Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Culture of Cancer Cells
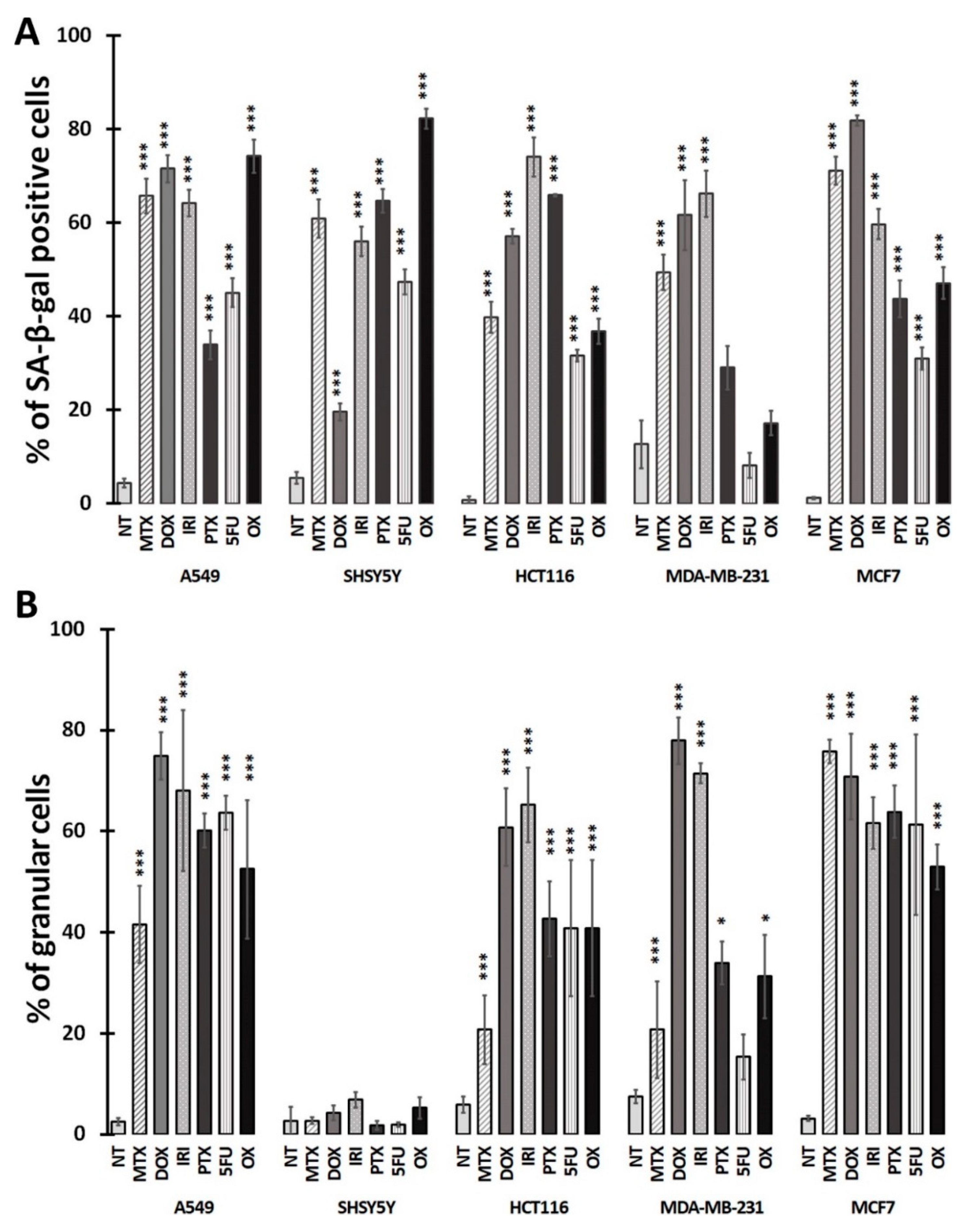
2.3. Quantification of Senescence-Associated β-galactosidase-Positive Cells
2.4. Measurement of Granularity
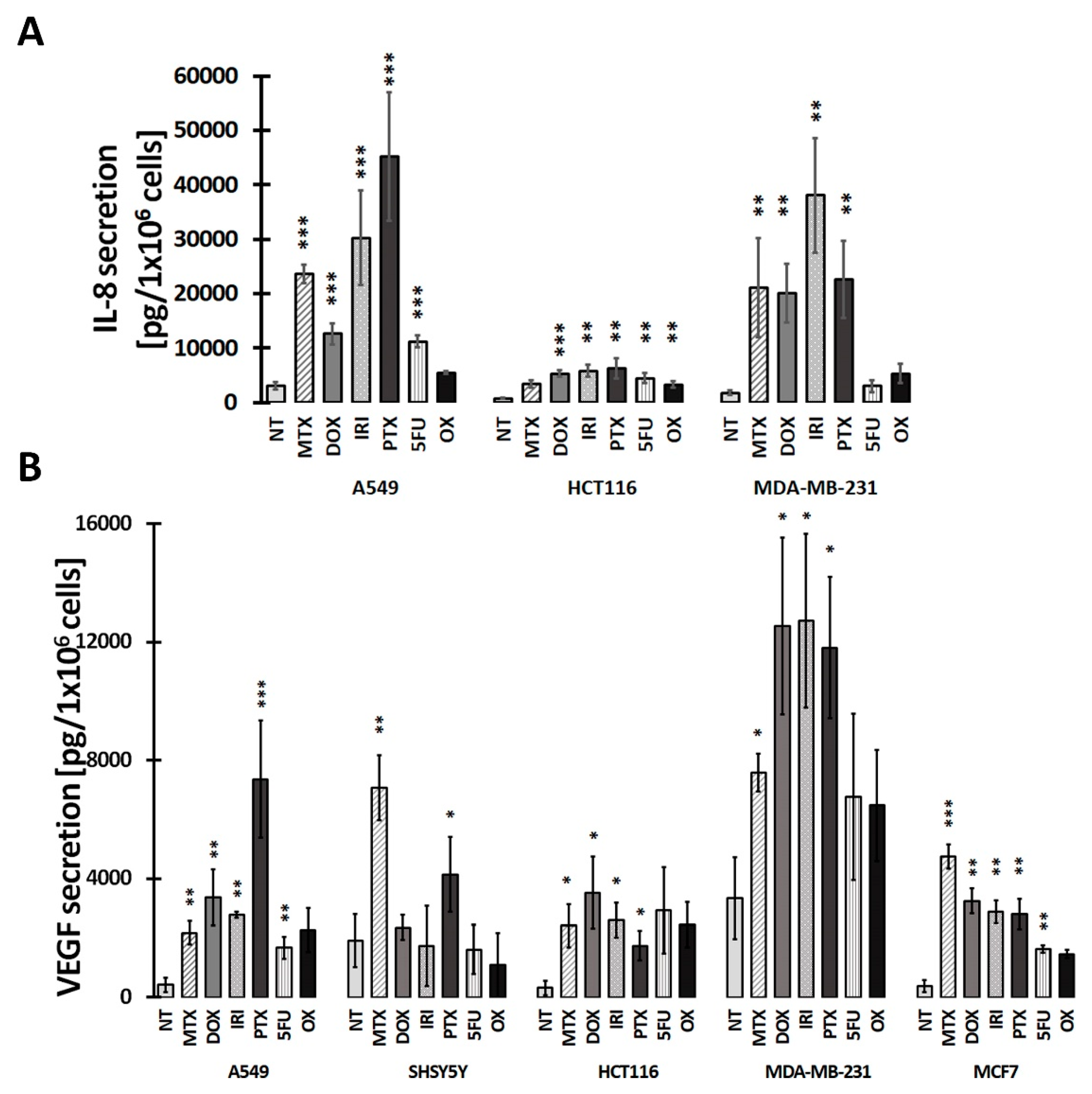
2.5. Measurements of Secreted Factors
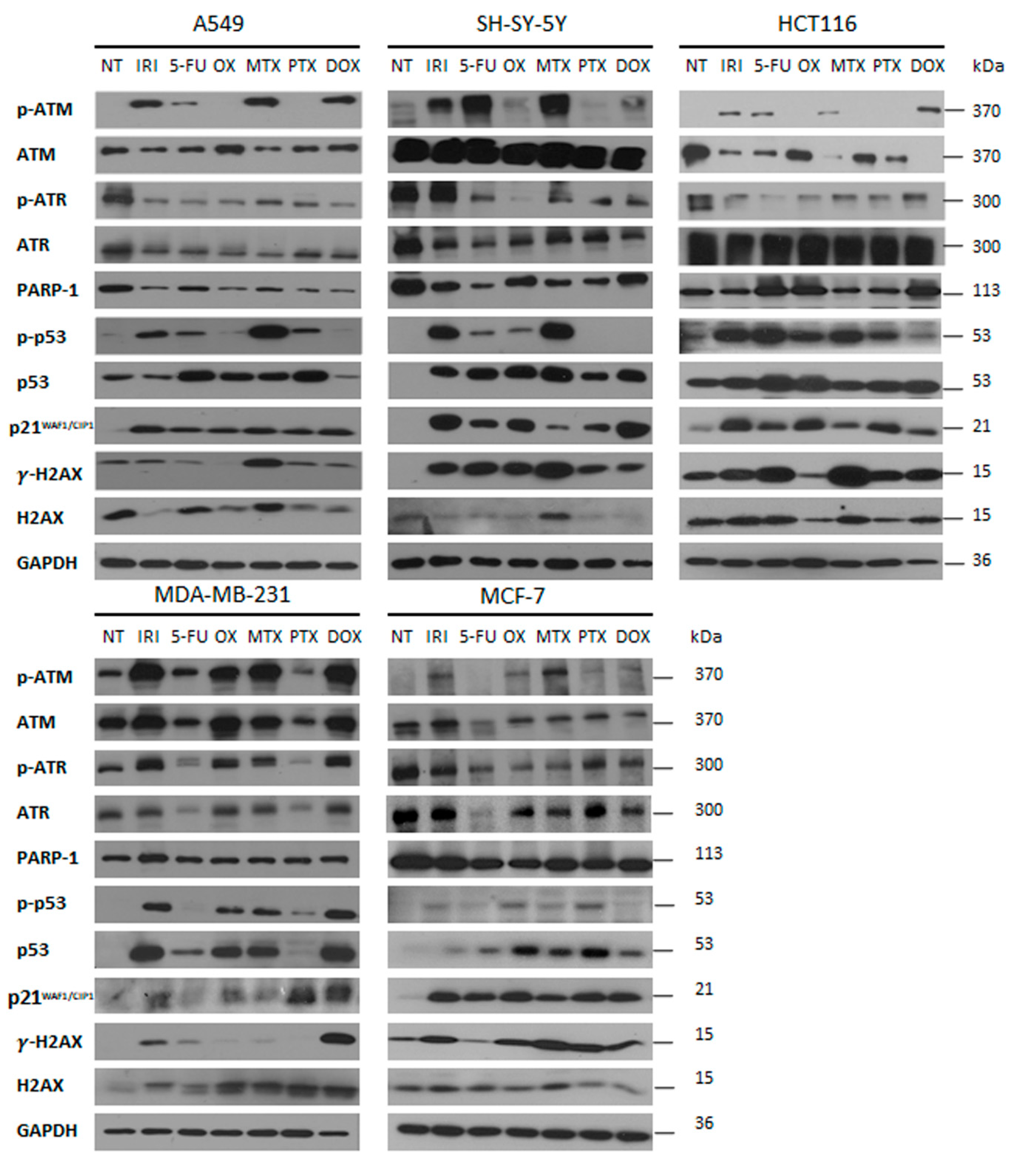
2.6. Western Blot Analysis
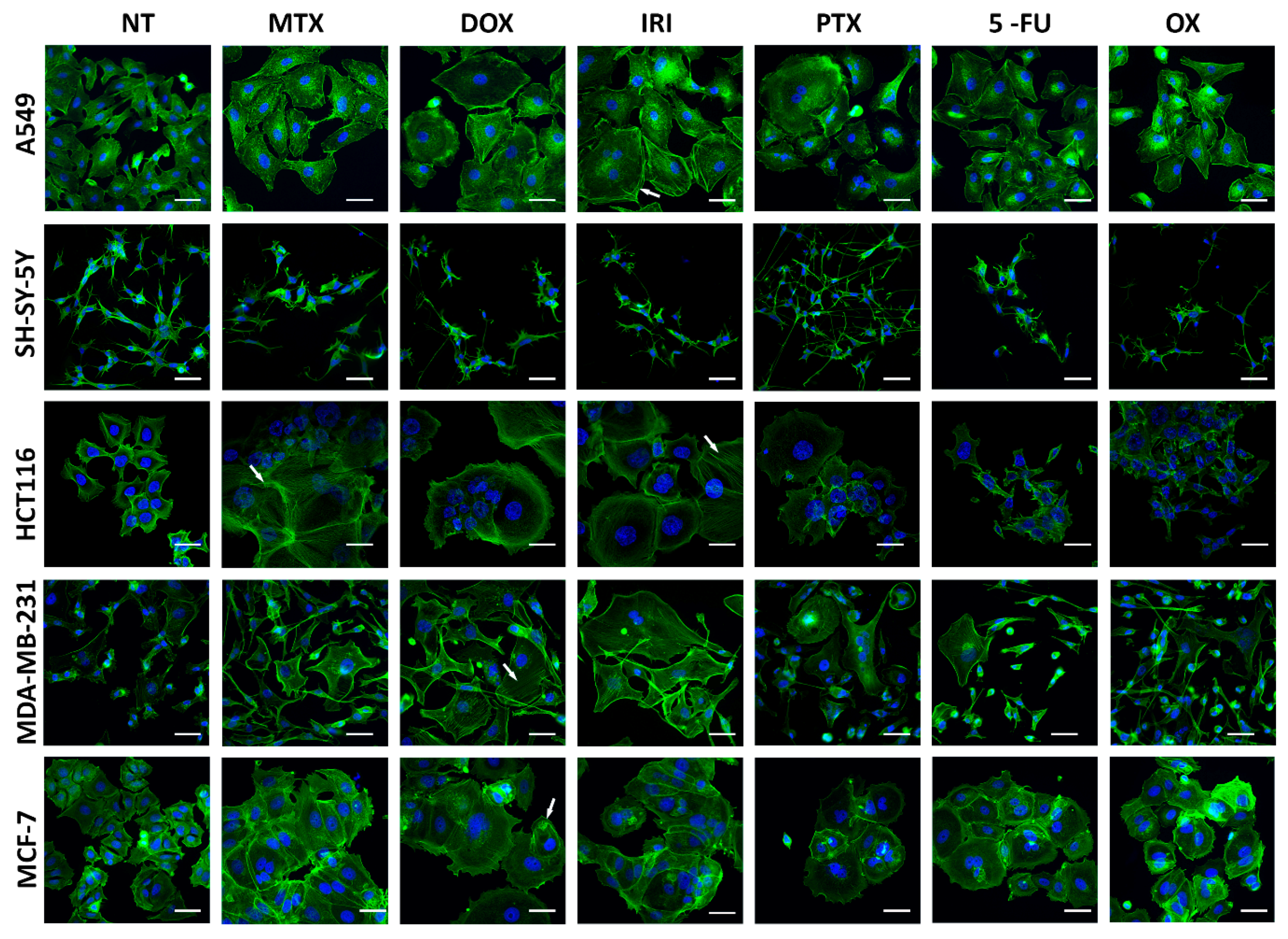
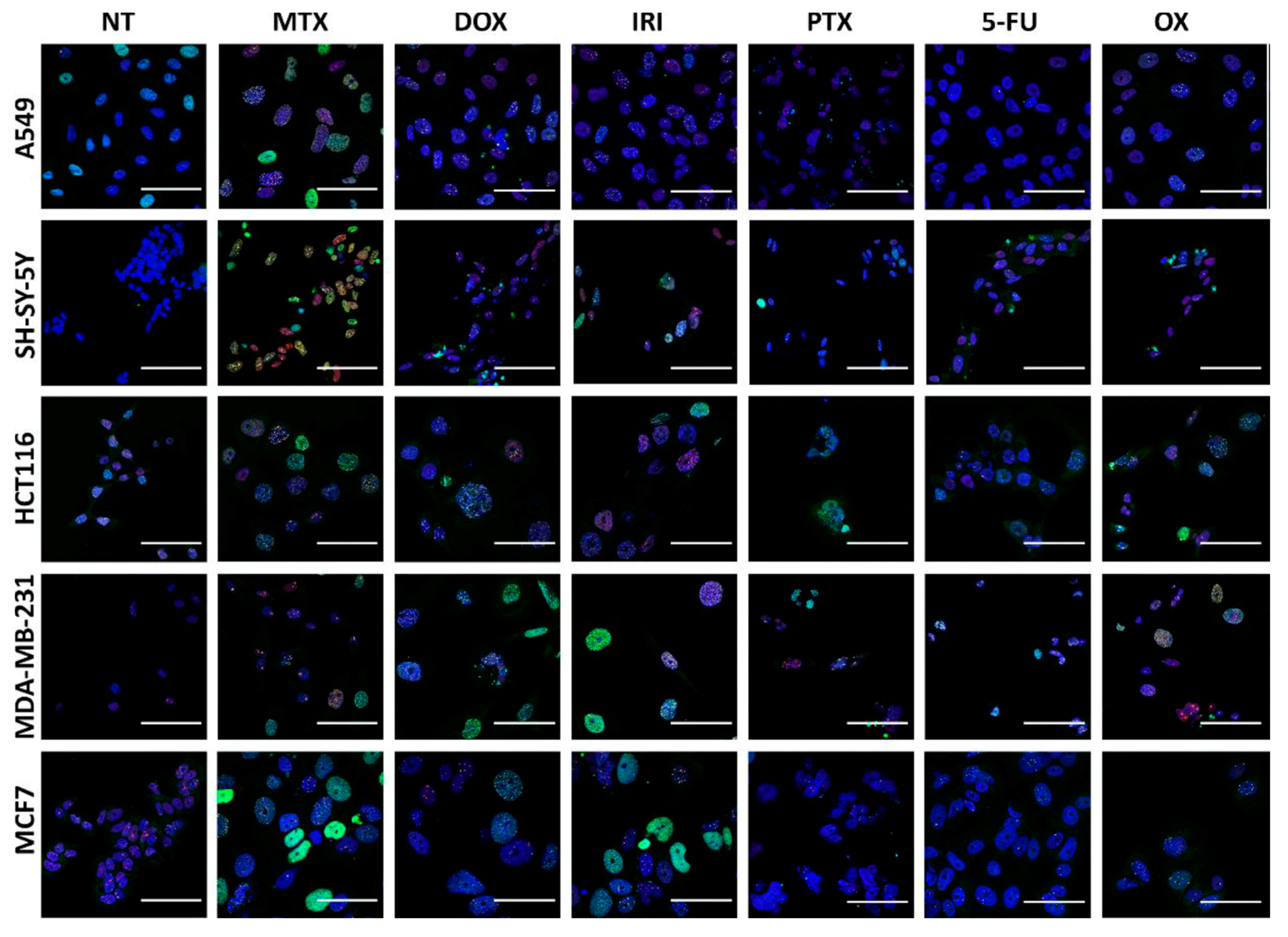
2.7. Immunocytochemistry
2.8. Statistical Analysis
3. Results
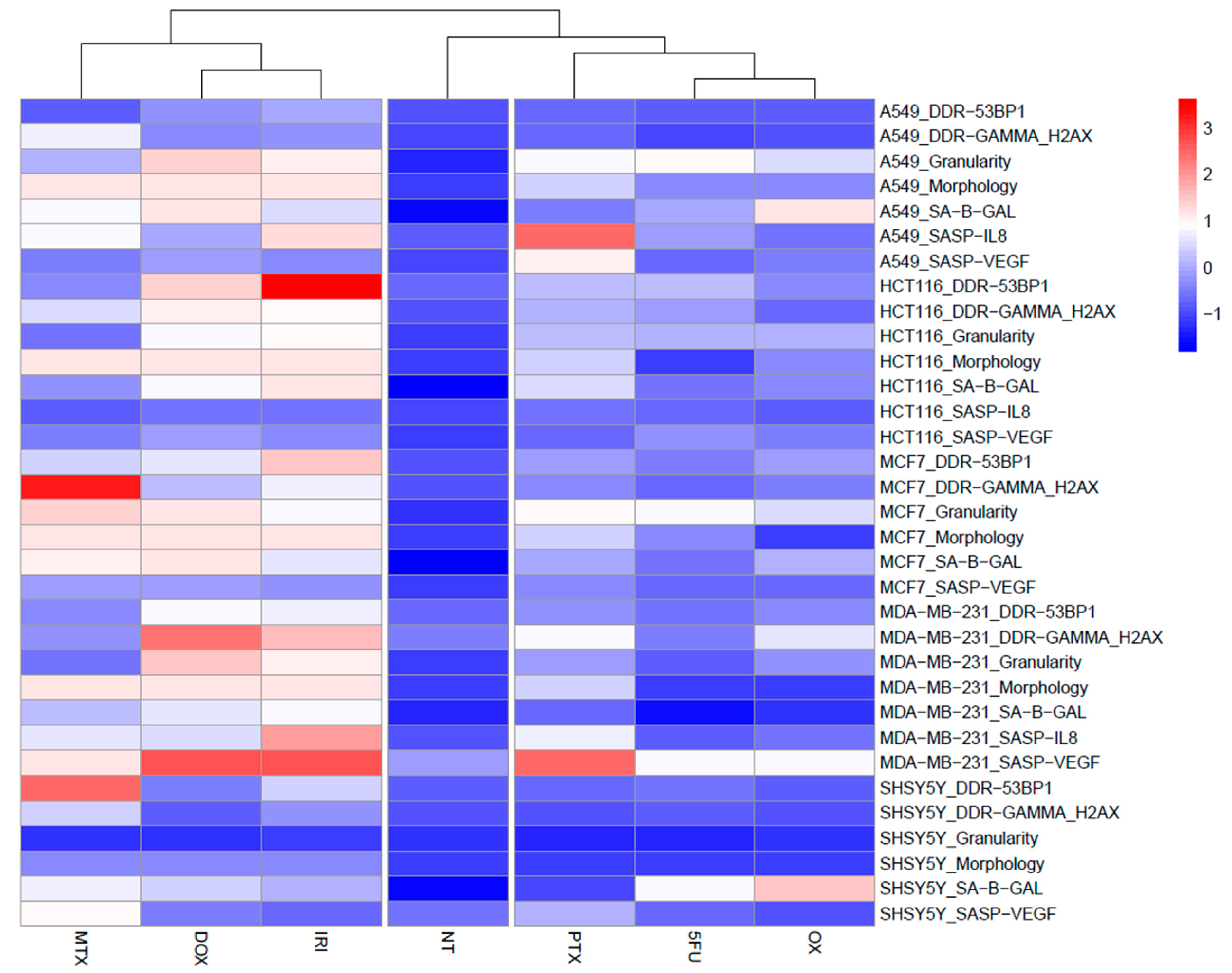
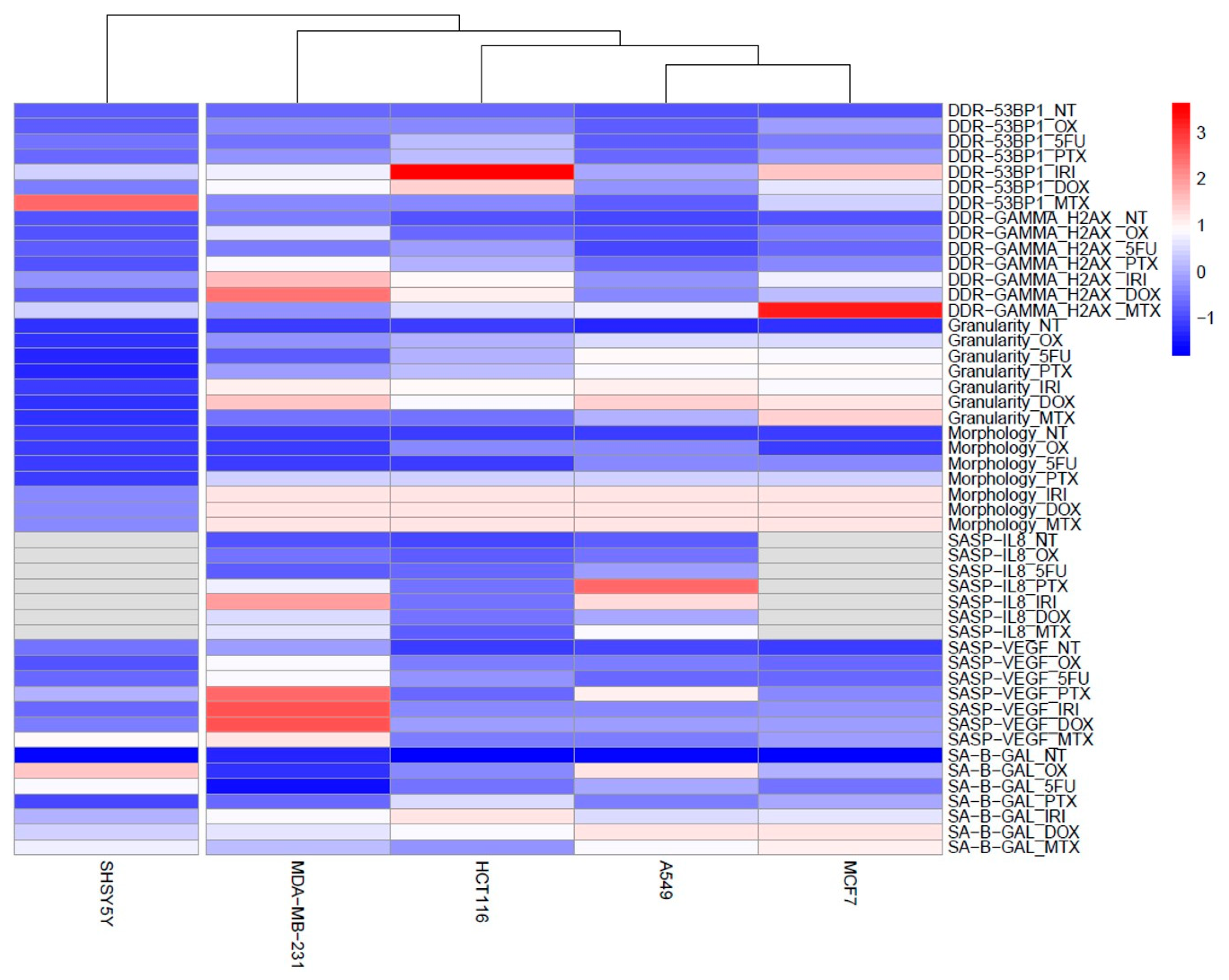
3.1. Cell Senescence Screening by SA-β-gal Activity and Cell Morphology
3.2. SASP in Therapy-Induced Senescence of Cancer Cells
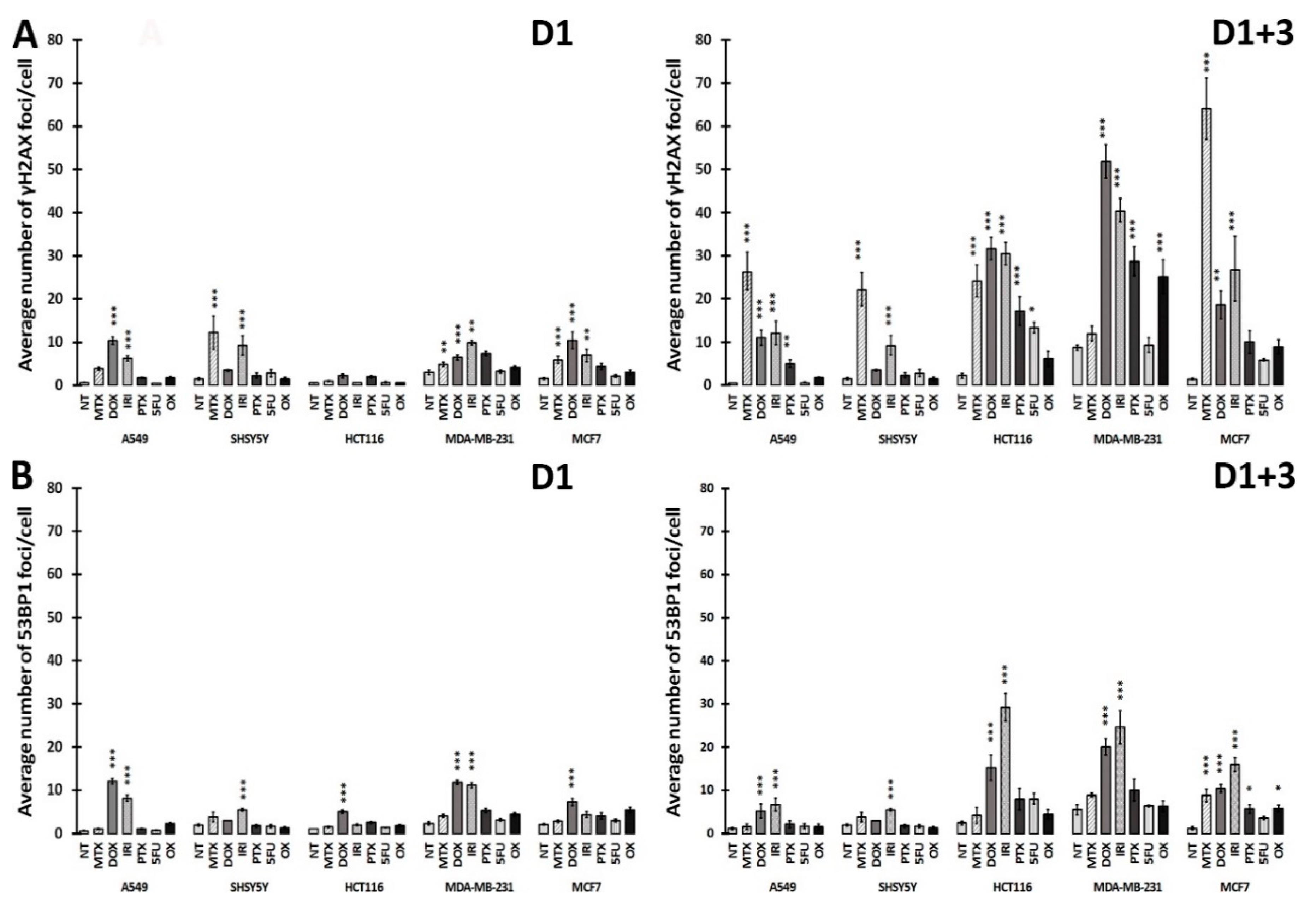
3.3. DNA Damage and DNA Damage Response (DDR) in Therapy-Induced Senescence
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chang, B.D.; Xuan, Y.; Broude, E.V.; Zhu, H.; Schott, B.; Fang, J.; Roninson, I.B. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 1999, 18, 4808–4818. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Gil, J. Senescence: A new weapon for cancer therapy. Trends Cell Biol. 2012, 22, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Mosieniak, G.; Sliwinska, M.A. Morphological and Functional Characteristic of Senescent Cancer Cells. Curr. Drug Targets 2016, 17, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front. Oncol. 2017, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Leite de Oliveira, R.; Wang, C.; Fernandes Neto, J.M.; Mainardi, S.; Evers, B.; Lieftink, C.; Morris, B.; Jochems, F.; Willemsen, L.; et al. High-Throughput Functional Genetic and Compound Screens Identify Targets for Senescence Induction in Cancer. Cell Rep. 2017, 21, 773–783. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Toussaint, O.; Medrano, E.E.; von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef]
- De Magalhaes, J.P.; Passos, J.F. Stress, cell senescence and organismal ageing. Mech. Ageing Dev. 2018, 170, 2–9. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Bielak-Zmijewska, A.; Mosieniak, G.; Sikora, E. Is DNA damage indispensable for stress-induced senescence? Mech. Ageing Dev. 2018, 170, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Strzeszewska, A.; Alster, O.; Mosieniak, G.; Ciolko, A.; Sikora, E. Insight into the role of PIKK family members and NF-κB in DNAdamage-induced senescence and senescence-associated secretory phenotype of colon cancer cells. Cell Death Dis. 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Bothmer, A.; Robbiani, D.F.; Di Virgilio, M.; Bunting, S.F.; Klein, I.A.; Feldhahn, N.; Barlow, J.; Chen, H.-T.; Bosque, D.; Callen, E.; et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol. Cell 2011, 42, 319–329. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.D.; Broude, E.V.; Fang, J.; Kalinichenko, T.V.; Abdryashitov, R.; Poole, J.C.; Roninson, I.B. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene 2000, 19, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Ferrandiz, N.; Caraballo, J.M.; Garcia-Gutierrez, L.; Devgan, V.; Rodriguez-Paredes, M.; Lafita, M.C.; Bretones, G.; Quintanilla, A.; Muñoz-Alonso, M.J.; Blanco, R.; et al. p21 as a transcriptional co-repressor of S-phase and mitotic control genes. PLoS ONE 2012, 7, e37759. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharm. Thera. 2015, 149, 124–138. [Google Scholar] [CrossRef]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2015, 128, 4255–4262. [Google Scholar] [CrossRef]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, T.J. Cellular senescence in cancer. BMB Rep. 2019, 52, 42–46. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.S. Cellular senescence: A promising strategy for cancer therapy. BMB Reports 2019, 52, 35–41. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. Targeting normal and cancer senescent cells as a strategy of senotherapy. Ageing Res. Rev. 2019, 55, 100941. [Google Scholar] [CrossRef]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends. Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef] [Green Version]
- Mosieniak, G.; Sliwinska, M.A.; Przybylska, D.; Grabowska, W.; Sunderland, P.; Bielak-Zmijewska, A.; Sikora, E. Curcumin-treated cancer cells show mitotic disturbances leading to growth arrest and induction of senescence phenotype. Int. J. Biochem. Cell Biol. 2016, 74, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Junk, D.J.; Vrba, L.; Watts, G.S.; Oshiro, M.M.; Martinez, J.D.; Futscher, B.W. Different mutant/wild-type p53 combinations cause a spectrum of increased invasive potential in nonmalignant immortalized human mammary epithelial cells. Neoplasia 2008, 10, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.H.; Gao, Y.; Moten, A.; Lin, H.K. Novel ARF/p53-independent senescence pathways in cancer repression. J. Mol. Med. 2011, 89, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieur, A.; Peeper, D.S. Cellular senescence in vivo: A barrier to tumorigenesis. Curr. Opin. Cell Biol. 2008, 20, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, E.; Palumbo, G.; de Boer, J.; Brady, H.J. Ataxia telangiectasia mutated and p21CIP1 modulate cell survival of drug-induced senescent tumor cells: Implications for chemotherapy. Clin. Cancer Res. 2008, 14, 1877–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korwek, Z.; Sewastianik, T.; Bielak-Zmijewska, A.; Mosieniak, G.; Alster, O.; Moreno-Villanueva, M.; Burkle, A.; Sikora, E. Inhibition of ATM blocks the etoposide-induced DNA damage response and apoptosis of resting human T cells. DNA Repair (Amst.) 2012, 11, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Wettersten, H.I.; Park, S.H.; Weiss, R.H. Small-molecule inhibitors of p21 as novel therapeutics for chemotherapy-resistant kidney cancer. Future Med. Chem. 2013, 5, 991–994. [Google Scholar] [CrossRef] [Green Version]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.-M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef]
- Lawless, C.; Wang, C.; Jurk, D.; Merz, A.; Zglinicki, T.; Passos, J.F. Quantitative assessment of markers for cell senescence. Exp. Gerontol 2010, 45, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Dis. 2017, 3, 17049. [Google Scholar] [CrossRef]
- Xie, L.; Zhao, T.; Cai, J.; Su, Y.; Wang, Z.; Dong, W. Methotrexate induces DNA damage and inhibits homologous recombination repair in choriocarcinoma cells. Onco.Targets Ther. 2016, 9, 7115–7122. [Google Scholar] [CrossRef] [Green Version]
- Dabrowska, M.; Uram, L.; Zielinski, Z.; Rode, W.; Sikora, E. Oxidative stress and inhibition of nitric oxide generation underlie methotrexate-induced senescence in human colon cancer cells. Mech. Ageing Dev. 2018, 170, 22–29. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Lee, J.J.; Schmitz, J.C. ATM Is Required for the Repair of Oxaliplatin-Induced DNA Damage in Colorectal Cancer. Clin Colorectal Cancer 2018, 17, 255–257. [Google Scholar] [CrossRef]
- Manfredi, J.J.; Horwitz, S.B. Taxol: An antimitotic agent with a new mechanism of action. Pharm. Thera. 1984, 25, 83–125. [Google Scholar] [CrossRef]
- Jakhar, R.; Luijten, M.N.H.; Wong, A.X.F.; Cheng, B.; Guo, K.; Neo, S.P.; Au, B.; Kulkarni, M.; Lim, K.J.; Maimaiti, J.; et al. Autophagy Governs Protumorigenic Effects of Mitotic Slippage-induced Senescence. Mol. Cancer Res. 2018, 16, 1625–1640. [Google Scholar] [CrossRef] [Green Version]
- Mirza-Aghazadeh-Attari, M.; Mohammadzadeh, A.; Yousefi, B.; Mihanfar, A.; Karimian, A.; Majidinia, M. 53BP1: A key player of DNA damage response with critical functions in cancer. DNA Repair (Amst.) 2019, 73, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johmura, Y.; Nakanishi, M. Multiple facets of p53 in senescence induction and maintenance. Cancer Sci. 2016, 107, 1550–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piechota, M.; Sunderland, P.; Wysocka, A.; Nalberczak, M.; Sliwinska, M.A.; Radwanska, K.; Sikora, E. Is senescence-associated beta-galactosidase a marker of neuronal senescence? Oncotarget 2016, 7, 81099–81109. [Google Scholar] [CrossRef] [PubMed]
- Sliwinska, M.A.; Mosieniak, G.; Wolanin, K.; Babik, A.; Piwocka, K.; Magalska, A.; Szczepanowska, J.; Fronk, J.; Sikora, E. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech. Ageing Dev. 2009, 130, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Czarnecka, J.; Kominek, A.; Barszcz, K.; Bernas, T.; Piwocka, K.; Kaminska, B. Some chemotherapeutics-treated colon cancer cells display a specific phenotype being a combination of stem-like and senescent cell features. Cancer Biol. Ther. 2018, 19, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.H.; Altschuler, S.J.; Wu, L.F. Patterns of Early p21 Dynamics Determine Proliferation-Senescence Cell Fate after Chemotherapy. Cell 2019, 178, 361–373. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Triana-Martinez, F.; Picallos-Rabina, P.; Da Silva-Alvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreiros, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Birch, J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Senescence Inducing Concentration | ||||
|---|---|---|---|---|---|
| A549 | SH-SY-5Y | HCT116 | MDA-MB-231 | MCF-7 | |
| MTX | 32 µM | 32 µM | 1.25 µM | 30 µM | 2.5 µM |
| DOX | 132 nM | 200 nM | 100 nM | 100 nM | 100 nM |
| IRI | 21.26 µM | 4 µM | 2.5 µM | 5 µM | 5 µM |
| PTX | 48 nM | 150 nM | 7.4 nM | 28 nM | 56 nM |
| 5-FU | 50 µM | 41.24 µM | 50 µM | 50 µM | 50 µM |
| OX | 7.32 µM | 7.5 µM | 5 µM | 10 µM | 10 µM |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501. https://doi.org/10.3390/cells8121501
Bojko A, Czarnecka-Herok J, Charzynska A, Dabrowski M, Sikora E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells. 2019; 8(12):1501. https://doi.org/10.3390/cells8121501
Chicago/Turabian StyleBojko, Agnieszka, Joanna Czarnecka-Herok, Agata Charzynska, Michal Dabrowski, and Ewa Sikora. 2019. "Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents" Cells 8, no. 12: 1501. https://doi.org/10.3390/cells8121501
APA StyleBojko, A., Czarnecka-Herok, J., Charzynska, A., Dabrowski, M., & Sikora, E. (2019). Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells, 8(12), 1501. https://doi.org/10.3390/cells8121501