MicroRNA—A Tumor Trojan Horse for Tumor-Associated Macrophages


Abstract
:1. Introduction
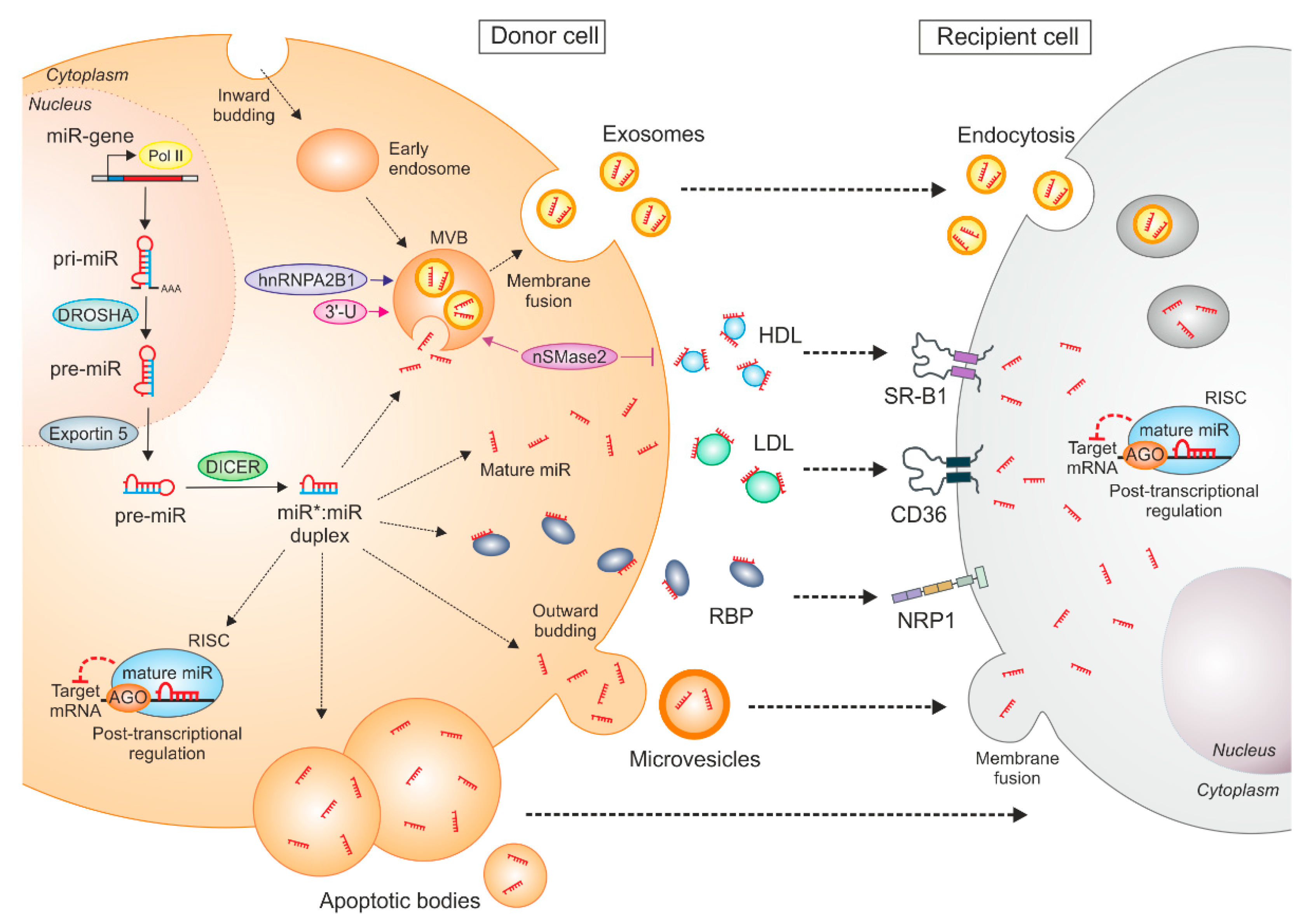
2. Tumor-Derived miRs—Packaging Matters!
3. Macrophage Uptake of Tumor-Derived miRs—A Toll-Free Highway
3.1. Extracellular Vesicle-Mediated miR Transfer
3.2. RNA-Binding Protein-Mediated miR Transfer
3.3. Lipoprotein-Mediated miR Transfer
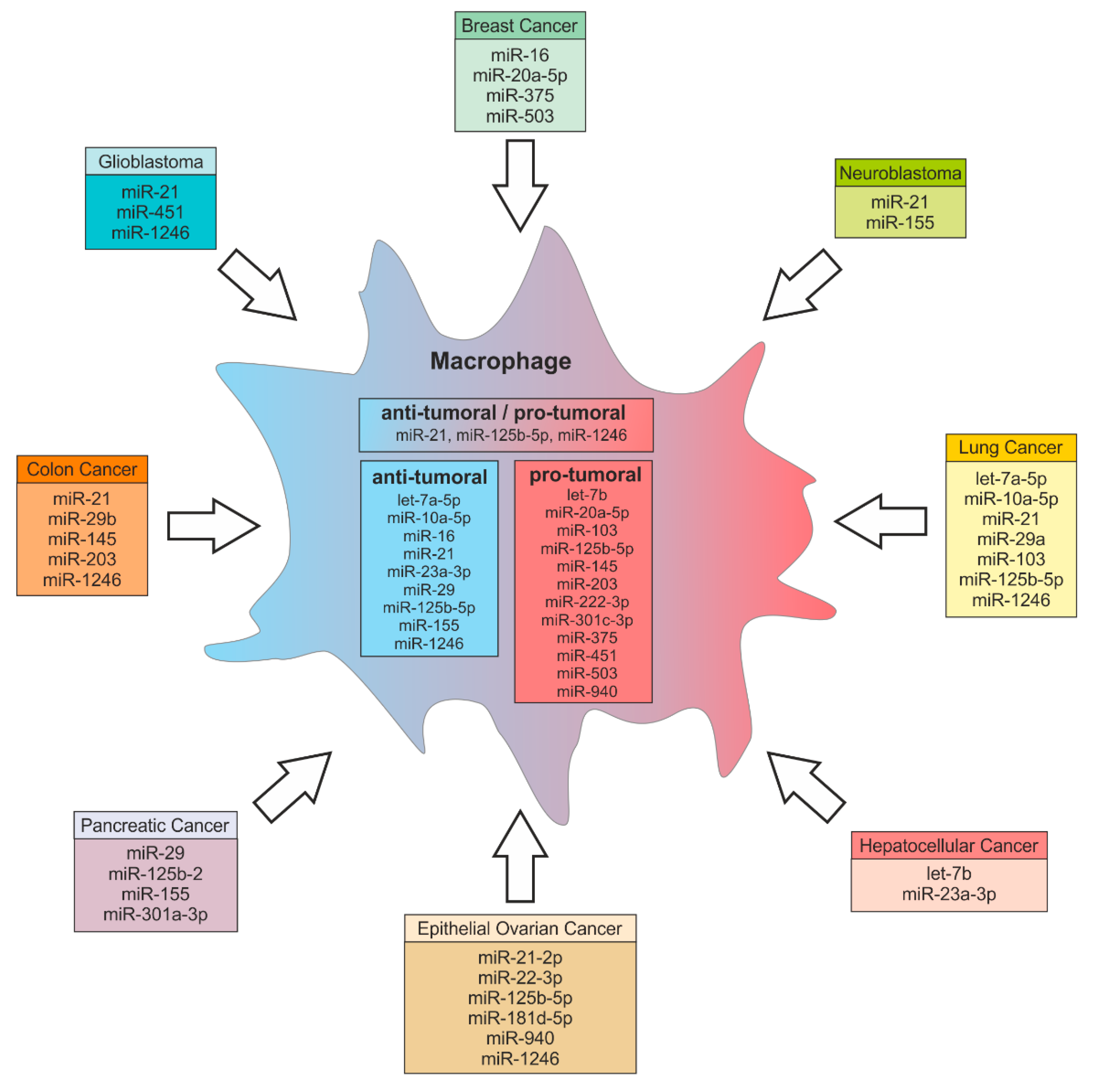
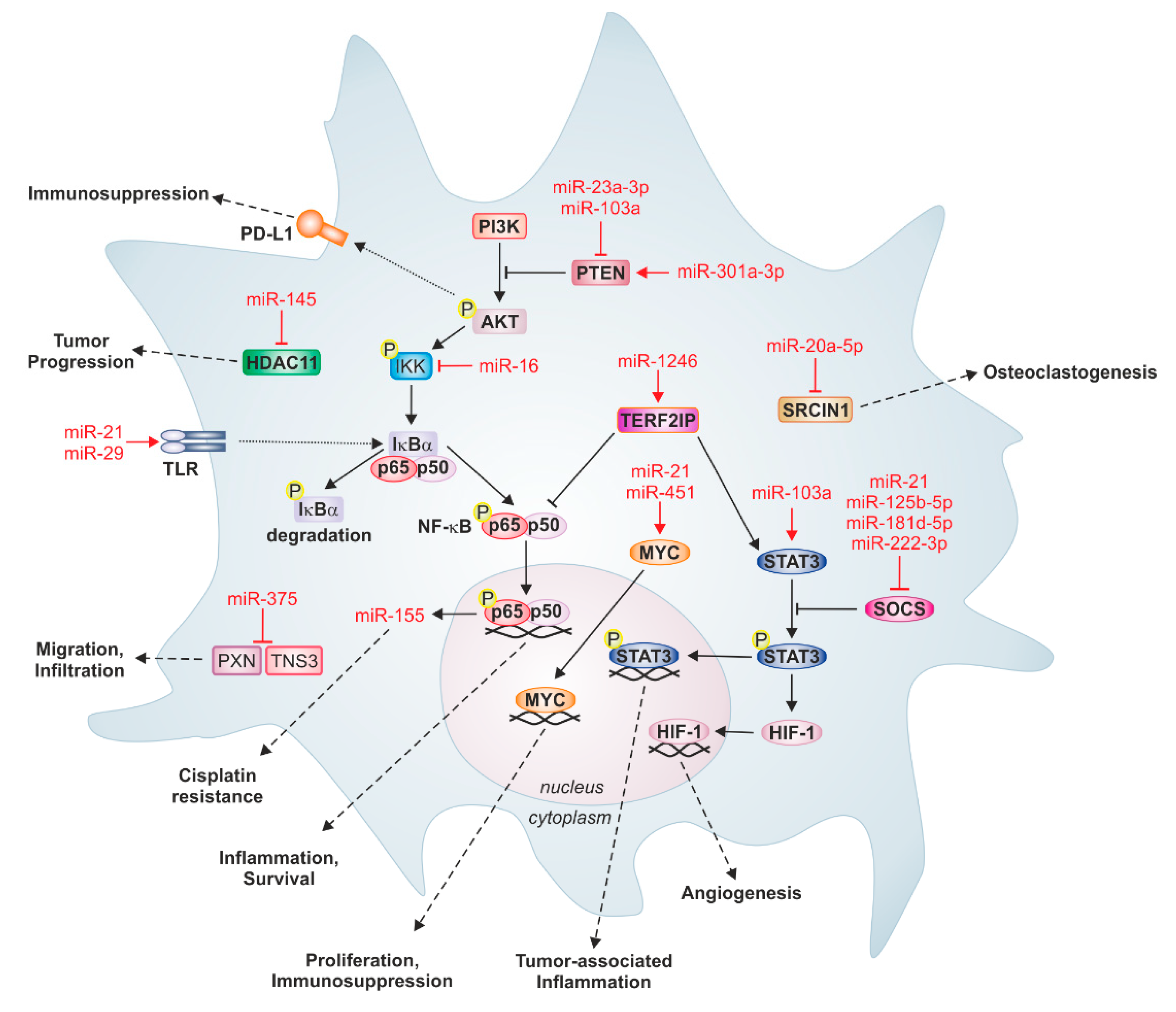
4. Functions of Tumor-Derived miRs in Macrophages—The Trojan Horse at Work
4.1. Colon Cancer
4.2. Glioblastoma
4.3. Lung Cancer
4.4. Hepatocellular Carcinoma
4.5. Breast Cancer
4.6. Epithelial Ovarian Cancer
4.7. Pancreatic Cancer
4.8. Neuroblastoma
4.9. Other Cancers
5. Macrophage-Derived miRs—An Arm of a Sentinel
6. Macrophage-Derived miRs—Agent Gone Rogue
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Hollmén, M.; Roudnicky, F.; Karaman, S.; Detmar, M. Characterization of macrophage—Cancer cell crosstalk in estrogen receptor positive and triple-negative breast cancer. Sci. Rep. 2015, 5, 9188. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Solhjoo, S.; Toolee, H.; Kharazinejad, E.; Mortezaee, K. Tumor microenvironment: Interactions and therapy. J. Cell. Physiol. 2019, 234, 5700–5721. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Brüne, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Jenkins, S.J.; Hume, D.A. Homeostasis in the mononuclear phagocyte system. Trends Immunol. 2014, 35, 358–367. [Google Scholar] [CrossRef]
- Weigert, A.; Tzieply, N.; von Knethen, A.; Johann, A.M.; Schmidt, H.; Geisslinger, G.; Brüne, B. Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol. Biol. Cell 2007, 18, 3810–3819. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Sekar, D.; Brüne, B. Tumor-associated macrophages as targets for tumor immunotherapy. Immunotherapy 2009, 1, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Turkowski, K.; Mora, J.; Brüne, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Denli, A.M.; Tops, B.B.J.; Plasterk, R.H.A.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef]
- Lee, H.Y.; Zhou, K.; Smith, A.M.; Noland, C.L.; Doudna, J.A. Differential roles of human Dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res. 2013, 41, 6568–6576. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Boettcher, S.; Caudy, A.A.; Kobayashi, R.; Hannon, G.J. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 2001, 293, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: microRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef]
- Zhang, X.; Zuo, X.; Yang, B.; Li, Z.; Xue, Y.; Zhou, Y.; Huang, J.; Zhao, X.; Zhou, J.; Yan, Y.; et al. MicroRNA Directly Enhances Mitochondrial Translation during Muscle Differentiation. Cell 2014, 158, 607–619. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- Luo, C.; Tetteh, P.W.; Merz, P.R.; Dickes, E.; Abukiwan, A.; Hotz-Wagenblatt, A.; Holland-Cunz, S.; Sinnberg, T.; Schittek, B.; Schadendorf, D.; et al. miR-137 inhibits the invasion of melanoma cells through downregulation of multiple oncogenic target genes. J. Invest. Dermatol. 2013, 133, 768–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frixa, T.; Donzelli, S.; Blandino, G. Oncogenic MicroRNAs: Key Players in Malignant Transformation. Cancers (Basel) 2015, 7, 2466–2485. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Kashima, R. Dysregulation of microRNA biogenesis machinery in cancer. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Weber, C.E.M.; Luo, C.; Hotz-Wagenblatt, A.; Gardyan, A.; Kordaß, T.; Holland-Letz, T.; Osen, W.; Eichmüller, S.B. miR-339-3p Is a Tumor Suppressor in Melanoma. Cancer Res. 2016, 76, 3562–3571. [Google Scholar] [CrossRef]
- Squadrito, M.L.; Etzrodt, M.; de Palma, M.; Pittet, M.J. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol. 2013, 34, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Weigert, A.; Mora, J.; Sekar, D.; Syed, S.; Brüne, B. Killing Is Not Enough: How Apoptosis Hijacks Tumor-Associated Macrophages to Promote Cancer Progression. Adv. Exp. Med. Biol. 2016, 930, 205–239. [Google Scholar] [CrossRef]
- Frank, A.-C.; Ebersberger, S.; Fink, A.F.; Lampe, S.; Weigert, A.; Schmid, T.; Ebersberger, I.; Syed, S.N.; Brüne, B. Apoptotic tumor cell-derived microRNA-375 uses CD36 to alter the tumor-associated macrophage phenotype. Nat. Commun. 2019, 10, 1135. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Schoepp, M.; Ströse, A.J.; Haier, J. Dysregulation of miRNA Expression in Cancer Associated Fibroblasts (CAFs) and Its Consequences on the Tumor Microenvironment. Cancers (Basel) 2017, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Lawrie, C.H.; Gal, S.; Dunlop, H.M.; Pushkaran, B.; Liggins, A.P.; Pulford, K.; Banham, A.H.; Pezzella, F.; Boultwood, J.; Wainscoat, J.S.; et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br. J. Haematol. 2008, 141, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Hagiwara, K.; Takeshita, F.; Ochiya, T. Competitive interactions of cancer cells and normal cells via secretory microRNAs. J. Biol. Chem. 2012, 287, 1397–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soon, P.; Kiaris, H. MicroRNAs in the tumour microenvironment: Big role for small players. Endocr. Relat. Cancer 2013, 20, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Inoue, K.; Fujiwara, A.; Hatakeyama, K.; Kanto, K.; Watanabe, Y.; Muramatsu, K.; Fukuda, Y.; Ogura, S.-i.; Yamaguchi, K.; et al. Let-7 microRNA family is selectively secreted into the extracellular environment via exosomes in a metastatic gastric cancer cell line. PLoS ONE 2010, 5, e13247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pigati, L.; Yaddanapudi, S.C.S.; Iyengar, R.; Kim, D.-J.; Hearn, S.A.; Danforth, D.; Hastings, M.L.; Duelli, D.M. Selective release of microRNA species from normal and malignant mammary epithelial cells. PLoS ONE 2010, 5, e13515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chim, S.S.C.; Shing, T.K.F.; Hung, E.C.W.; Leung, T.-Y.; Lau, T.-K.; Chiu, R.W.K.; Lo, Y.M.D. Detection and characterization of placental microRNAs in maternal plasma. Clin. Chem. 2008, 54, 482–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef]
- Hunter, M.P.; Ismail, N.; Zhang, X.; Aguda, B.D.; Lee, E.J.; Yu, L.; Xiao, T.; Schafer, J.; Lee, M.-L.T.; Schmittgen, T.D.; et al. Detection of microRNA expression in human peripheral blood microvesicles. PLoS ONE 2008, 3, e3694. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Mercado, M.; Manterola, L.; Larrea, E.; Goicoechea, I.; Arestin, M.; Armesto, M.; Otaegui, D.; Lawrie, C.H. The circulating transcriptome as a source of non-invasive cancer biomarkers: Concepts and controversies of non-coding and coding RNA in body fluids. J. Cell. Mol. Med. 2015, 19, 2307–2323. [Google Scholar] [CrossRef]
- Ortiz-Quintero, B. Cell-free microRNAs in blood and other body fluids, as cancer biomarkers. Cell Prolif. 2016, 49, 281–303. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Ochiya, T. Circulating microRNAs and extracellular vesicles as potential cancer biomarkers: A systematic review. Int. J. Clin. Oncol. 2017, 22, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Teng, Y.; Ye, Z.; Zhou, Y.; Tan, W.-S. The effect of gap junction-mediated transfer of miR-200b on osteogenesis and angiogenesis in a co-culture of MSCs and HUVECs. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemcke, H.; Steinhoff, G.; David, R. Gap junctional shuttling of miRNA--A novel pathway of intercellular gene regulation and its prospects in clinical application. Cell. Signal. 2015, 27, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Zhu, Y.; Liang, R.; Zhao, H.-B. Gap junction mediated miRNA intercellular transfer and gene regulation: A novel mechanism for intercellular genetic communication. Sci. Rep. 2016, 6, 19884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, I.K.H.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Atkin-Smith, G.K.; Poon, I.K.H. Disassembly of the Dying: Mechanisms and Functions. Trends Cell Biol. 2017, 27, 151–162. [Google Scholar] [CrossRef]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Lee, Y.; El Andaloussi, S.; Wood, M.J.A. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Yim, N.; Ryu, S.-W.; Choi, K.; Lee, K.R.; Lee, S.; Choi, H.; Kim, J.; Shaker, M.R.; Sun, W.; Park, J.-H.; et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat. Commun. 2016, 7, 12277. [Google Scholar] [CrossRef]
- Staubach, S.; Razawi, H.; Hanisch, F.-G. Proteomics of MUC1-containing lipid rafts from plasma membranes and exosomes of human breast carcinoma cells MCF-7. Proteomics 2009, 9, 2820–2835. [Google Scholar] [CrossRef] [PubMed]
- Di Vizio, D.; Kim, J.; Hager, M.H.; Morello, M.; Yang, W.; Lafargue, C.J.; True, L.D.; Rubin, M.A.; Adam, R.M.; Beroukhim, R.; et al. Oncosome formation in prostate cancer: Association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009, 69, 5601–5609. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Weiz, L.; Burwinkel, B. Extracellular miRNAs: The mystery of their origin and function. Trends Biochem. Sci. 2012, 37, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, S.; Chiba, M.; Watanabe, M.; Sakamoto, M.; Watanabe, N. Secretion of small/microRNAs including miR-638 into extracellular spaces by sphingomyelin phosphodiesterase 3. Oncol. Rep. 2015, 33, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janas, T.; Janas, M.M.; Sapoń, K.; Janas, T. Mechanisms of RNA loading into exosomes. FEBS Lett. 2015, 589, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Guduric-Fuchs, J.; O’Connor, A.; Camp, B.; O’Neill, C.L.; Medina, R.J.; Simpson, D.A. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC Genom. 2012, 13, 357. [Google Scholar] [CrossRef] [Green Version]
- Kogure, T.; Lin, W.-L.; Yan, I.K.; Braconi, C.; Patel, T. Intercellular nanovesicle-mediated microRNA transfer: A mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology 2011, 54, 1237–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- JIN, Y.; BIAN, T. Nontemplated nucleotide addition prior to polyadenylation: A comparison of Arabidopsis cDNA and genomic sequences. RNA 2004, 10, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.C.Y.; Eaton, S.A.; Young, P.E.; Lee, M.; Shuttleworth, R.; Humphreys, D.T.; Grau, G.E.; Combes, V.; Bebawy, M.; Gong, J.; et al. Glioma microvesicles carry selectively packaged coding and non-coding RNAs which alter gene expression in recipient cells. RNA Biol. 2013, 10, 1333–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittelbrunn, M.; Gutiérrez-Vázquez, C.; Villarroya-Beltri, C.; González, S.; Sánchez-Cabo, F.; González, M.Á.; Bernad, A.; Sánchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef] [Green Version]
- Nolte-’t Hoen, E.N.M.; Buermans, H.P.J.; Waasdorp, M.; Stoorvogel, W.; Wauben, M.H.M.; ’t Hoen, P.A.C. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res. 2012, 40, 9272–9285. [Google Scholar] [CrossRef] [Green Version]
- Squadrito, M.L.; Baer, C.; Burdet, F.; Maderna, C.; Gilfillan, G.D.; Lyle, R.; Ibberson, M.; de Palma, M. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep. 2014, 8, 1432–1446. [Google Scholar] [CrossRef] [Green Version]
- Villarroya-Beltri, C.; Gutiérrez-Vázquez, C.; Sánchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liang, H.; Zhang, J.; Zen, K.; Zhang, C.-Y. Secreted microRNAs: A new form of intercellular communication. Trends Cell Biol. 2012, 22, 125–132. [Google Scholar] [CrossRef]
- Xu, J.; Chen, Q.; Zen, K.; Zhang, C.; Zhang, Q. Synaptosomes secrete and uptake functionally active microRNAs via exocytosis and endocytosis pathways. J. Neurochem. 2013, 124, 15–25. [Google Scholar] [CrossRef]
- Tian, T.; Zhu, Y.-L.; Zhou, Y.-Y.; Liang, G.-F.; Wang, Y.-Y.; Hu, F.-H.; Xiao, Z.-D. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Ma, C.; Zhou, T.; Dong, X.; Luo, Q.; Geng, L.; Ding, L.; Zhang, Y.; Zhang, L.; Li, N.; et al. Exosomes derived from gemcitabine-resistant cells transfer malignant phenotypic traits via delivery of miRNA-222-3p. Mol. Cancer 2017, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, S.; Weber, J.; Baxter, D.; Galas, D.J. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010, 38, 7248–7259. [Google Scholar] [CrossRef] [Green Version]
- Prud’homme, G.J.; Glinka, Y.; Lichner, Z.; Yousef, G.M. Neuropilin-1 is a receptor for extracellular miRNA and AGO2/miRNA complexes and mediates the internalization of miRNAs that modulate cell function. Oncotarget 2016, 7, 68057–68071. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Bag, A.K.; Dutta, S.; Polavaram, N.S.; Islam, R.; Schellenburg, S.; Banwait, J.; Guda, C.; Ran, S.; Hollingsworth, M.A.; et al. Macrophage-Derived Neuropilin-2 Exhibits Novel Tumor-Promoting Functions. Cancer Res. 2018, 78, 5600–5617. [Google Scholar] [CrossRef] [Green Version]
- Akat, K.M.; Moore-McGriff, D.V.; Morozov, P.; Brown, M.; Gogakos, T.; Correa Da Rosa, J.; Mihailovic, A.; Sauer, M.; Ji, R.; Ramarathnam, A.; et al. Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc. Natl. Acad. Sci. USA 2014, 111, 11151–11156. [Google Scholar] [CrossRef] [Green Version]
- Banzet, S.; Chennaoui, M.; Girard, O.; Racinais, S.; Drogou, C.; Chalabi, H.; Koulmann, N. Changes in circulating microRNAs levels with exercise modality. J. Appl. Physiol. 2013, 115, 1237–1244. [Google Scholar] [CrossRef]
- Corsten, M.F.; Dennert, R.; Jochems, S.; Kuznetsova, T.; Devaux, Y.; Hofstra, L.; Wagner, D.R.; Staessen, J.A.; Heymans, S.; Schroen, B. Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circ. Cardiovasc. Genet. 2010, 3, 499–506. [Google Scholar] [CrossRef]
- Laterza, O.F.; Lim, L.; Garrett-Engele, P.W.; Vlasakova, K.; Muniappa, N.; Tanaka, W.K.; Johnson, J.M.; Sina, J.F.; Fare, T.L.; Sistare, F.D.; et al. Plasma MicroRNAs as sensitive and specific biomarkers of tissue injury. Clin. Chem. 2009, 55, 1977–1983. [Google Scholar] [CrossRef] [Green Version]
- Uhlemann, M.; Möbius-Winkler, S.; Fikenzer, S.; Adam, J.; Redlich, M.; Möhlenkamp, S.; Hilberg, T.; Schuler, G.C.; Adams, V. Circulating microRNA-126 increases after different forms of endurance exercise in healthy adults. Eur. J. Prev. Cardiol. 2014, 21, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jia, Y.; Zheng, R.; Guo, Y.; Wang, Y.; Guo, H.; Fei, M.; Sun, S. Plasma microRNA-122 as a biomarker for viral-, alcohol-, and chemical-related hepatic diseases. Clin. Chem. 2010, 56, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Michell, D.L.; Vickers, K.C. Lipoprotein carriers of microRNAs. Biochim. Biophys. Acta 2016, 1861, 2069–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Tabet, F.; Vickers, K.C.; Cuesta Torres, L.F.; Wiese, C.B.; Shoucri, B.M.; Lambert, G.; Catherinet, C.; Prado-Lourenco, L.; Levin, M.G.; Thacker, S.; et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat. Commun. 2014, 5, 3292. [Google Scholar] [CrossRef] [Green Version]
- Peiser, L.; Gordon, S. The function of scavenger receptorsexpressed by macrophages and their rolein the regulation of inflammation. Microbes Infect. 2001, 3, 149–159. [Google Scholar] [CrossRef]
- Fogelman, A.M.; van Lenten, B.J.; Warden, C.; Haberland, M.E.; Edwards, P.A. Macrophage lipoprotein receptors. J. Cell Sci. 1988, 1988, 135–149. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.; Riwanto, M.; Besler, C.; Knau, A.; Fichtlscherer, S.; Röxe, T.; Zeiher, A.M.; Landmesser, U.; Dimmeler, S. Characterization of levels and cellular transfer of circulating lipoprotein-bound microRNAs. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1392–1400. [Google Scholar] [CrossRef] [Green Version]
- Niculescu, L.S.; Simionescu, N.; Sanda, G.M.; Carnuta, M.G.; Stancu, C.S.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; Simionescu, M.; et al. MiR-486 and miR-92a Identified in Circulating HDL Discriminate between Stable and Vulnerable Coronary Artery Disease Patients. PLoS ONE 2015, 10, e0140958. [Google Scholar] [CrossRef]
- Szczyrba, J.; Nolte, E.; Wach, S.; Kremmer, E.; Stöhr, R.; Hartmann, A.; Wieland, W.; Wullich, B.; Grässer, F.A. Downregulation of Sec23A protein by miRNA-375 in prostate carcinoma. Mol. Cancer Res. 2011, 9, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Simonini, P.d.S.R.; Breiling, A.; Gupta, N.; Malekpour, M.; Youns, M.; Omranipour, R.; Malekpour, F.; Volinia, S.; Croce, C.M.; Najmabadi, H.; et al. Epigenetically deregulated microRNA-375 is involved in a positive feedback loop with estrogen receptor alpha in breast cancer cells. Cancer Res. 2010, 70, 9175–9184. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.F.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garikipati, V.N.S.; Verma, S.K.; Jolardarashi, D.; Cheng, Z.; Ibetti, J.; Cimini, M.; Tang, Y.; Khan, M.; Yue, Y.; Benedict, C.; et al. Therapeutic inhibition of miR-375 attenuates post-myocardial infarction inflammatory response and left ventricular dysfunction via PDK-1-AKT signalling axis. Cardiovasc. Res. 2017, 113, 938–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, Y.; Yasunaga, M.; Moriya, Y.; Akasu, T.; Fujita, S.; Yamamoto, S.; Matsumura, Y. Exosome can prevent RNase from degrading microRNA in feces. J. Gastrointest. Oncol. 2011, 2, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Diehl, P.; Fricke, A.; Sander, L.; Stamm, J.; Bassler, N.; Htun, N.; Ziemann, M.; Helbing, T.; El-Osta, A.; Jowett, J.B.M.; et al. Microparticles: Major transport vehicles for distinct microRNAs in circulation. Cardiovasc. Res. 2012, 93, 633–644. [Google Scholar] [CrossRef]
- Chang, M.K.; Bergmark, C.; Laurila, A.; Hörkkö, S.; Han, K.H.; Friedman, P.; Dennis, E.A.; Witztum, J.L. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: Evidence that oxidation-specific epitopes mediate macrophage recognition. Proc. Natl. Acad. Sci. USA 1999, 96, 6353–6358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, M.E.; Sun, M.; Zhang, R.; Febbraio, M.; Silverstein, R.; Hazen, S.L. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 2006, 203, 2613–2625. [Google Scholar] [CrossRef] [Green Version]
- Brundert, M.; Heeren, J.; Merkel, M.; Carambia, A.; Herkel, J.; Groitl, P.; Dobner, T.; Ramakrishnan, R.; Moore, K.J.; Rinninger, F. Scavenger receptor CD36 mediates uptake of high density lipoproteins in mice and by cultured cells. J. Lipid Res. 2011, 52, 745–758. [Google Scholar] [CrossRef] [Green Version]
- Thorne, R.F.; Mhaidat, N.M.; Ralston, K.J.; Burns, G.F. CD36 is a receptor for oxidized high density lipoprotein: Implications for the development of atherosclerosis. FEBS Lett. 2007, 581, 1227–1232. [Google Scholar] [CrossRef] [Green Version]
- Adachi, H.; Tsujimoto, M. Endothelial scavenger receptors. Prog. Lipid Res. 2006, 45, 379–404. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yin, Y.; Li, N.; Zhu, D.; Zhang, J.; Zhang, C.-Y.; Zen, K. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J. Mol. Cell Biol. 2012, 4, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Z.; Chen, C.; Liu, Y.; Si, Q.; Chuang, T.-H.; Li, N.; Gomez-Cabrero, A.; Reisfeld, R.A.; Xiang, R.; et al. MicroRNA-19a-3p inhibits breast cancer progression and metastasis by inducing macrophage polarization through downregulated expression of Fra-1 proto-oncogene. Oncogene 2014, 33, 3014–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhao, L.; Shi, B.; Ma, S.; Xu, Z.; Ge, Y.; Liu, Y.; Zheng, D.; Shi, J. Functions of miR-146a and miR-222 in Tumor-associated Macrophages in Breast Cancer. Sci. Rep. 2015, 5, 18648. [Google Scholar] [CrossRef] [PubMed]
- Sonda, N.; Simonato, F.; Peranzoni, E.; Calì, B.; Bortoluzzi, S.; Bisognin, A.; Wang, E.; Marincola, F.M.; Naldini, L.; Gentner, B.; et al. miR-142-3p prevents macrophage differentiation during cancer-induced myelopoiesis. Immunity 2013, 38, 1236–1249. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, A.A.; So, A.Y.-L.; Sinha, N.; Gibson, W.S.J.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. MicroRNA-125b potentiates macrophage activation. J. Immunol. 2011, 187, 5062–5068. [Google Scholar] [CrossRef]
- Squadrito, M.L.; Pucci, F.; Magri, L.; Moi, D.; Gilfillan, G.D.; Ranghetti, A.; Casazza, A.; Mazzone, M.; Lyle, R.; Naldini, L.; et al. miR-511-3p modulates genetic programs of tumor-associated macrophages. Cell Rep. 2012, 1, 141–154. [Google Scholar] [CrossRef] [Green Version]
- Imaoka, H.; Toiyama, Y.; Okigami, M.; Yasuda, H.; Saigusa, S.; Ohi, M.; Tanaka, K.; Inoue, Y.; Mohri, Y.; Kusunoki, M. Circulating microRNA-203 predicts metastases, early recurrence, and poor prognosis in human gastric cancer. Gastric Cancer 2016, 19, 744–753. [Google Scholar] [CrossRef] [Green Version]
- Hur, K.; Toiyama, Y.; Okugawa, Y.; Ide, S.; Imaoka, H.; Boland, C.R.; Goel, A. Circulating microRNA-203 predicts prognosis and metastasis in human colorectal cancer. Gut 2017, 66, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Takano, Y.; Masuda, T.; Iinuma, H.; Yamaguchi, R.; Sato, K.; Tobo, T.; Hirata, H.; Kuroda, Y.; Nambara, S.; Hayashi, N.; et al. Circulating exosomal microRNA-203 is associated with metastasis possibly via inducing tumor-associated macrophages in colorectal cancer. Oncotarget 2017, 8, 78598–78613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, H.; Kuranaga, Y.; Kumazaki, M.; Sugito, N.; Yoshikawa, Y.; Takai, T.; Taniguchi, K.; Ito, Y.; Akao, Y. Regulated Polarization of Tumor-Associated Macrophages by miR-145 via Colorectal Cancer-Derived Extracellular Vesicles. J. Immunol. 2017, 199, 1505–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.-H.; Tsao, C.-J. Emerging role of microRNA-21 in cancer. Biomed. Rep. 2016, 5, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krichevsky, A.M.; Gabriely, G. miR-21: A small multi-faceted RNA. J. Cell. Mol. Med. 2009, 13, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Liu, M.; Stribinskis, V.; Klinge, C.M.; Ramos, K.S.; Colburn, N.H.; Li, Y. MicroRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene 2008, 27, 4373–4379. [Google Scholar] [CrossRef] [Green Version]
- Rask, L.; Balslev, E.; Jørgensen, S.; Eriksen, J.; Flyger, H.; Møller, S.; Høgdall, E.; Litman, T.; Nielsen, B.S. High expression of miR-21 in tumor stroma correlates with increased cancer cell proliferation in human breast cancer. APMIS 2011, 119, 663–673. [Google Scholar] [CrossRef]
- Yan, L.X.; Wu, Q.N.; Zhang, Y.; Li, Y.Y.; Liao, D.Z.; Hou, J.H.; Fu, J.; Zeng, M.S.; Yun, J.P.; Wu, Q.L.; et al. Knockdown of miR-21 in human breast cancer cell lines inhibits proliferation, in vitro migration and in vivo tumor growth. Breast Cancer Res. 2011, 13, R2. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.A.A.; Gooderham, N.J. IL6 Mediates Immune and Colorectal Cancer Cell Cross-talk via miR-21 and miR-29b. Mol. Cancer Res. 2015, 13, 1502–1508. [Google Scholar] [CrossRef] [Green Version]
- Waldner, M.J.; Foersch, S.; Neurath, M.F. Interleukin-6--a key regulator of colorectal cancer development. Int. J. Biol. Sci. 2012, 8, 1248–1253. [Google Scholar] [CrossRef]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Vos, K.E.; Abels, E.R.; Zhang, X.; Lai, C.; Carrizosa, E.; Oakley, D.; Prabhakar, S.; Mardini, O.; Crommentuijn, M.H.W.; Skog, J.; et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro-oncology. 2016, 18, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.; Lewis, C.E. Macrophage migration and gene expression in response to tumor hypoxia. Int. J. Cancer 2005, 117, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Qian, M.; Wang, S.; Guo, X.; Wang, J.; Zhang, Z.; Qiu, W.; Gao, X.; Chen, Z.; Xu, J.; Zhao, R.; et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-κB pathways. Oncogene 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, 2110–2116. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.-L.; Hung, J.-Y.; Chang, W.-A.; Jian, S.-F.; Lin, Y.-S.; Pan, Y.-C.; Wu, C.-Y.; Kuo, P.-L. Hypoxic Lung-Cancer-Derived Extracellular Vesicle MicroRNA-103a Increases the Oncogenic Effects of Macrophages by Targeting PTEN. Mol. Ther. 2018, 26, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.-L.; Hung, J.-Y.; Chang, W.-A.; Lin, Y.-S.; Pan, Y.-C.; Tsai, P.-H.; Wu, C.-Y.; Kuo, P.-L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36, 4929–4942. [Google Scholar] [CrossRef]
- Trivedi, M.; Talekar, M.; Shah, P.; Ouyang, Q.; Amiji, M. Modification of tumor cell exosome content by transfection with wt-p53 and microRNA-125b expressing plasmid DNA and its effect on macrophage polarization. Oncogenesis 2016, 5, e250. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Fan, L.; Yu, H.; Zhang, J.; He, Y.; Feng, D.; Wang, F.; Li, X.; Liu, Q.; Li, Y.; et al. Endoplasmic Reticulum Stress Causes Liver Cancer Cells to Release Exosomal miR-23a-3p and Up-regulate Programmed Death Ligand 1 Expression in Macrophages. Hepatology 2019, 70, 241–258. [Google Scholar] [CrossRef]
- Li, D.; Jia, H.; Zhang, H.; Lv, M.; Liu, J.; Zhang, Y.; Huang, T.; Huang, B. TLR4 signaling induces the release of microparticles by tumor cells that regulate inflammatory cytokine IL-6 of macrophages via microRNA let-7b. Oncoimmunology 2012, 1, 687–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.-Y.; Lee, J.-K.; Jeon, Y.-K.; Kim, C.-W. Exosome derived from epigallocatechin gallate treated breast cancer cells suppresses tumor growth by inhibiting tumor-associated macrophage infiltration and M2 polarization. BMC Cancer 2013, 13, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, F.; Liu, Y.; Wu, S.-Y.; Wu, K.; Sharma, S.; Mo, Y.-Y.; Feng, J.; Sanders, S.; Jin, G.; Singh, R.; et al. Loss of XIST in Breast Cancer Activates MSN-c-Met and Reprograms Microglia via Exosomal miRNA to Promote Brain Metastasis. Cancer Res. 2018, 78, 4316–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Zhu, Y.; Li, L.; Zhou, S.; Yin, G.; Yu, G.; Cui, H. Breast cancer cell-derived exosomal miR-20a-5p promotes the proliferation and differentiation of osteoclasts by targeting SRCIN1. Cancer Med. 2019, 8, 5687–5701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stangé, G.; van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; de Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Ke, J.; Xu, J.; Wang, F.; Zhou, Y.; Jiang, Y.; Wang, Z. Downregulation of microRNA-132 by DNA hypermethylation is associated with cell invasion in colorectal cancer. Onco. Targets. Ther. 2015, 8, 3639–3648. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Ostergaard, H.L. A role for the protein tyrosine phosphatase CD45 in macrophage adhesion through the regulation of paxillin degradation. PLoS ONE 2013, 8, e71531. [Google Scholar] [CrossRef] [Green Version]
- Martuszewska, D.; Ljungberg, B.; Johansson, M.; Landberg, G.; Oslakovic, C.; Dahlbäck, B.; Hafizi, S. Tensin3 is a negative regulator of cell migration and all four Tensin family members are downregulated in human kidney cancer. PLoS ONE 2009, 4, e4350. [Google Scholar] [CrossRef]
- Veß, A.; Blache, U.; Leitner, L.; Kurz, A.R.M.; Ehrenpfordt, A.; Sixt, M.; Posern, G. A dual phenotype of MDA-MB-468 cancer cells reveals mutual regulation of tensin3 and adhesion plasticity. J. Cell Sci. 2017, 130, 2172–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, X.; Wu, Q.; Wu, X.; Zhu, Q.; Wang, X.; Jiang, L.; Chen, X.; Wang, X. Epithelial ovarian cancer-secreted exosomal miR-222-3p induces polarization of tumor-associated macrophages. Oncotarget 2016, 7, 43076–43087. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ying, X.; Wang, X.; Wu, X.; Zhu, Q.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer deliver microRNA-940 to induce macrophage M2 polarization. Oncol. Rep. 2017, 38, 522–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhou, J.; Li, X.; Wang, X.; Lin, Y.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer cells deliver microRNAs to macrophages and elicit a tumor-promoted phenotype. Cancer Lett. 2018, 435, 80–91. [Google Scholar] [CrossRef]
- Kanlikilicer, P.; Bayraktar, R.; Denizli, M.; Rashed, M.H.; Ivan, C.; Aslan, B.; Mitra, R.; Karagoz, K.; Bayraktar, E.; Zhang, X.; et al. Exosomal miRNA confers chemo resistance via targeting Cav1/p-gp/M2-type macrophage axis in ovarian cancer. EBioMedicine 2018, 38, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Su, M.-J.; Aldawsari, H.; Amiji, M. Pancreatic Cancer Cell Exosome-Mediated Macrophage Reprogramming and the Role of MicroRNAs 155 and 125b2 Transfection using Nanoparticle Delivery Systems. Sci. Rep. 2016, 6, 30110. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kγ to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018, 78, 4586–4598. [Google Scholar] [CrossRef] [Green Version]
- Salama, A.; Fichou, N.; Allard, M.; Dubreil, L.; de Beaurepaire, L.; Viel, A.; Jégou, D.; Bösch, S.; Bach, J.-M. MicroRNA-29b modulates innate and antigen-specific immune responses in mouse models of autoimmunity. PLoS ONE 2014, 9, e106153. [Google Scholar] [CrossRef]
- Challagundla, K.B.; Wise, P.M.; Neviani, P.; Chava, H.; Murtadha, M.; Xu, T.; Kennedy, R.; Ivan, C.; Zhang, X.; Vannini, I.; et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [Green Version]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef]
- Torrisani, J.; Parmentier, L.; Buscail, L.; Cordelier, P. Enjoy the Silence: The Story of let-7 MicroRNA and Cancer. CG 2007, 8, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhao, L.; Li, D.; Yin, Y.; Zhang, C.-Y.; Li, J.; Zhang, Y. Microvesicle-delivery miR-150 promotes tumorigenesis by up-regulating VEGF, and the neutralization of miR-150 attenuate tumor development. Protein Cell 2013, 4, 932–941. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-H.; Tai, S.-K.; Yang, M.-H. Snail-overexpressing Cancer Cells Promote M2-Like Polarization of Tumor-Associated Macrophages by Delivering MiR-21-Abundant Exosomes. Neoplasia 2018, 20, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Chao, M.P.; Majeti, R.; Weissman, I.L. Macrophages as mediators of tumor immunosurveillance. Trends Immunol. 2010, 31, 212–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aucher, A.; Rudnicka, D.; Davis, D.M. MicroRNAs transfer from human macrophages to hepato-carcinoma cells and inhibit proliferation. J. Immunol. 2013, 191, 6250–6260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shan, W.-F.; Jin, T.-T.; Wu, G.-Q.; Xiong, X.-X.; Jin, H.-Y.; Zhu, S.-M. Propofol exerts anti-hepatocellular carcinoma by microvesicle-mediated transfer of miR-142-3p from macrophage to cancer cells. J. Transl. Med. 2014, 12, 279. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Li, D.; Wu, A.; Qiu, X.; Di, W.; Huang, L.; Qiu, L. TWEAK-stimulated macrophages inhibit metastasis of epithelial ovarian cancer via exosomal shuttling of microRNA. Cancer Lett. 2017, 393, 60–67. [Google Scholar] [CrossRef]
- Wang, J.-J.; Wang, Z.-Y.; Chen, R.; Xiong, J.; Yao, Y.-L.; Wu, J.-H.; Li, G.-X. Macrophage-secreted Exosomes Delivering miRNA-21 Inhibitor can Regulate BGC-823 Cell Proliferation. Asian Pac. J. Cancer Prev. 2015, 16, 4203–4209. [Google Scholar] [CrossRef] [Green Version]
- Akao, Y.; Iio, A.; Itoh, T.; Noguchi, S.; Itoh, Y.; Ohtsuki, Y.; Naoe, T. Microvesicle-mediated RNA molecule delivery system using monocytes/macrophages. Mol. Ther. 2011, 19, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Kaduka, Y.; Takeda, K.; Nakayama, M.; Kinoshita, K.; Yagita, H.; Okumura, K. TWEAK mediates anti-tumor effect of tumor-infiltrating macrophage. Biochem. Biophys. Res. Commun. 2005, 331, 384–390. [Google Scholar] [CrossRef]
- Maecker, H.; Varfolomeev, E.; Kischkel, F.; Lawrence, D.; LeBlanc, H.; Lee, W.; Hurst, S.; Danilenko, D.; Li, J.; Filvaroff, E.; et al. TWEAK Attenuates the Transition from Innate to Adaptive Immunity. Cell 2005, 123, 931–944. [Google Scholar] [CrossRef] [Green Version]
- Johnnidis, J.B.; Harris, M.H.; Wheeler, R.T.; Stehling-Sun, S.; Lam, M.H.; Kirak, O.; Brummelkamp, T.R.; Fleming, M.D.; Camargo, F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008, 451, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.; Wang, Y.; Dakhlallah, D.; Moldovan, L.; Agarwal, K.; Batte, K.; Shah, P.; Wisler, J.; Eubank, T.D.; Tridandapani, S.; et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood 2013, 121, 984–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Shen, H.; Yin, X.; Yang, M.; Wei, H.; Chen, Q.; Feng, F.; Liu, Y.; Xu, W.; Li, Y. Macrophages derived exosomes deliver miR-223 to epithelial ovarian cancer cells to elicit a chemoresistant phenotype. J. Exp. Clin. Cancer Res. 2019, 38, 81. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, J.; Su, F.; Yu, B.; Su, F.; Lin, L.; Liu, Y.; Huang, J.-D.; Song, E. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol. Cancer 2011, 10, 117. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Ma, T.; Huang, B.; Lin, L.; Zhou, Y.; Yan, J.; Zou, Y.; Chen, S. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-β signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 310. [Google Scholar] [CrossRef] [Green Version]
- Binenbaum, Y.; Fridman, E.; Yaari, Z.; Milman, N.; Schroeder, A.; Ben David, G.; Shlomi, T.; Gil, Z. Transfer of miRNA in Macrophage-Derived Exosomes Induces Drug Resistance in Pancreatic Adenocarcinoma. Cancer Res. 2018, 78, 5287–5299. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef] [Green Version]
- Korabecna, M.; Koutova, L.; Tesarova, P. The potential roles of vesicle-enclosed miRNAs in communication between macrophages and cancer cells in tumor microenvironment. Neoplasma 2017, 64, 406–411. [Google Scholar] [CrossRef]
- Mercatelli, N.; Coppola, V.; Bonci, D.; Miele, F.; Costantini, A.; Guadagnoli, M.; Bonanno, E.; Muto, G.; Frajese, G.V.; de Maria, R.; et al. The inhibition of the highly expressed miR-221 and miR-222 impairs the growth of prostate carcinoma xenografts in mice. PLoS ONE 2008, 3, e4029. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, D.-Y.; Huang, L. In vivo delivery of miRNAs for cancer therapy: Challenges and strategies. Adv. Drug Deliv. Rev. 2015, 81, 128–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, G.; Kao, S.C.; Pavlakis, N.; Brahmbhatt, H.; MacDiarmid, J.; Clarke, S.; Boyer, M.; van Zandwijk, N. Clinical development of TargomiRs, a miRNA mimic-based treatment for patients with recurrent thoracic cancer. Epigenomics 2016, 8, 1079–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| miR | Donor cells | Acceptor cells | Function | Ref. |
|---|---|---|---|---|
| miR-203 | Colon cancer cells | Monocytes | Induction of TAM differentiation | [121] |
| miR-1246 | Colon cancer cells | Macrophages | Immunosuppression | [122] |
| Hypoxic glioma cells | Macrophages | M2 macrophage polarization | [135] | |
| EOC cells | M2 macrophages | M2 macrophage polarization | [155] | |
| miR-145 | Colorectal cancer cells | Macrophages | M2 macrophage polarization | [123] |
| miR-103a | Hypoxic lung cancer cells | Macrophages | Cancer progression, angiogenesis | [138] |
| miR let-7a-5p, miR-10a-5p, miR-1246, miR-125b-5p | Lung adenocarcinoma cells | Macrophages | M1 macrophage reprogramming | [139] |
| miR let-7a | Hypoxic melanoma B16-F0 cells | Bone marrow macrophages | Enhanced oxidative phosphorylation activity, M2 macrophage polarization | [161] |
| miR let-7b | Hepato-carcinoma cells | Macrophages | Attenuation of tumor inflammation | [141] |
| miR-23a-3p | Liver cancer cells | Macrophages | Immune evasion | [140] |
| miR-16 | EGCG-treated 4T1 breast cancer cells | TAMs | Repolarization to M1 macrophages | [142] |
| miR-503 | Breast cancer cells | Microglia | M2 macrophage polarization | [143] |
| miR-20a-5p | MDA-MB-231 cells | Bone marrow macrophages | Stimulation of osteoclastogenesis | [144] |
| miR-375 | Breast cancer cells | Macrophages | Macrophage migration/infiltration | [38] |
| miR-222-3p | EOC cells | Macrophages | TAM polarization | [152] |
| miR-940 | Hypoxic EOC cells | Macrophages | TAM polarization | [153] |
| miR-21 | Head and neck cancer cells | CD14+ human monocytes | M2 macrophage polarization | [163] |
| Neuroblastoma cells | TAMs | Activation of TLR8, upregulation of miR-155 | [159] | |
| miR-21-3p, miR-125b-5p, miR-181d-5p | EOC cells | PMA-treated U937 cells | TAM polarization | [154] |
| miR-21, miR-29a | NSCLC cells | Macrophages | TLR activation, activation of NF-kB signaling and pro-inflammatory cytokine secretion | [136] |
| miR-21, miR-29b | Colorectal cancer cells | Macrophages | TLR activation, activation of NF-kB signaling and pro-inflammatory cytokine secretion | [129] |
| miR-21, miR-451 | Glioma cells | Microglia, monocytes/macrophages | Increased proliferation, immunosuppression | [132] |
| miR-301a-3p | Hypoxic pancreatic cancer cells | Macrophages | Macrophage M2 polarization | [157] |
| miR-150 | THP-1 cells | TAMs | Promotion of tumorigenesis | [162] |
| miR-29 | Pancreatic beta cells | Macrophages | Increased TNFα secretion | [158] |
| exogenous miR-155, miR-125b-2 | Transfected pancreatic cancer cells | Macrophages | Repolarization to M1 macrophages | [156] |
| miR-155 | TAMs | Neuroblastoma cells | Cisplatin resistance | [159] |
| miR-223 | IL-4 activated macrophages | Breast cancer cells | Increased invasiveness | [175] |
| Hypoxic macrophages | EOC | Enhanced drug resistance | [174] | |
| miR-223, miR-142-3p | Macrophages | Hepato-carcinoma cells | Inhibited cancer cell proliferation | [165] |
| miR-142-3p | TAMs | Hepato-carcinoma cells | Conveys propofol effect | [166] |
| miR-21 | TAM-like M2 macrophage | Gastric cancer cells | Drug resistance, reduced apoptosis | [178] |
| miR-501-3p | M2 macrophages | PDAC cells | Metastasis | [176] |
| miR-365 | Macrophages | PDAC cells | Gemcitabine resistance | [177] |
| miR-7 | TWEAK-stimulated macrophages | EOC cells | Inhibits metastasis and invasiveness | [167] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Syed, S.N.; Frank, A.-C.; Raue, R.; Brüne, B. MicroRNA—A Tumor Trojan Horse for Tumor-Associated Macrophages. Cells 2019, 8, 1482. https://doi.org/10.3390/cells8121482
Syed SN, Frank A-C, Raue R, Brüne B. MicroRNA—A Tumor Trojan Horse for Tumor-Associated Macrophages. Cells. 2019; 8(12):1482. https://doi.org/10.3390/cells8121482
Chicago/Turabian StyleSyed, Shahzad Nawaz, Ann-Christin Frank, Rebecca Raue, and Bernhard Brüne. 2019. "MicroRNA—A Tumor Trojan Horse for Tumor-Associated Macrophages" Cells 8, no. 12: 1482. https://doi.org/10.3390/cells8121482
APA StyleSyed, S. N., Frank, A. -C., Raue, R., & Brüne, B. (2019). MicroRNA—A Tumor Trojan Horse for Tumor-Associated Macrophages. Cells, 8(12), 1482. https://doi.org/10.3390/cells8121482