Jiang, J.; Tan, Z.; Yim, G.; Ng, J.; Göke, J.; Kraus, P.; Liang, H.; Gonzales, K.A.U.; Chong, H.; et al. Klf2 is an essential factor that sustains ground state pluripotency. Cell Stem Cell 2014, 14, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.; Pereira, L.; Hoffman, J.A.; Shy, B.R.; Yuen, C.M.; Liu, D.R.; Merrill, B.J. Opposing effects of Tcf3 and Tcf1 control Wnt stimulation of embryonic stem cell self-renewal. Nat. Cell Biol. 2011, 13, 762–770. [Google Scholar] [CrossRef]
- Martello, G.; Sugimoto, T.; Diamanti, E.; Joshi, A.; Hannah, R.; Ohtsuka, S.; Göttgens, B.; Niwa, H.; Smith, A. Esrrb is a pivotal target of the Gsk3/Tcf3 axis regulating embryonic stem cell self-renewal. Cell Stem Cell 2012, 11, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Festuccia, N.; Osorno, R.; Halbritter, F.; Karwacki-Neisius, V.; Navarro, P.; Colby, D.; Wong, F.; Yates, A.; Tomlinson, S.R.; Chambers, I. Esrrb is a direct Nanog target gene that can substitute for Nanog function in pluripotent cells. Cell Stem Cell 2012, 11, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Wray, J.; Kalkan, T.; Smith, A.G. The ground state of pluripotency. Biochem. Soc. Trans. 2010, 38, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Yang, J.; Nichols, J.; Hall, J.S.; Eyres, I.; Mansfield, W.; Smith, A. Klf4 reverts developmentally programmed restriction of ground state pluripotency. Development 2009, 136, 1063–1069. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.; Guo, G.; Wray, J.; Eyres, I.; Nichols, J.; Grotewold, L.; Morfopoulou, S.; Humphreys, P.; Mansfield, W.; Walker, R.; et al. Oct4 and LIF/Stat3 additively induce Krüppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell 2009, 5, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Nichols, J.; Theunissen, T.W.; Guo, G.; van Oosten, A.L.; Barrandon, O.; Wray, J.; Yamanaka, S.; Chambers, I.; Smith, A. Nanog is the gateway to the pluripotent ground state. Cell 2009, 138, 722–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, J.; Cheng, A.W.; Saha, K.; Kim, J.; Lengner, C.J.; Soldner, F.; Cassady, J.P.; Muffat, J.; Carey, B.W.; Jaenisch, R. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc. Natl. Acad. Sci. USA 2010, 107, 9222–9227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashima, Y.; Guo, G.; Loos, R.; Nichols, J.; Ficz, G.; Krueger, F.; Oxley, D.; Santos, F.; Clarke, J.; Mansfield, W.; et al. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell 2014, 158, 1254–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theunissen, T.W.; Powell, B.E.; Wang, H.; Mitalipova, M.; Faddah, D.A.; Reddy, J.; Fan, Z.P.; Maetzel, D.; Ganz, K.; Shi, L.; et al. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell 2014, 15, 471–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.; Göke, J.; Ng, J.; Lu, X.; Gonzales, K.A.U.; Tan, C.; Tng, W.; Hong, Z.; Lim, Y.; Ng, H. Induction of a human pluripotent state with distinct regulatory circuitry that resembles preimplantation epiblast. Cell Stem Cell 2013, 13, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Gafni, O.; Weinberger, L.; Mansour, A.A.; Manor, Y.S.; Chomsky, E.; Ben-Yosef, D.; Kalma, Y.; Viukov, S.; Maza, I.; Zviran, A.; et al. Derivation of novel human ground state naive pluripotent stem cells. Nature 2013, 504, 282–286. [Google Scholar] [CrossRef]
- Valamehr, B.; Robinson, M.; Abujarour, R.; Rezner, B.; Vranceanu, F.; Le, T.; Medcalf, A.; Lee, T.T.; Fitch, M.; Robbins, D.; et al. Platform for induction and maintenance of transgene-free hiPSCs resembling ground state pluripotent stem cells. Stem Cell Rep. 2014, 2, 366–381. [Google Scholar] [CrossRef] [Green Version]
- Ware, C.B.; Nelson, A.M.; Mecham, B.; Hesson, J.; Zhou, W.; Jonlin, E.C.; Jimenez-Caliani, A.J.; Deng, X.; Cavanaugh, C.; Cook, S.; et al. Derivation of naive human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 4484–4489. [Google Scholar] [CrossRef] [Green Version]
- Duggal, G.; Warrier, S.; Ghimire, S.; Broekaert, D.; Van der Jeught, M.; Lierman, S.; Deroo, T.; Peelman, L.; Van Soom, A.; Cornelissen, R.; et al. Alternative Routes to Induce Naïve Pluripotency in Human Embryonic Stem Cells. Stem Cells 2015, 33, 2686–2698. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, X.; Yi, L.; Hou, Z.; Chen, J.; Kou, X.; Zhao, Y.; Wang, H.; Sun, X.; Jiang, C.; et al. Naïve Induced Pluripotent Stem Cells Generated From β-Thalassemia Fibroblasts Allow Efficient Gene Correction With CRISPR/Cas9. Stem Cells Transl. Med. 2016, 5, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, F.; Yong, J.; Zhang, P.; Hou, P.; Li, H.; Jiang, W.; Cai, J.; Liu, M.; Cui, K.; et al. Generation of induced pluripotent stem cells from adult rhesus monkey fibroblasts. Cell Stem Cell 2008, 3, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Resnick, J.L.; Bixler, L.S.; Cheng, L.; Donovan, P.J. Long-term proliferation of mouse primordial germ cells in culture. Nature 1992, 359, 550–551. [Google Scholar] [CrossRef]
- Shamblott, M.J.; Axelman, J.; Wang, S.; Bugg, E.M.; Littlefield, J.W.; Donovan, P.J.; Blumenthal, P.D.; Huggins, G.R.; Gearhart, J.D. Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc. Natl. Acad. Sci. USA 1998, 95, 13726–13731. [Google Scholar] [CrossRef] [Green Version]
- Shamblott, M.J.; Axelman, J.; Littlefield, J.W.; Blumenthal, P.D.; Huggins, G.R.; Cui, Y.; Cheng, L.; Gearhart, J.D. Human embryonic germ cell derivatives express a broad range of developmentally distinct markers and proliferate extensively in vitro. Proc. Natl. Acad. Sci. USA 2001, 98, 113–118. [Google Scholar] [CrossRef]
- McLaren, A.; Durcova-Hills, G. Germ cells and pluripotent stem cells in the mouse. Reprod. Fertil. Dev. 2001, 13, 661–664. [Google Scholar] [CrossRef]
- Durcova-Hills, G.; Wianny, F.; Merriman, J.; Zernicka-Goetz, M.; McLaren, A. Developmental fate of embryonic germ cells (EGCs), in vivo and in vitro. Differentiation 2003, 71, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Turnpenny, L.; Brickwood, S.; Spalluto, C.M.; Piper, K.; Cameron, I.T.; Wilson, D.I.; Hanley, N.A. Derivation of human embryonic germ cells: An alternative source of pluripotent stem cells. Stem Cells 2003, 21, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004, 430, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Pauklin, S.; Vallier, L. The cell-cycle state of stem cells determines cell fate propensity. Cell 2013, 155, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaki-Yumoto, M.; Liu, J.; Ramalho-Santos, M.; Yoshida, N.; Derynck, R. Smad2 is essential for maintenance of the human and mouse primed pluripotent stem cell state. J. Biol. Chem. 2013, 288, 18546–18560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005, 23, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Gadue, P.; Huber, T.L.; Paddison, P.J.; Keller, G.M. Wnt and TGF-beta signaling are required for the induction of an in vitro model of primitive streak formation using embryonic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16806–16811. [Google Scholar] [CrossRef] [Green Version]
- Teo, A.K.K.; Ali, Y.; Wong, K.Y.; Chipperfield, H.; Sadasivam, A.; Poobalan, Y.; Tan, E.K.; Wang, S.T.; Abraham, S.; Tsuneyoshi, N.; et al. Activin and BMP4 synergistically promote formation of definitive endoderm in human embryonic stem cells. Stem Cells 2012, 30, 631–642. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Xi, Q.; Wang, Z.; Zaromytidou, A.; Zhang, X.H.; Chow-Tsang, L.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-Todorova, K.; Kaartinen, V.; et al. A poised chromatin platform for TGF-β access to master regulators. Cell 2011, 147, 1511–1524. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Yoon, S.; Chuong, E.; Oyolu, C.; Wills, A.E.; Gupta, R.; Baker, J. Chromatin and transcriptional signatures for Nodal signaling during endoderm formation in hESCs. Dev. Biol. 2011, 357, 492–504. [Google Scholar] [CrossRef] [Green Version]
- Chia, C.Y.; Madrigal, P.; Denil, S.L.I.J.; Martinez, I.; Garcia-Bernardo, J.; El-Khairi, R.; Chhatriwala, M.; Shepherd, M.H.; Hattersley, A.T.; Dunn, N.R.; et al. GATA6 Cooperates with EOMES/SMAD2/3 to Deploy the Gene Regulatory Network Governing Human Definitive Endoderm and Pancreas Formation. Stem Cell Rep. 2019, 12, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, Z.; Teo, A.; Pedersen, R.A.; Vallier, L. SIP1 mediates cell-fate decisions between neuroectoderm and mesendoderm in human pluripotent stem cells. Cell Stem Cell 2010, 6, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, L.C.; Little, C.C. Spontaneous Testicular Teratomas in an Inbred Strain of Mice. Proc. Natl. Acad. Sci. USA 1954, 40, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinsmith, L.J.; Pierce, G.B.J. Multipotentiality of single embryonal carcinoma cells. Cancer Res. 1964, 24, 1544–1551. [Google Scholar] [PubMed]
- Solter, D.; Skreb, N.; Damjanov, I. Extrauterine growth of mouse egg-cylinders results in malignant teratoma. Nature 1970, 227, 503–504. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.C. The development of teratomas from intratesticular grafts of tubal mouse eggs. Development 1968, 20, 329–341. [Google Scholar]
- Stevens, L.C.; Varnum, D.S. The development of teratomas from parthenogenetically activated ovarian mouse eggs. Dev. Biol. 1974, 37, 369–380. [Google Scholar] [CrossRef]
- Zeuthen, J.; Nørgaard, J.O.; Avner, P.; Fellous, M.; Wartiovaara, J.; Vaheri, A.; Rosén, A.; Giovanella, B.C. Characterization of a human ovarian teratocarcinoma-derived cell line. Int. J. Cancer 1980, 25, 19–32. [Google Scholar] [CrossRef]
- McBurney, M.W.; Rogers, B.J. Isolation of male embryonal carcinoma cells and their chromosome replication patterns. Dev. Biol. 1982, 89, 503–508. [Google Scholar] [CrossRef]
- Andrews, P.W.; Damjanov, I.; Simon, D.; Banting, G.S.; Carlin, C.; Dracopoli, N.C.; Føgh, J. Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2. Differentiation in vivo and in vitro. Lab. Invest. 1984, 50, 147–162. [Google Scholar]
- Andrews, P.W. Human teratocarcinomas. Biochim. Biophys. Acta 1988, 948, 17–36. [Google Scholar] [CrossRef]
- Teshima, S.; Shimosato, Y.; Hirohashi, S.; Tome, Y.; Hayashi, I.; Kanazawa, H.; Kakizoe, T. Four new human germ cell tumor cell lines. Lab. Invest. 1988, 59, 328–336. [Google Scholar] [PubMed]
- Pera, M.F.; Cooper, S.; Mills, J.; Parrington, J.M. Isolation and characterization of a multipotent clone of human embryonal carcinoma cells. Differentiation 1989, 42, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Breuer, B.; Steuer, B.; Fischer, J. The F9-EC cell line as a model for the analysis of differentiation. Int. J. Dev. Biol. 1991, 35, 389–397. [Google Scholar]
- Wang, N.; Trend, B.; Bronson, D.L.; Fraley, E.E. Nonrandom abnormalities in chromosome 1 in human testicular cancers. Cancer Res. 1980, 40, 796–802. [Google Scholar]
- Tainsky, M.A.; Cooper, C.S.; Giovanella, B.C.; Vande Woude, G.F. An activated rasN gene: Detected in late but not early passage human PA1 teratocarcinoma cells. Science 1984, 225, 643–645. [Google Scholar] [CrossRef]
- Tainsky, M.A.; Shamanski, F.; Blair, D.; Giovanella, B.C. Causal role for an activated N-ras oncogene in the induction of tumorigenicity acquired by a human cell line. Cancer Res. 1987, 47, 3235–3238. [Google Scholar]
- Wang, L.C.; Vass, W.; Gao, C.L.; Chang, K.S. Amplification and enhanced expression of the c-Ki-ras2 protooncogene in human embryonal carcinomas. Cancer Res. 1987, 47, 4192–4198. [Google Scholar]
- Jakobovits, A.; Schwab, M.; Bishop, J.M.; Martin, G.R. Expression of N-myc in teratocarcinoma stem cells and mouse embryos. Nature 1985, 318, 188–191. [Google Scholar] [CrossRef]
- Tobaly-Tapiero, J.; Saal, F.; Peries, J.; Emanoil-Ravier, R. Amplification and rearrangement of Ki-ras oncogene in human teratocarcinoma-derived cell lines. Biochimie 1986, 68, 1019–1023. [Google Scholar] [CrossRef]
- Brinster, R.L. The effect of cells transferred into the mouse blastocyst on subsequent development. J. Exp. Med. 1974, 140, 1049–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaioannou, V.E.; McBurney, M.W.; Gardner, R.L.; Evans, M.J. Fate of teratocarcinoma cells injected into early mouse embryos. Nature 1975, 258, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Illmensee, K.; Mintz, B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc. Natl. Acad. Sci. USA 1976, 73, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaioannou, V.E.; Gardner, R.L.; McBurney, M.W.; Babinet, C.; Evans, M.J. Participation of cultured teratocarcinoma cells in mouse embryogenesis. Development 1978, 44, 93–104. [Google Scholar]
- Rossant, J.; McBurney, M.W. The developmental potential of a euploid male teratocarcinoma cell line after blastocyst injection. Development 1982, 70, 99–112. [Google Scholar]
- Andrews, P.W.; Damjanov, I.; Simon, D.; Dignazio, M. A pluripotent human stem-cell clone isolated from the TERA-2 teratocarcinoma line lacks antigens SSEA-3 and SSEA-4 in vitro, but expresses these antigens when grown as a xenograft tumor. Differentiation 1985, 29, 127–135. [Google Scholar] [CrossRef]
- Blelloch, R.H.; Hochedlinger, K.; Yamada, Y.; Brennan, C.; Kim, M.; Mintz, B.; Chin, L.; Jaenisch, R. Nuclear cloning of embryonal carcinoma cells. Proc. Natl. Acad. Sci. USA 2004, 101, 13985–13990. [Google Scholar]
- Gordeeva, O.F.; Nikonova, T.M. Development of experimental tumors formed by mouse and human embryonic stem and teratocarcinoma cells after subcutaneous and intraperitoneal transplantations into immunodeficient and immunocompetent mice. Cell Transplant. 2013, 22, 1901–1914. [Google Scholar] [CrossRef]
- Andrews, P.W. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev. Biol. 1984, 103, 285–293. [Google Scholar] [CrossRef]
- Gordeeva, O.F. Normal and pathological development of pluripotent stem cells. J. Stem Cells 2011, 6, 129–154. [Google Scholar]
- Josephson, R.; Ording, C.J.; Liu, Y.; Shin, S.; Lakshmipathy, U.; Toumadje, A.; Love, B.; Chesnut, J.D.; Andrews, P.W.; Rao, M.S.; et al. Qualification of embryonal carcinoma 2102Ep as a reference for human embryonic stem cell research. Stem Cells 2007, 25, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Lin, G.; Deng, L.; Lu, G. Tumourigenic characteristics of embryonal carcinoma cells as a model for studying tumour progression of human embryonic stem cells. Cell Prolif. 2012, 45, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Chaerkady, R.; Kerr, C.L.; Kandasamy, K.; Marimuthu, A.; Gearhart, J.D.; Pandey, A. Comparative proteomics of human embryonic stem cells and embryonal carcinoma cells. Proteomics 2010, 10, 1359–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lifantseva, N.; Koltsova, A.; Krylova, T.; Yakovleva, T.; Poljanskaya, G.; Gordeeva, O. Expression patterns of cancer-testis antigens in human embryonic stem cells and their cell derivatives indicate lineage tracks. Stem Cells Int. 2011, 2011, 795239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordeeva, O.; Gordeev, A.; Khaydukov, S. Expression dynamics of Mage family genes during self-renewal and differentiation of mouse pluripotent stem and teratocarcinoma cells. Oncotarget 2019, 10, 3248–3266. [Google Scholar]
- Sperger, J.M.; Chen, X.; Draper, J.S.; Antosiewicz, J.E.; Chon, C.H.; Jones, S.B.; Brooks, J.D.; Andrews, P.W.; Brown, P.O.; Thomson, J.A. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 13350–13355. [Google Scholar] [CrossRef] [Green Version]
- Skotheim, R.I.; Lind, G.E.; Monni, O.; Nesland, J.M.; Abeler, V.M.; Fosså, S.D.; Duale, N.; Brunborg, G.; Kallioniemi, O.; Andrews, P.W.; et al. Differentiation of human embryonal carcinomas in vitro and in vivo reveals expression profiles relevant to normal development. Cancer Res. 2005, 65, 5588–5598. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Peterson, H.; Chavez, L.; Kahlem, P.; Lehrach, H.; Vilo, J.; Adjaye, J. A data integration approach to mapping OCT4 gene regulatory networks operative in embryonic stem cells and embryonal carcinoma cells. PLoS ONE 2010, 5, e10709. [Google Scholar] [CrossRef]
- International Stem Cell Initiative; Amps, K.; Andrews, P.W.; Anyfantis, G.; Armstrong, L.; Avery, S.; Baharvand, H.; Baker, J.; Baker, D.; Munoz, M.B.; et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011, 29, 1132–1144. [Google Scholar]
- Van Puijenbroek, A.A.; van der Saag, P.T.; Coffer, P.J. Cytokine signal transduction in P19 embryonal carcinoma cells: Regulation of Stat3-mediated transactivation occurs independently of p21ras-Erk signaling. Exp. Cell Res. 1999, 251, 465–476. [Google Scholar] [CrossRef]
- Schuringa, J.J.; van der Schaaf, S.; Vellenga, E.; Eggen, B.J.L.; Kruijer, W. LIF-induced STAT3 signaling in murine versus human embryonal carcinoma (EC) cells. Exp. Cell Res. 2002, 274, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawazoe, S.; Ikeda, N.; Miki, K.; Shibuya, M.; Morikawa, K.; Nakano, S.; Oshimura, M.; Hisatome, I.; Shirayoshi, Y. Extrinsic factors derived from mouse embryonal carcinoma cell lines maintain pluripotency of mouse embryonic stem cells through a novel signal pathway. Dev. Growth Differ. 2009, 51, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordeeva, O.F.; Nikonova, T.M.; Lifantseva, N.V. Regulation of in vitro and in vivo differentiation of mouse embryonic stem cells, embryonic germ cells, and teratocarcinoma cells by TGFb family signaling factors. Ontogenez 2009, 40, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.S.; Brown, M.A.; Hilton, D.; Gough, N.M.; Sleigh, M.J. Inhibition of differentiation in a murine F9 embryonal carcinoma cell subline by leukemia inhibitory factor (LIF). Growth Factors 1992, 7, 41–52. [Google Scholar] [CrossRef]
- Hirayoshi, K.; Tsuru, A.; Yamashita, M.; Tomida, M.; Yamamoto-Yamaguchi, Y.; Yasukawa, K.; Hozumi, M.; Goeddel, D.V.; Nagata, K. Both D factor/LIF and IL-6 inhibit the differentiation of mouse teratocarcinoma F9 cells. FEBS Lett. 1991, 282, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Pacherník, J.; Bryja, V.; Esner, M.; Hampl, A.; Dvorák, P. Retinoic acid-induced neural differentiation of P19 embryonal carcinoma cells is potentiated by leukemia inhibitory factor. Physiol. Res. 2005, 54, 257–262. [Google Scholar]
- Pacherník, J.; Horváth, V.; Kubala, L.; Dvorák, P.; Kozubík, A.; Hampl, A. Neural differentiation potentiated by the leukaemia inhibitory factor through STAT3 signalling in mouse embryonal carcinoma cells. Folia Biol. 2007, 53, 157–163. [Google Scholar]
- Bastien, J.; Plassat, J.; Payrastre, B.; Rochette-Egly, C. The phosphoinositide 3-kinase/Akt pathway is essential for the retinoic acid-induced differentiation of F9 cells. Oncogene 2006, 25, 2040–2047. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Shin, S.Y.; Kim, S.; Choo, J.; Lee, Y.H. Suppression of PTEN expression during aggregation with retinoic acid in P19 mouse embryonal carcinoma cells. Biochem. Biophys. Res. Commun. 2006, 347, 715–722. [Google Scholar] [CrossRef]
- Lin, Y.; Yang, Y.; Li, W.; Chen, Q.; Li, J.; Pan, X.; Zhou, L.; Liu, C.; Chen, C.; He, J.; et al. Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol. Cell 2012, 48, 627–640. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Xue, Z.; Yang, G.; Shi, B.; Yang, B.; Yan, Y.; Wang, X.; Han, D.; Huang, Y.; Dong, W. Akt-signal integration is involved in the differentiation of embryonal carcinoma cells. PLoS ONE 2013, 8, e64877. [Google Scholar] [CrossRef] [PubMed]
- Gordeeva, O.F.; Lifantseva, N.V.; Khaĭdukov, S.V. Expression patterns of germ line specific genes in mouse and human pluripotent stem cells are associated with regulation of ground and primed state of pluripotency. Ontogenez 2011, 42, 403–424. [Google Scholar] [CrossRef]
- Mummery, C.L.; van den Eijnden-van Raaij, A.J. Type beta transforming growth factors and activins in differentiating embryonal carcinoma cells, embryonic stem cells and early embryonic development. Int. J. Dev. Biol. 1993, 37, 169–182. [Google Scholar]
- Gordeeva, O.F. Low expression of activin A in mouse and human embryonic teratocarcinoma cells. Ontogenez 2014, 45, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.F.; Khoury, R.H.; Smith, P.C.; McConnell, D.S.; Padmanahban, V.; Midgley, A.R.J.; Schneyer, A.L.; Crowley, W.F.J.; Sluss, P.M. A two-site monoclonal antibody immunoradiometric assay for human follistatin: Secretion by a human ovarian teratocarcinoma-derived cell line (PA-1). J. Clin. Endocrinol. Metab. 1996, 81, 1434–1441. [Google Scholar] [PubMed] [Green Version]
- Delbaere, A.; Sidis, Y.; Schneyer, A.L. Differential response to exogenous and endogenous activin in a human ovarian teratocarcinoma-derived cell line (PA-1): Regulation by cell surface follistatin. Endocrinology 1999, 140, 2463–2470. [Google Scholar] [CrossRef]
- Rizzino, A. Appearance of high affinity receptors for type beta transforming growth factor during differentiation of murine embryonal carcinoma cells. Cancer Res. 1987, 47, 4386–4390. [Google Scholar]
- van den Eijnden-van Raaij, A.J.; Feijen, A.; Lawson, K.A.; Mummery, C.L. Differential expression of inhibin subunits and follistatin, but not of activin receptor type II, during early murine embryonic development. Dev. Biol. 1992, 154, 356–365. [Google Scholar] [CrossRef]
- de Jong, F.H.; de Winter, J.P.; Wesseling, J.G.; Timmerman, M.A.; van Genesen, S.; van den Eijnden-van Raaij, A.J.; van Zoelen, E.J. Inhibin subunits, follistatin and activin receptors in the human teratocarcinoma cell line Tera-2. Biochem. Biophys. Res. Commun. 1993, 192, 1334–1339. [Google Scholar] [CrossRef]
- Mern, D.S.; Hasskarl, J.; Burwinkel, B. Inhibition of Id proteins by a peptide aptamer induces cell-cycle arrest and apoptosis in ovarian cancer cells. Br. J. Cancer 2010, 103, 1237–1244. [Google Scholar] [CrossRef] [Green Version]
- Monzen, K.; Shiojima, I.; Hiroi, Y.; Kudoh, S.; Oka, T.; Takimoto, E.; Hayashi, D.; Hosoda, T.; Habara-Ohkubo, A.; Nakaoka, T.; et al. Bone morphogenetic proteins induce cardiomyocyte differentiation through the mitogen-activated protein kinase kinase kinase TAK1 and cardiac transcription factors Csx/Nkx-2.5 and GATA-4. Mol. Cell. Biol. 1999, 19, 7096–7105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamali, M.; Karamboulas, C.; Rogerson, P.J.; Skerjanc, I.S. BMP signaling regulates Nkx2-5 activity during cardiomyogenesis. FEBS Lett. 2001, 509, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Du, Z.; Yao, Z. Roles of the Nanog protein in murine F9 embryonal carcinoma cells and their endoderm-differentiated counterparts. Cell Res. 2006, 16, 641–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Wang, H.; Pan, G.; Geng, Y.; Guo, Y.; Pei, D. Regulation of the pluripotency marker Rex-1 by Nanog and Sox2. J. Biol. Chem. 2006, 281, 23319–23325. [Google Scholar] [CrossRef] [Green Version]
- Gokhale, P.J.; Giesberts, A.M.; Andrews, P.W. Brachyury is expressed by human teratocarcinoma cells in the absence of mesodermal differentiation. Cell Growth Differ. 2000, 11, 157–162. [Google Scholar]
- Van der Heyden, M.A.G.; Defize, L.H.K. Twenty one years of P19 cells: What an embryonal carcinoma cell line taught us about cardiomyocyte differentiation. Cardiovasc. Res. 2003, 58, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Kelly, G.M.; Gatie, M.I. Mechanisms Regulating Stemness and Differentiation in Embryonal Carcinoma Cells. Stem Cells Int. 2017, 2017, 3684178. [Google Scholar] [CrossRef] [Green Version]
- van den Eijnden-van Raaij, A.J.; van Achterberg, T.A.; van der Kruijssen, C.M.; Piersma, A.H.; Huylebroeck, D.; de Laat, S.W.; Mummery, C.L. Differentiation of aggregated murine P19 embryonal carcinoma cells is induced by a novel visceral endoderm-specific FGF-like factor and inhibited by activin A. Mech. Dev. 1991, 33, 157–165. [Google Scholar] [CrossRef]
- Andrews, P.W.; Damjanov, I.; Berends, J.; Kumpf, S.; Zappavigna, V.; Mavilio, F.; Sampath, K. Inhibition of proliferation and induction of differentiation of pluripotent human embryonal carcinoma cells by osteogenic protein-1 (or bone morphogenetic protein-7). Lab. Invest. 1994, 71, 243–251. [Google Scholar]
- Pera, M.F.; Herszfeld, D. Differentiation of human pluripotent teratocarcinoma stem cells induced by bone morphogenetic protein-2. Reprod. Fertil. Dev. 1998, 10, 551–555. [Google Scholar] [CrossRef]
- Chadalavada, R.S.V.; Houldsworth, J.; Olshen, A.B.; Bosl, G.J.; Studer, L.; Chaganti, R.S.K. Transcriptional program of bone morphogenetic protein-2-induced epithelial and smooth muscle differentiation of pluripotent human embryonal carcinoma cells. Funct. Integr. Genomics 2005, 5, 59–69. [Google Scholar] [CrossRef] [PubMed]
- van der Kruijssen, C.M.; Feijen, A.; Huylebroeck, D.; van den Eijnden-van Raaij, A.J. Modulation of activin expression by type beta transforming growth factors. Exp. Cell Res. 1993, 207, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Gordeeva, O.; Khaydukov, S. Tumorigenic and Differentiation Potentials of Embryonic Stem Cells Depend on TGFβ Family Signaling: Lessons from Teratocarcinoma Cells Stimulated to Differentiate with Retinoic Acid. Stem Cells Int 2017, 2017, 7284872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.C.; Chae, C.B. Demethylation of somatic and testis-specific histone H2A and H2B genes in F9 embryonal carcinoma cells. Mol. Cell. Biol. 1993, 13, 5538–5548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teubner, B.; Schulz, W.A. Regulation of DNA methyltransferase during differentiation of F9 mouse embryonal carcinoma cells. J. Cell. Physiol. 1995, 165, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Frostesjö, L.; Holm, I.; Grahn, B.; Page, A.W.; Bestor, T.H.; Heby, O. Interference with DNA methyltransferase activity and genome methylation during F9 teratocarcinoma stem cell differentiation induced by polyamine depletion. J. Biol. Chem. 1997, 272, 4359–4366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb-Rinker, P.; Ly, D.; Jezierski, A.; Sikorska, M.; Walker, P.R. Sequential DNA methylation of the Nanog and Oct-4 upstream regions in human NT2 cells during neuronal differentiation. J. Biol. Chem. 2005, 280, 6257–6260. [Google Scholar] [CrossRef] [Green Version]
- Hatada, I.; Morita, S.; Kimura, M.; Horii, T.; Yamashita, R.; Nakai, K. Genome-wide demethylation during neural differentiation of P19 embryonal carcinoma cells. J. Hum. Genet. 2008, 53, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Shahhoseini, M.; Taei, A.; Mehrjardi, N.Z.; Salekdeh, G.H.; Baharvand, H. Epigenetic analysis of human embryonic carcinoma cells during retinoic acid-induced neural differentiation. Biochem. Cell Biol. 2010, 88, 527–538. [Google Scholar] [CrossRef]
- Abbey, D.; Seshagiri, P.B. Aza-induced cardiomyocyte differentiation of P19 EC-cells by epigenetic co-regulation and ERK signaling. Gene 2013, 526, 364–373. [Google Scholar] [CrossRef]
- Takahashi, K.; Mitsui, K.; Yamanaka, S. Role of ERas in promoting tumour-like properties in mouse embryonic stem cells. Nature 2003, 423, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Blum, B.; Benvenisty, N. The tumorigenicity of diploid and aneuploid human pluripotent stem cells. Cell Cycle 2009, 8, 3822–3830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, B.; Bar-Nur, O.; Golan-Lev, T.; Benvenisty, N. The anti-apoptotic gene survivin contributes to teratoma formation by human embryonic stem cells. Nat. Biotechnol. 2009, 27, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 2011, 11, 268–277. [Google Scholar] [CrossRef]
- Werbowetski-Ogilvie, T.E.; Bossé, M.; Stewart, M.; Schnerch, A.; Ramos-Mejia, V.; Rouleau, A.; Wynder, T.; Smith, M.; Dingwall, S.; Carter, T.; et al. Characterization of human embryonic stem cells with features of neoplastic progression. Nat. Biotechnol. 2009, 27, 91–97. [Google Scholar] [CrossRef]
- Kooreman, N.G.; Wu, J.C. Tumorigenicity of pluripotent stem cells: Biological insights from molecular imaging. J. R. Soc. Interface 2010, 7, S753–S763. [Google Scholar] [CrossRef] [Green Version]
- Brederlau, A.; Correia, A.S.; Anisimov, S.V.; Elmi, M.; Paul, G.; Roybon, L.; Morizane, A.; Bergquist, F.; Riebe, I.; Nannmark, U.; et al. Transplantation of human embryonic stem cell-derived cells to a rat model of Parkinson’s disease: Effect of in vitro differentiation on graft survival and teratoma formation. Stem Cells 2006, 24, 1433–1440. [Google Scholar] [CrossRef] [Green Version]
- Cooke, M.J.; Stojkovic, M.; Przyborski, S.A. Growth of teratomas derived from human pluripotent stem cells is influenced by the graft site. Stem Cells Dev. 2006, 15, 254–259. [Google Scholar] [CrossRef]
- Dressel, R. Effects of histocompatibility and host immune responses on the tumorigenicity of pluripotent stem cells. Semin. Immunopathol. 2011, 33, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Moore, E.E.; Moritz, E.A.; Mitra, N.S. A variant F9 embryonal carcinoma cell line which undergoes incomplete differentiation in retinoic acid. Cancer Res. 1985, 45, 4387–4396. [Google Scholar]
- Strickland, S.; Mahdavi, V. The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell 1978, 15, 393–403. [Google Scholar] [CrossRef]



© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordeeva, O. TGFβ Family Signaling Pathways in Pluripotent and Teratocarcinoma Stem Cells’ Fate Decisions: Balancing Between Self-Renewal, Differentiation, and Cancer. Cells 2019, 8, 1500. https://doi.org/10.3390/cells8121500
Gordeeva O. TGFβ Family Signaling Pathways in Pluripotent and Teratocarcinoma Stem Cells’ Fate Decisions: Balancing Between Self-Renewal, Differentiation, and Cancer. Cells. 2019; 8(12):1500. https://doi.org/10.3390/cells8121500
Chicago/Turabian StyleGordeeva, Olga. 2019. "TGFβ Family Signaling Pathways in Pluripotent and Teratocarcinoma Stem Cells’ Fate Decisions: Balancing Between Self-Renewal, Differentiation, and Cancer" Cells 8, no. 12: 1500. https://doi.org/10.3390/cells8121500
APA StyleGordeeva, O. (2019). TGFβ Family Signaling Pathways in Pluripotent and Teratocarcinoma Stem Cells’ Fate Decisions: Balancing Between Self-Renewal, Differentiation, and Cancer. Cells, 8(12), 1500. https://doi.org/10.3390/cells8121500