Gene Expression Profiles Controlled by the Alternative Splicing Factor Nova2 in Endothelial Cells

 , , , , ,
, , , , , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and Transfection
2.3. SiRNA-Mediated RNA Interference
2.4. Lentivirus Production and Transduction
2.5. Immunoblot Analysis
2.6. Co-Immunoprecipitation (Co-IP)
2.7. RNA Extraction, RT–PCR, and RT–qPCR
2.8. RNA-seq and Analysis of Differentially Expressed Genes (DEGs)
2.9. Functional Enrichment Analysis
2.10. Identification of Nova2 Target Genes Implicated in Transcriptional Regulation
3. Results
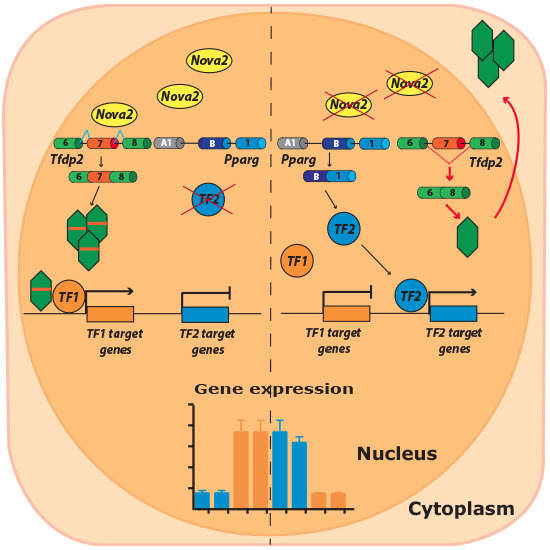
3.1. Nova2 is a Regulator of EC Transcriptome
3.2. Nova2 Affects the Expression Levels of Genes Governing Key EC Functions
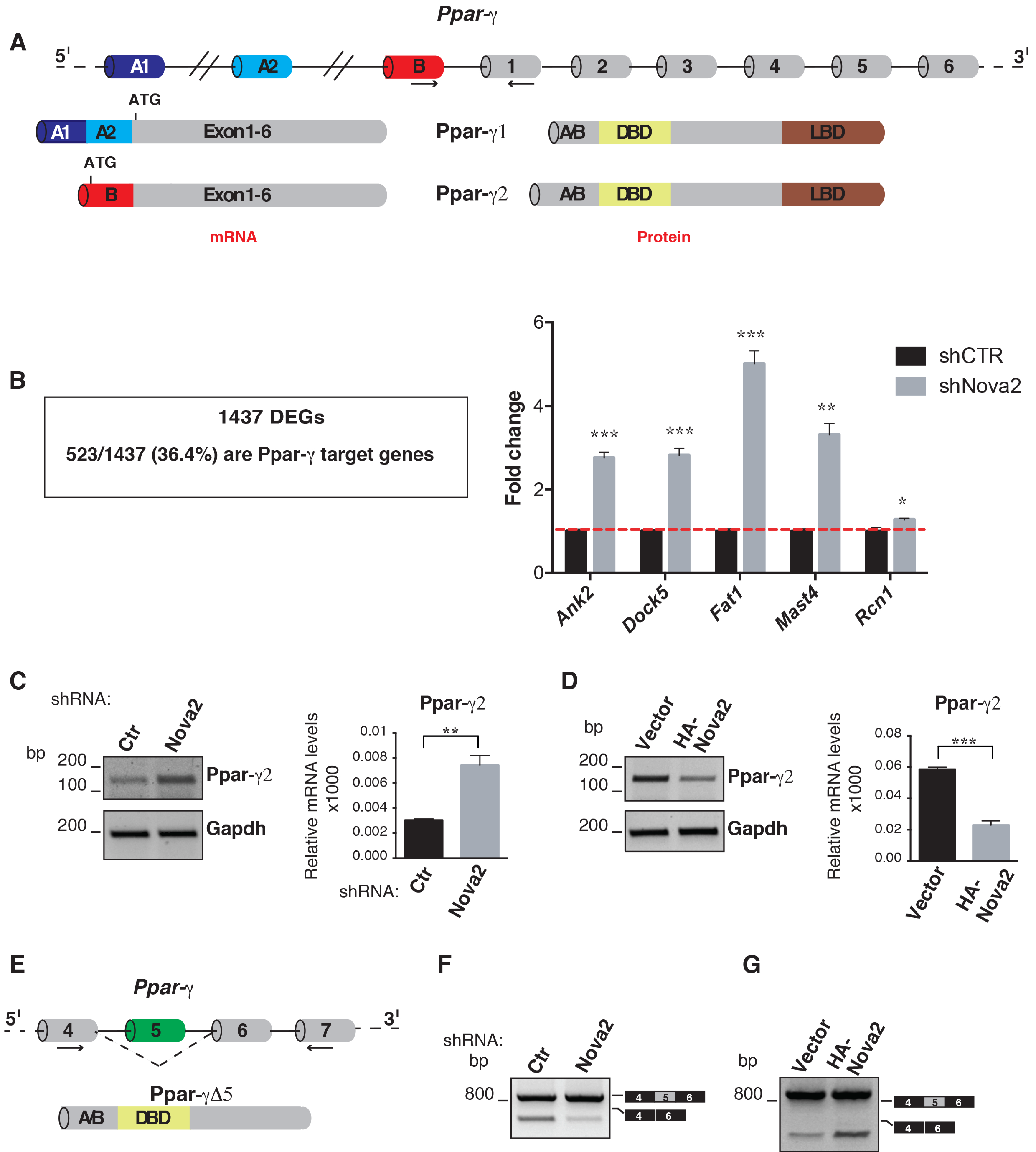
3.3. DEGs are Enriched for Ppar-γ Target Genes
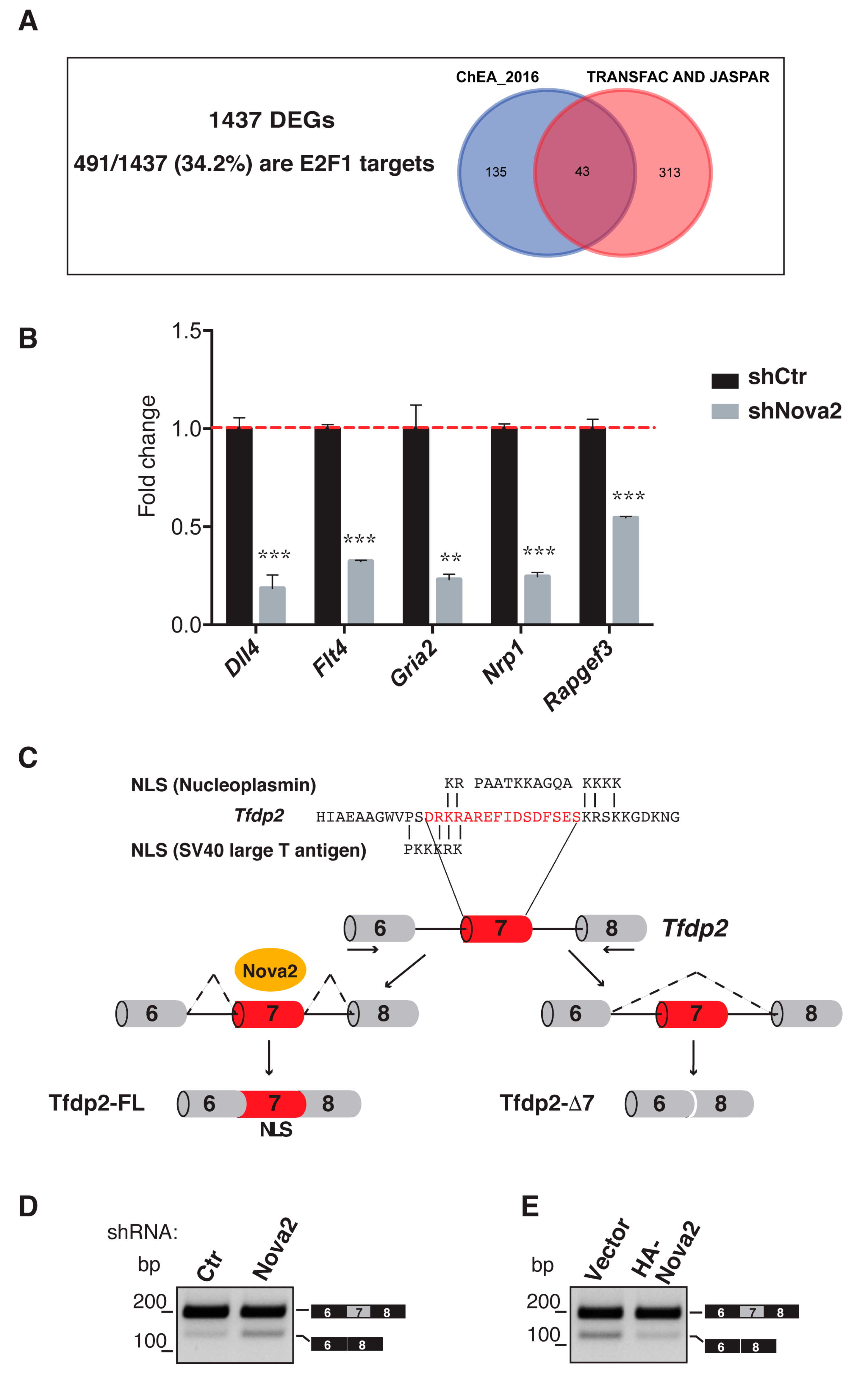
3.4. Nova2 Controls the Nuclear Localization of E2F Dimerization Partner 2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Baralle, F.E.; Giudice, J. Alternative Splicing as a Regulator of Development and Tissue Identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep Surveying of Alternative Splicing Complexity in the Human Transcriptome by High-Throughput Sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative Isoform Regulation in Human Tissue Transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Sebestyén, E.; Singh, B.; Miñana, B.; Pagès, A.; Mateo, F.; Pujana, M.A.; Valcárcel, J.; Eyras, E. Large-Scale Analysis of Genome and Transcriptome Alterations in Multiple Tumors Unveils Novel Cancer-Relevant Splicing Networks. Genome Res. 2016, 26, 732–744. [Google Scholar] [CrossRef]
- Ghigna, C.; Valacca, C.; Biamonti, G. Alternative Splicing and Tumor Progression. Curr. Genomics 2008, 9, 556–570. [Google Scholar] [CrossRef]
- Bonomi, S.; Gallo, S.; Catillo, M.; Pignataro, D.; Biamonti, G.; Ghigna, C. Oncogenic Alternative Splicing Switches: Role in Cancer Progression and Prospects for Therapy. Int. J. Cell Biol. 2013, 2013, 962038. [Google Scholar] [CrossRef]
- Biamonti, G.; Catillo, M.; Pignataro, D.; Montecucco, A.; Ghigna, C. The Alternative Splicing Side of Cancer. Semin. Cell Dev. Biol. 2014, 32, 30–36. [Google Scholar] [CrossRef]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The Gene Encoding the Splicing Factor SF2/ASF is a Proto-Oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef]
- Golan-Gerstl, R.; Cohen, M.; Shilo, A.; Suh, S.-S.; Bakàcs, A.; Coppola, L.; Karni, R. Splicing Factor HnRNP A2/B1 Regulates Tumor Suppressor Gene Splicing and Is an Oncogenic Driver in Glioblastoma. Cancer Res. 2011, 71, 4464–4472. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, D.; Qian, H.; Tsai, Y.S.; Shao, S.; Liu, Q.; Dominguez, D.; Wang, Z. The Splicing Factor RBM4 Controls Apoptosis, Proliferation, and Migration to Suppress Tumor Progression. Cancer Cell 2014, 26, 374–389. [Google Scholar] [CrossRef]
- Zong, F.-Y.; Fu, X.; Wei, W.-J.; Luo, Y.-G.; Heiner, M.; Cao, L.-J.; Fang, Z.; Fang, R.; Lu, D.; Ji, H.; et al. The RNA-Binding Protein QKI Suppresses Cancer-Associated Aberrant Splicing. PLoS Genet. 2014, 10, e1004289. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Bates, D.O. Hallmarks of Alternative Splicing in Cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef] [PubMed]
- Ladomery, M. Aberrant Alternative Splicing is Another Hallmark of Cancer. Int. J. Cell Biol. 2013, 2013, 463786. [Google Scholar] [CrossRef] [PubMed]
- Stricker, T.P.; Brown, C.D.; Bandlamudi, C.; McNerney, M.; Kittler, R.; Montoya, V.; Peterson, A.; Grossman, R.; White, K.P. Robust Stratification of Breast Cancer Subtypes Using Differential Patterns of Transcript Isoform Expression. PLoS Genet. 2017, 13, e1006589. [Google Scholar] [CrossRef] [PubMed]
- Trincado, J.L.; Sebestyén, E.; Pagés, A.; Eyras, E. The Prognostic Potential of Alternative Transcript Isoforms across Human Tumors. Genome Med. 2016, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Neri, D.; Bicknell, R. Tumour Vascular Targeting. Nat. Rev. Cancer 2005, 5, 436–446. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; van Beijnum, J.R.; Huijbers, E.J.M.; Gasull, P.C.; Mans, L.; Bex, A.; Griffioen, A.W. Oncofoetal Insulin Receptor Isoform A Marks the Tumour Endothelium; an Underestimated Pathway during Tumour Angiogenesis and Angiostatic Treatment. Br. J. Cancer 2019, 120, 218–228. [Google Scholar] [CrossRef]
- Roudnicky, F.; Yoon, S.Y.; Poghosyan, S.; Schwager, S.; Poyet, C.; Vella, G.; Bachmann, S.B.; Karaman, S.; Shin, J.W.; Otto, V.I.; et al. Alternative Transcription of a Shorter, Non-Anti-Angiogenic Thrombospondin-2 Variant in Cancer-Associated Blood Vessels. Oncogene 2018, 37, 2573–2585. [Google Scholar] [CrossRef]
- Rybak, J.-N.; Roesli, C.; Kaspar, M.; Villa, A.; Neri, D. The Extra-Domain A of Fibronectin is a Vascular Marker of Solid Tumors and Metastases. Cancer Res. 2007, 67, 10948–10957. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.-P.; Sieuwerts, A.M.; Luider, T.M.; van der Weiden, M.; Sillevis-Smitt, P.A.E.; Kros, J.M. Differential Expression of Splicing Variants of the Human Caldesmon Gene (CALD1) in Glioma Neovascularization versus Normal Brain Microvasculature. Am. J. Pathol. 2004, 164, 2217–2228. [Google Scholar] [CrossRef]
- Steiner, M.; Neri, D. Antibody-Radionuclide Conjugates for Cancer Therapy: Historical Considerations and New Trends. Clin. Cancer Res. 2011, 17, 6406–6416. [Google Scholar] [CrossRef] [PubMed]
- Carnemolla, B.; Borsi, L.; Balza, E.; Castellani, P.; Meazza, R.; Berndt, A.; Ferrini, S.; Kosmehl, H.; Neri, D.; Zardi, L. Enhancement of the Antitumor Properties of Interleukin-2 by Its Targeted Delivery to the Tumor Blood Vessel Extracellular Matrix. Blood 2002, 99, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Stefani, G.; Mele, A.; Ruggiu, M.; Wang, X.; Taneri, B.; Gaasterland, T.; Blencowe, B.J.; Darnell, R.B. An RNA Map Predicting Nova-Dependent Splicing Regulation. Nature 2006, 444, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Ule, A.; Spencer, J.; Williams, A.; Hu, J.-S.; Cline, M.; Wang, H.; Clark, T.; Fraser, C.; Ruggiu, M.; et al. Nova Regulates Brain-Specific Splicing to Shape the Synapse. Nat. Genet. 2005, 37, 844–852. [Google Scholar] [CrossRef]
- Saito, Y.; Miranda-Rottmann, S.; Ruggiu, M.; Park, C.Y.; Fak, J.J.; Zhong, R.; Duncan, J.S.; Fabella, B.A.; Junge, H.J.; Chen, Z.; et al. NOVA2-Mediated RNA Regulation is Required for Axonal Pathfinding during Development. Elife 2016, 5, e14371. [Google Scholar] [CrossRef]
- Giampietro, C.; Deflorian, G.; Gallo, S.; Di Matteo, A.; Pradella, D.; Bonomi, S.; Belloni, E.; Nyqvist, D.; Quaranta, V.; Confalonieri, S.; et al. The Alternative Splicing Factor Nova2 Regulates Vascular Development and Lumen Formation. Nat. Commun. 2015, 6, 8479. [Google Scholar] [CrossRef]
- Baek, S.; Oh, T.G.; Secker, G.; Sutton, D.L.; Okuda, K.S.; Paterson, S.; Bower, N.I.; Toubia, J.; Koltowska, K.; Capon, S.J.; et al. The Alternative Splicing Regulator Nova2 Constrains Vascular Erk Signaling to Limit Specification of the Lymphatic Lineage. Dev. Cell 2019, 49, 279–292.e5. [Google Scholar] [CrossRef]
- Gallo, S.; Arcidiacono, M.V.; Tisato, V.; Piva, R.; Penolazzi, L.; Bosi, C.; Feo, C.V.; Gafà, R.; Secchiero, P. Upregulation of the Alternative Splicing factor NOVA2 in Colorectal Cancer Vasculature. Onco. Targets. Ther. 2018, 11, 6049–6056. [Google Scholar] [CrossRef]
- Angiolini, F.; Belloni, E.; Giordano, M.; Campioni, M.; Forneris, F.; Paronetto, M.P.; Lupia, M.; Brandas, C.; Pradella, D.; Di Matteo, A.; et al. A Novel L1CAM Isoform with Angiogenic Activity Generated by NOVA2-Mediated Alternative Splicing. Elife 2019, 8, e44305. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP Yields Genome-Wide Insights into Brain Alternative RNA Processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racca, C.; Gardiol, A.; Eom, T.; Ule, J.; Triller, A.; Darnell, R.B. The Neuronal Splicing Factor Nova Co-Localizes with Target RNAs in the Dendrite. Front. Neural Circuits 2010, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, T.; Zhang, C.; Wang, H.; Lay, K.; Fak, J.; Noebels, J.L.; Darnell, R.B. NOVA-Dependent Regulation of Cryptic NMD Exons Controls Synaptic Protein Levels after Seizure. Elife 2013, 2, e00178. [Google Scholar] [CrossRef]
- Lampugnani, M.G.; Zanetti, A.; Breviario, F.; Balconi, G.; Orsenigo, F.; Corada, M.; Spagnuolo, R.; Betson, M.; Braga, V.; Dejana, E. VE-Cadherin Regulates Endothelial Actin Activating Rac and Increasing Membrane Association of Tiam. Mol. Biol. Cell 2002, 13, 1175–1189. [Google Scholar] [CrossRef]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial Adherens Junctions Control Tight Junctions by VE-Cadherin-Mediated Upregulation of Claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef]
- Irimia, M.; Weatheritt, R.J.; Ellis, J.D.; Parikshak, N.N.; Gonatopoulos-Pournatzis, T.; Babor, M.; Quesnel-Vallières, M.; Tapial, J.; Raj, B.; O’Hanlon, D.; et al. A Highly Conserved Program of Neuronal Microexons Is Misregulated in Autistic Brains. Cell 2014, 159, 1511–1523. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinformatics 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape Plug-In to Decipher Functionally Gouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Moulton, K.S.; Khan, M.K.; Vineberg, S.; Boye, E.; Davis, V.M.; O’Donnell, P.E.; Bischoff, J.; Milstone, D.S. E-selectin is Required for the Antiangiogenic Activity of Endostatin. Proc. Natl. Acad. Sci. USA 2004, 101, 8005–8010. [Google Scholar] [CrossRef] [Green Version]
- Lasorella, A.; Rothschild, G.; Yokota, Y.; Russell, R.G.; Iavarone, A. Id2 Mediates Tumor Initiation, Proliferation, and Angiogenesis in Rb Mutant Mice. Mol. Cell. Biol. 2005, 25, 3563–3574. [Google Scholar] [CrossRef] [Green Version]
- Debreczeni, M.L.; Németh, Z.; Kajdácsi, E.; Schwaner, E.; Makó, V.; Masszi, A.; Doleschall, Z.; Rigó, J.; Walter, F.R.; Deli, M.A.; et al. MASP-1 Increases Endothelial Permeability. Front. Immunol. 2019, 10, 991. [Google Scholar] [CrossRef]
- Radisavljevic, Z.; Avraham, H.; Avraham, S. Vascular Endothelial Growth Factor Up-Regulates ICAM-1 Expression via the Phosphatidylinositol 3 OH-Kinase/AKT/Nitric Oxide Pathway and Modulates Migration of Brain Microvascular Endothelial Cells. J. Biol. Chem. 2000, 275, 20770–20774. [Google Scholar] [CrossRef] [Green Version]
- Cébe-Suarez, S.; Zehnder-Fjällman, A.; Ballmer-Hofer, K. The Role of VEGF Receptors in Angiogenesis; Complex Partnerships. Cell. Mol. Life Sci. 2006, 63, 601–615. [Google Scholar] [CrossRef] [Green Version]
- Kotlinowski, J.; Jozkowicz, A. PPAR Gamma and Angiogenesis: Endothelial Cells Perspective. J. Diabetes Res. 2016, 2016, 8492353. [Google Scholar] [CrossRef] [Green Version]
- Mueller, E.; Drori, S.; Aiyer, A.; Yie, J.; Sarraf, P.; Chen, H.; Hauser, S.; Rosen, E.D.; Ge, K.; Roeder, R.G.; et al. Genetic Analysis of Adipogenesis Through Peroxisome Proliferator-Activated Receptor Gamma Isoforms. J. Biol. Chem. 2002, 277, 41925–41930. [Google Scholar] [CrossRef] [Green Version]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR Receptor in Different Diseases and Their Ligands: Physiological Importance and Clinical Implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef]
- Chen, Y.; Jimenez, A.R.; Medh, J.D. Identification and Regulation of Novel PPAR-gamma Splice Variants in Human THP-1 Macrophages. Biochim. Biophys. Acta 2006, 1759, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christodoulides, C.; Vidal-Puig, A. PPARs and Adipocyte Function. Mol. Cell. Endocrinol. 2010, 318, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, T.; Koga, H.; Shimotohno, K. Transcriptional Activity of Peroxisome Proliferator-Activated Receptor γ Is Modulated by SUMO-1 Modification. J. Biol. Chem. 2004, 279, 29551–29557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juge-Aubry, C.E.; Hammar, E.; Siegrist-Kaiser, C.; Pernin, A.; Takeshita, A.; Chin, W.W.; Burger, A.G.; Meier, C.A. Regulation of the Transcriptional Activity of the Peroxisome Proliferator-activated Receptor α by Phosphorylation of a Ligand-independent trans -Activating Domain. J. Biol. Chem. 1999, 274, 10505–10510. [Google Scholar] [CrossRef] [Green Version]
- Gelman, L.; Zhou, G.; Fajas, L.; Raspé, E.; Fruchart, J.-C.; Auwerx, J. p300 Interacts with the N- and C-Terminal Part of PPARγ2 in a Ligand-Independent and -Dependent Manner, Respectively. J. Biol. Chem. 1999, 274, 7681–7688. [Google Scholar] [CrossRef] [Green Version]
- Strand, D.W.; Jiang, M.; Murphy, T.A.; Yi, Y.; Konvinse, K.C.; Franco, O.E.; Wang, Y.; Young, J.D.; Hayward, S.W. PPARγ Isoforms Differentially Regulate Metabolic Networks to Mediate Mouse Prostatic Epithelial Differentiation. Cell Death Dis. 2012, 3, e361. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zhang, F.; Zhang, X.; Xue, C.; Namwanje, M.; Fan, L.; Reilly, M.P.; Hu, F.; Qiang, L. Distinct Functions of PPARγ Isoforms in Regulating Adipocyte Plasticity. Biochem. Biophys. Res. Commun. 2016, 481, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Paz, I.; Akerman, M.; Dror, I.; Kosti, I.; Mandel-Gutfreund, Y. SFmap: A Web Server for Motif Analysis and Prediction of Splicing Factor Binding Sites. Nucleic Acids Res. 2010, 38, W281–W285. [Google Scholar] [CrossRef] [Green Version]
- Akerman, M.; David-Eden, H.; Pinter, R.Y.; Mandel-Gutfreund, Y. A Computational Approach for Genome-Wide Mapping of Splicing Factor Binding Sites. Genome Biol. 2009, 10, R30. [Google Scholar] [CrossRef] [Green Version]
- Aprile, M.; Cataldi, S.; Ambrosio, M.R.; D’Esposito, V.; Lim, K.; Dietrich, A.; Blüher, M.; Savage, D.B.; Formisano, P.; Ciccodicola, A.; et al. PPARγΔ5, a Naturally Occurring Dominant-Negative Splice Isoform, Impairs PPARγ Function and Adipocyte Differentiation. Cell Rep. 2018, 25, 1577–1592.e6. [Google Scholar] [CrossRef] [Green Version]
- Murphy, P.A.; Butty, V.L.; Boutz, P.L.; Begum, S.; Kimble, A.L.; Sharp, P.A.; Burge, C.B.; Hynes, R.O. Alternative RNA Splicing in the Endothelium Mediated in Part by Rbfox2 Regulates the Arterial Response to Low Flow. Elife 2018, 7, e29494. [Google Scholar] [CrossRef] [PubMed]
- Braeutigam, C.; Rago, L.; Rolke, A.; Waldmeier, L.; Christofori, G.; Winter, J. The RNA-Binding Protein Rbfox2: An Essential Regulator of EMT-Driven Alternative Splicing and a Mediator of Cellular Invasion. Oncogene 2014, 33, 1082–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, N. The Regulation of E2F by pRB-Family Proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, W.J.; Weijts, B.G.M.W.; Westendorp, B.; de Bruin, A. HIF Proteins Connect the RB-E2F Factors to Angiogenesis. Transcription 2013, 4, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-Z.; Tsai, S.-Y.; Leone, G. Emerging Roles of E2Fs in Cancer: An Exit from Cell Cycle Control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Biyashev, D.; Qin, G. E2F and MicroRNA Regulation of Angiogenesis. Am. J. Cardiovasc. Dis. 2011, 1, 110–118. [Google Scholar]
- Zhou, J.; Zhu, Y.; Cheng, M.; Dinesh, D.; Thorne, T.; Poh, K.K.; Liu, D.; Botros, C.; Tang, Y.L.; Reisdorph, N.; et al. Regulation of Vascular Contractility and Blood Pressure by the E2F2 Transcription Factor. Circulation 2009, 120, 1213–1221. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-C.; Day, R.M. Angiotensin II Regulates Activation of Bim Via Rb/E2F1 During Apoptosis: Involvement of Interaction between AMPKβ1/2 and Cdk4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L228–L238. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, M.; Zhang, W.; Hua, H.; Wang, N.; Zhao, J.; Ge, J.; Jiang, X.; Zhang, Z.; Ye, D.; et al. Growth Differentiation Factor 15 Promotes Blood Vessel Growth by Stimulating Cell Cycle Progression in Repair of Critical-Sized Calvarial Defect. Sci. Rep. 2017, 7, 9027. [Google Scholar] [CrossRef] [Green Version]
- Hollern, D.P.; Honeysett, J.; Cardiff, R.D.; Andrechek, E.R. The E2F Transcription Factors Regulate Tumor Development and Metastasis in a Mouse Model of Metastatic Breast Cancer. Mol. Cell. Biol. 2014, 34, 3229–3243. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.-J.; Lee, J.-H.; Kim, Y.-M.; Oh, G.T.; Lee, H. Macrophage Inhibitory Cytokine-1 Stimulates Proliferation of Human Umbilical Vein Endothelial Cells by Up-Regulating Cyclins D1 and E through the PI3K/Akt-, ERK-, and JNK-Dependent AP-1 and E2F Activation Signaling Pathways. Cell. Signal. 2012, 24, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Kovacs, M.; Chellappan, S. Regulation of Vascular Endothelial Growth Factor Receptors by Rb and E2F1: Role of Acetylation. Cancer Res. 2010, 70, 4931–4940. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, S.; Hasebe, M.; Matsumura, N.; Takashima, H.; Miyamoto-Sato, E.; Tomita, M.; Yanagawa, H. Six Classes of Nuclear Localization Signals Specific to Different Binding Grooves of Importin Alpha. J. Biol. Chem. 2009, 284, 478–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Wang, N.; Zhang, L.-N.; Huang, N.; Song, T.-F.; Li, Z.-Z.; Li, M.; Luo, X.-G.; Zhou, H.; He, H.-P.; et al. Knockdown of DNMT1 and DNMT3a Promotes the Angiogenesis of Human Mesenchymal Stem Cells Leading to Arterial Specific Differentiation. Stem Cells 2016, 34, 1273–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejana, E.; Taddei, A.; Randi, A.M. Foxs and Ets in the Transcriptional Regulation of Endothelial Cell Differentiation and Angiogenesis. Biochim. Biophys. Acta 2007, 1775, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Dimmeler, S. Epigenetic Regulation of Cardiovascular Differentiation. Cardiovasc. Res. 2011, 90, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.; Miscianinov, V.; Sluimer, J.C.; Newby, D.E.; Baker, A.H. Targeting Non-coding RNA in Vascular Biology and Disease. Front. Physiol. 2018, 9, 1655. [Google Scholar] [CrossRef] [Green Version]
- Arif, M.; Sadayappan, S.; Becker, R.C.; Martin, L.J.; Urbina, E.M. Epigenetic Modification: A Regulatory Mechanism in Essential Hypertension. Hypertens. Res. 2019, 42, 1099–1113. [Google Scholar] [CrossRef]
- Bonomi, S.; Di Matteo, A.; Buratti, E.; Cabianca, D.S.; Baralle, F.E.; Ghigna, C.; Biamonti, G. HnRNP A1 Controls a Splicing Regulatory Circuit Promoting Mesenchymal-to-Epithelial Transition. Nucleic Acids Res. 2013, 41, 8665–8679. [Google Scholar] [CrossRef] [Green Version]
- Isken, O.; Maquat, L.E. The Multiple Lives of NMD Factors: Balancing Roles in Gene and Genome Regulation. Nat. Rev. Genet. 2008, 9, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Scarpato, M.; Federico, A.; Ciccodicola, A.; Costa, V. Novel Transcription Factor Variants through RNA-Sequencing: The Importance of Being “Alternative”. Int. J. Mol. Sci. 2015, 16, 1755–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ Signaling and Metabolism: The Good, the Bad and the Future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, A.N.; Yang, L.; Duff, M.O.; Hansen, K.D.; Park, J.W.; Dudoit, S.; Brenner, S.E.; Graveley, B.R. Conservation of an RNA Regulatory Map Between Drosophila and Mammals. Genome Res. 2011, 21, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, D.; Hu, L.; Kong, X. Alternative Promoters Influence Alternative Splicing at the Genomic Level. PLoS ONE 2008, 3, e2377. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Bayha, C.; Klein, K.; Müller, S.; Weiss, T.S.; Schwab, M.; Zanger, U.M. The Truncated Splice Variant of Peroxisome Proliferator-Activated Receptor Alpha, PPARα-tr, Autonomously Regulates Proliferative and Pro-Inflammatory Genes. BMC Cancer 2015, 15, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasula, S.M.; Ahmad, M.; Guo, Y.; Zhan, Y.; Lazebnik, Y.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of an Endogenous Dominant-Negative Short Isoform of Caspase-9 that Can Regulate Apoptosis. Cancer Res. 1999, 59, 999–1002. [Google Scholar] [PubMed]
- Patel, B.K.; Pierce, J.H.; LaRochelle, W.J. Regulation of Interleukin 4-Mediated Signaling by Naturally Occurring Dominant Negative and Attenuated Forms of Human Stat6. Proc. Natl. Acad. Sci. USA 1998, 95, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Stravopodis, D.; Teglund, S.; Kitazawa, J.; Ihle, J.N. Naturally Occurring Dominant Negative Variants of Stat5. Mol. Cell. Biol. 1996, 16, 6141–6148. [Google Scholar] [CrossRef] [Green Version]
- Bugge, A.; Grøntved, L.; Aagaard, M.M.; Borup, R.; Mandrup, S. The PPARgamma2 A/B-Domain Plays a Gene-Specific Role in Transactivation and Cofactor Recruitment. Mol. Endocrinol. 2009, 23, 794–808. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Markan, K.; Temple, K.A.; Deplewski, D.; Brady, M.J.; Cohen, R.N. The Nuclear Receptor Corepressors NCoR and SMRT Decrease Peroxisome Proliferator-Activated Receptor γ Transcriptional Activity and Repress 3T3-L1 Adipogenesis. J. Biol. Chem. 2005, 280, 13600–13605. [Google Scholar] [CrossRef] [Green Version]
- Vernia, S.; Edwards, Y.J.; Han, M.S.; Cavanagh-Kyros, J.; Barrett, T.; Kim, J.K.; Davis, R.J. An Alternative Splicing Program Promotes Adipose Tissue Thermogenesis. Elife 2016, 5, e17672. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Huang, C.; Meng, X.M.; Song, Y.; Wu, X.Q.; Miu, C.G.; Zhan, X.S.; Li, J. Promising Roles of Mammalian E2Fs in Hepatocellular Carcinoma. Cell. Signal. 2014, 26, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Altiok, S.; Xu, M.; Spiegelman, B.M. PPARgamma induces Cell Cycle Withdrawal: Inhibition of E2F/DP DNA-Binding Activity via Down-Regulation of PP2A. Genes Dev. 1997, 11, 1987–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellström, M.; Phng, L.-K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.-K.; Karlsson, L.; Gaiano, N.; et al. Dll4 Signalling through Notch1 Regulates Formation of Tip Cells During Angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef]
- Suchting, S.; Freitas, C.; le Noble, F.; Benedito, R.; Breant, C.; Duarte, A.; Eichmann, A. The Notch Ligand Delta-Like 4 Negatively Regulates Endothelial Tip Cell Formation and Vessel Branching. Proc. Natl. Acad. Sci. USA 2007, 104, 3225–3230. [Google Scholar] [CrossRef] [Green Version]
- Dufraine, J.; Funahashi, Y.; Kitajewski, J. Notch Signaling Regulates Tumor Angiogenesis by Diverse Mechanisms. Oncogene 2008, 27, 5132–5137. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Huang, X.; Zhang, J.; Shao, N.; Chen, L.O.; Ma, D.; Ji, C. The Expression of VEGF and Dll4/Notch Pathway Molecules in Ovarian Cancer. Clin. Chim. Acta. 2014, 436, 243–248. [Google Scholar] [CrossRef]
- Hu, W.; Lu, C.; Dong, H.H.; Huang, J.; Shen, D.; Stone, R.L.; Nick, A.M.; Shahzad, M.M.K.; Mora, E.; Jennings, N.B.; et al. Biological Roles of the Delta Family Notch Ligand Dll4 in Tumor and Endothelial Cells in Ovarian Cancer. Cancer Res. 2011, 71, 6030–6039. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belloni, E.; Di Matteo, A.; Pradella, D.; Vacca, M.; Wyatt, C.D.R.; Alfieri, R.; Maffia, A.; Sabbioneda, S.; Ghigna, C. Gene Expression Profiles Controlled by the Alternative Splicing Factor Nova2 in Endothelial Cells. Cells 2019, 8, 1498. https://doi.org/10.3390/cells8121498
Belloni E, Di Matteo A, Pradella D, Vacca M, Wyatt CDR, Alfieri R, Maffia A, Sabbioneda S, Ghigna C. Gene Expression Profiles Controlled by the Alternative Splicing Factor Nova2 in Endothelial Cells. Cells. 2019; 8(12):1498. https://doi.org/10.3390/cells8121498
Chicago/Turabian StyleBelloni, Elisa, Anna Di Matteo, Davide Pradella, Margherita Vacca, Christopher D. R. Wyatt, Roberta Alfieri, Antonio Maffia, Simone Sabbioneda, and Claudia Ghigna. 2019. "Gene Expression Profiles Controlled by the Alternative Splicing Factor Nova2 in Endothelial Cells" Cells 8, no. 12: 1498. https://doi.org/10.3390/cells8121498
APA StyleBelloni, E., Di Matteo, A., Pradella, D., Vacca, M., Wyatt, C. D. R., Alfieri, R., Maffia, A., Sabbioneda, S., & Ghigna, C. (2019). Gene Expression Profiles Controlled by the Alternative Splicing Factor Nova2 in Endothelial Cells. Cells, 8(12), 1498. https://doi.org/10.3390/cells8121498