The Impact of Kinases in Amyotrophic Lateral Sclerosis at the Neuromuscular Synapse: Insights into BDNF/TrkB and PKC Signaling

 , and
, and 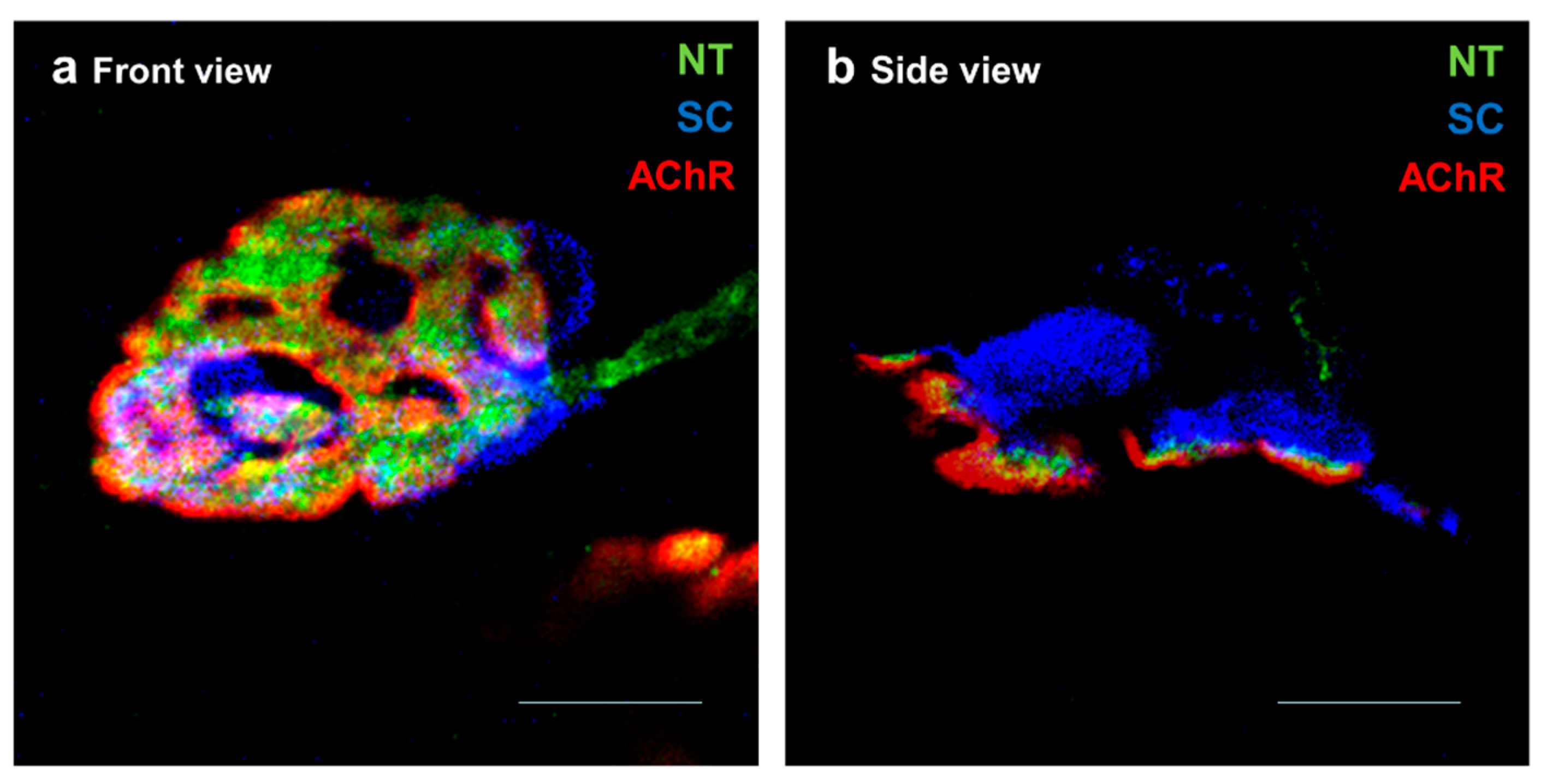{kind=link}
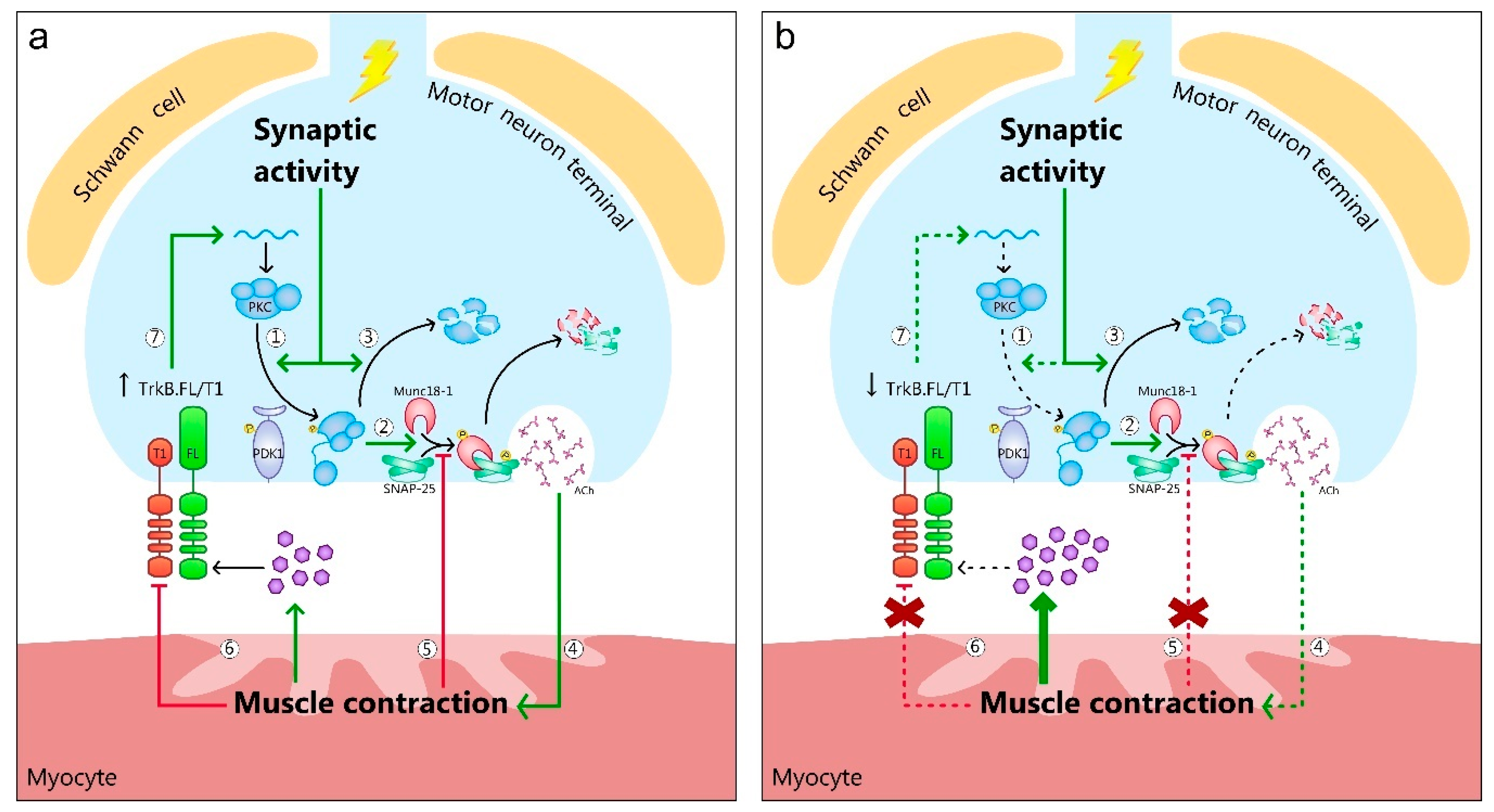{kind=link}
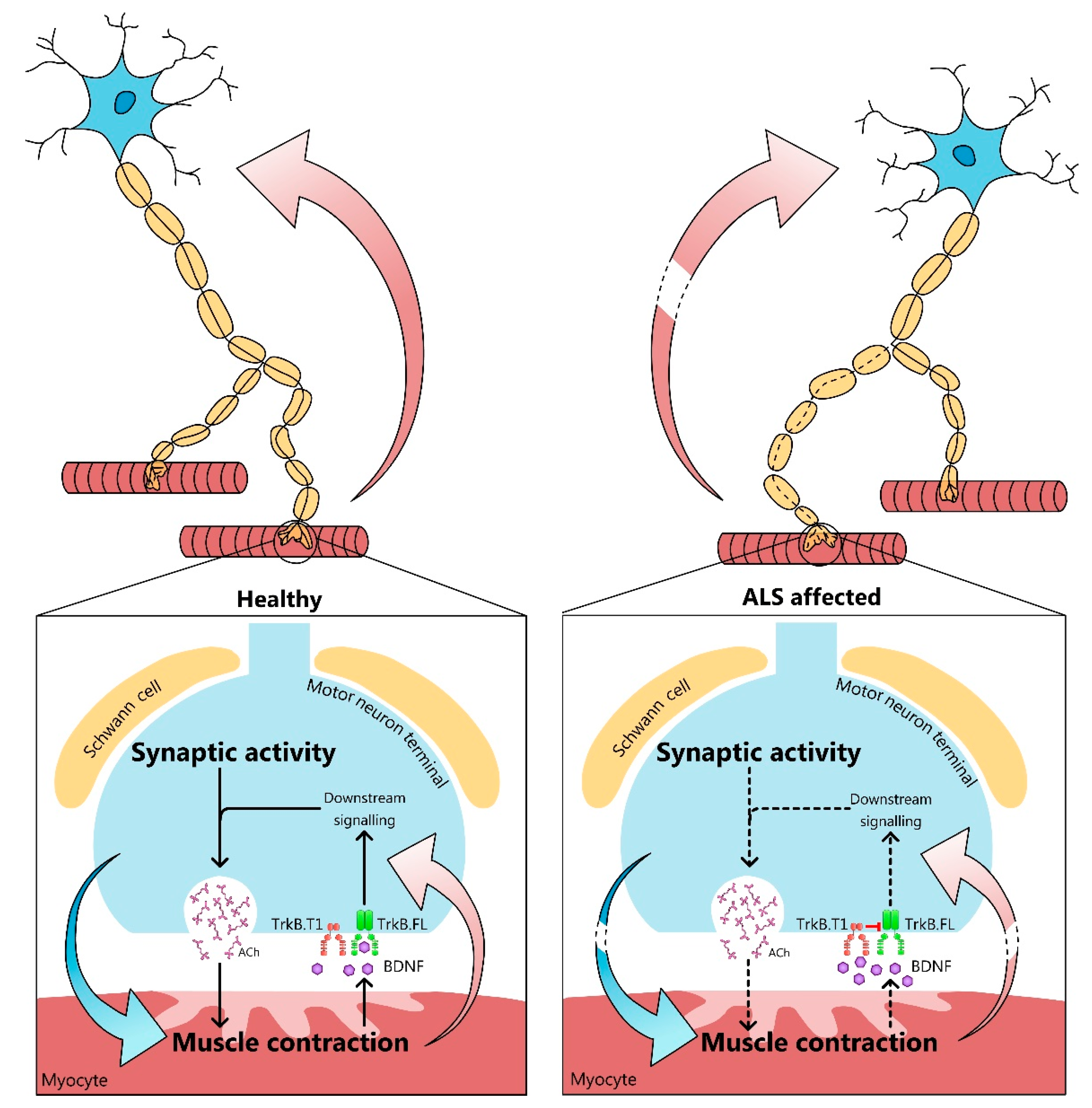{kind=link}
Abstract
:1. Introduction
2. The NMJ Is Essential in Nerve–Muscle Bidirectional Communication
3. BDNF/TrkB Signaling in the NMJ Is Regulated by Pre and Postsynaptic Activity
4. BDNF/TrkB Signaling Is Impaired in ALS NMJ
5. BDNF/TrkB Downstream Protein Kinases and Targets Are Impaired in the ALS NMJ
6. Exercise Reduces the BDNF/TrkB/PKC Signaling Impairment in ALS NMJ
7. Other Kinases Involved in NMJ Health in ALS
7.1. TBK1 and Ripk
7.2. MuSK
7.3. ErbB
7.4. PINK1
7.5. PI3K and AKT
8. Conclusions
Funding
Conflicts of Interest
References
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. ALS as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 2014, 8, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pun, S.; Santos, A.F.; Saxena, S.; Xu, L.; Caroni, P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 2006, 9, 408–419. [Google Scholar] [CrossRef]
- Schaefer, A.M.; Sanes, J.R.; Lichtman, J.W. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J. Comp. Neurol. 2005, 490, 209–219. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Williamson, T.L. Slowing of axonal transport is a very early event in the toxicity ofALS–linked SOD1 mutants to motor neurons. Nat. Neurosci. 1999, 2, 50–56. [Google Scholar]
- Baldwin, K.M.; Haddad, F.; Pandorf, C.E.; Roy, R.R.; Edgerton, V.R. Alterations in muscle mass and contractile phenotype in response to unloading models: Role of transcriptional/pretranslational mechanisms. Front. Physiol. 2013, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, K.; Machida, T.; Hirafuji, M. Skeletal muscle is an endocrine organ. J. Pharm. Sci. 2014, 125, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef]
- Wu, H.; Xiong, W.C.; Mei, L. To build a synapse: Signaling pathways in neuromuscular junction assembly. Development 2010, 137, 1017–1033. [Google Scholar] [CrossRef] [Green Version]
- Campanelli, J.T.; Hoch, W.; Rupp, F.; Kreiner, T.; Scheller, R.H. Agrin mediates cell contact-induced acetylcholine receptor clustering. Cell 1991, 67, 909–916. [Google Scholar] [CrossRef]
- Gautam, M.; Noakes, P.G.; Moscoso, L.; Rupp, F.; Scheller, R.H.; Merlie, J.P.; Sanes, J.R. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 1996, 85, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Fox, M.A.; Sanes, J.R.; Borza, D.-B.; Eswarakumar, V.P.; Fässler, R.; Hudson, B.G.; John, S.W.M.; Ninomiya, Y.; Pedchenko, V.; Pfaff, S.L.; et al. Distinct Target-Derived Signals Organize Formation, Maturation, and Maintenance of Motor Nerve Terminals. Cell 2007, 129, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimune, H.; Valdez, G.; Jarad, G.; Moulson, C.L.; Müller, U.; Miner, J.H.; Sanes, J.R. Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J. Cell Biol. 2008, 182, 1201–1215. [Google Scholar] [CrossRef] [Green Version]
- Nishimune, H.; Sanes, J.R.; Carlson, S.S. A synaptic laminin–calcium channel interaction organizes active zones in motor nerve terminals. Nature 2004, 432, 580–587. [Google Scholar] [CrossRef]
- Singhal, N.; Martin, P.T. Role of extracellular matrix proteins and their receptors in the development of the vertebrate neuromuscular junction. Dev. Neurobiol. 2011, 71, 982–1005. [Google Scholar] [CrossRef] [Green Version]
- Polo-Parada, L.; Bose, C.M.; Landmesser, L.T. Alterations in Transmission, Vesicle Dynamics, and Transmitter Release Machinery at NCAM-Deficient Neuromuscular Junctions. Neuron 2001, 32, 815–828. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Sanchez, H.B.; Deerinck, T.; Morris, J.K.; Ellisman, M.; Lee, K.-F. Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc. Natl. Acad. Sci. USA 2000, 97, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- Buonanno, A.; Fischbach, G.D. Neuregulin and ErbB receptor signaling pathways in the nervous system. Curr. Opin. Neurobiol. 2001, 11, 287–296. [Google Scholar] [CrossRef]
- Li, L.; Xiong, W.-C.; Mei, L. Neuromuscular Junction Formation, Aging, and Disorders. Annu. Rev. Physiol. 2018, 80, 159–188. [Google Scholar] [CrossRef]
- Liu, W.; Chakkalakal, J.V. The Composition, Development, and Regeneration of Neuromuscular Junctions. In Current Topics in Developmental Biology; Academic Press: New York, NY, USA, 2018; pp. 99–124. [Google Scholar]
- Baudet, C.; Pozas, E.; Adameyko, I.; Andersson, E.; Ericson, J.; Ernfors, P. Retrograde Signaling onto Ret during Motor Nerve Terminal Maturation. J. Neurosci. 2008, 28, 963–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Je, H.S.; Yang, F.; Ji, Y.; Potluri, S.; Fu, X.-Q.; Luo, Z.-G.; Nagappan, G.; Chan, J.P.; Hempstead, B.; Son, Y.-J.; et al. ProBDNF and mature BDNF as punishment and reward signals for synapse elimination at mouse neuromuscular junctions. J. Neurosci. 2013, 33, 9957–9962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, Q.T. Hyperinnervation of Neuromuscular Junctions Caused by GDNF Overexpression in Muscle. Science 1998, 279, 1725–1729. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Ko, C.-P. Schwann cells promote synaptogenesis at the neuromuscular junction via transforming growth factor-beta1. J. Neurosci. 2008, 28, 9599–9609. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Medel, Y.; Ashley, J.; Barria, R.; Maloney, R.; Freeman, M.; Budnik, V. Integration of a Retrograde Signal during Synapse Formation by Glia-Secreted TGF-β Ligand. Curr. Biol. 2012, 22, 1831–1838. [Google Scholar] [CrossRef] [Green Version]
- Matthews, V.B.; Åström, M.-B.; Chan, M.H.S.; Bruce, C.R.; Krabbe, K.S.; Prelovsek, O.; Åkerström, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef] [Green Version]
- Pitts, E.V.; Potluri, S.; Hess, D.M.; Balice-Gordon, R.J. Neurotrophin and Trk-mediated signaling in the neuromuscular system. Int. Anesth. Clin. 2006, 44, 21–76. [Google Scholar] [CrossRef]
- Funakoshi, H. Differential expression of mRNAs for neurotrophins and their receptors after axotomy of the sciatic nerve. J. Cell Biol. 1993, 123, 455–465. [Google Scholar] [CrossRef]
- Cisterna, B.A.; Cardozo, C.; Saez, J.C. Neuronal involvement in muscular atrophy. Front. Cell. Neurosci. 2014, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Minic, J.; Molgó, J.; Karlsson, E.; Krejci, E. Regulation of acetylcholine release by muscarinic receptors at the mouse neuromuscular junction depends on the activity of acetylcholinesterase. Eur. J. Neurosci. 2002, 15, 439–448. [Google Scholar] [CrossRef]
- Santafé, M.M.; Salon, I.; Garcia, N.; Lanuza, M.A.; Uchitel, O.D.; Tomàs, J. Modulation of ACh release by presynaptic muscarinic autoreceptors in the neuromuscular junction of the newborn and adult rat. Eur. J. Neurosci. 2003, 17, 119–127. [Google Scholar] [CrossRef]
- Santafé, M.M.; Salon, I.; Garcia, N.; Lanuza, M.A.; Uchitel, O.D.; Tomàs, J. Muscarinic autoreceptors related with calcium channels in the strong and weak inputs at polyinnervated developing rat neuromuscular junctions. Neuroscience 2004, 123, 61–73. [Google Scholar] [CrossRef]
- Santafé, M.M.; Lanuza, M.A.; Garcia, N.; Tomàs, J. Calcium inflow-dependent protein kinase C activity is involved in the modulation of transmitter release in the neuromuscular junction of the adult rat. Synapse 2005, 57, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Slutsky, I.; Parnas, H.; Parnas, I. Presynaptic effects of muscarine on ACh release at the frog neuromuscular junction. J. Physiol. 1999, 514, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, E.; Cilleros, V.; Nadal, L.; Simó, A.; Obis, T.; Garcia, N.; Santafé, M.M.; Tomàs, M.; Halievski, K.; Jordan, C.L.; et al. Muscle Contraction Regulates BDNF/TrkB Signaling to Modulate Synaptic Function through Presynaptic cPKCα and cPKCβI. Front. Mol. Neurosci. 2017, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Song, W.-J.; Tkatch, T.; Surmeier, D.J. Adenosine Receptor Expression and Modulation of Ca2+ Channels in Rat Striatal Cholinergic Interneurons. J. Neurophysiol. 2000, 83, 322–332. [Google Scholar] [CrossRef]
- Tomàs, J.; Garcia, N.; Lanuza, M.A.; Santafé, M.M.; Tomàs, M.; Nadal, L.; Hurtado, E.; Simó-Ollé, A.; Cilleros-Mañé, V.; Just-Borràs, L. Adenosine Receptors in Developing and Adult Mouse Neuromuscular Junctions and Functional Links With Other Metabotropic Receptor Pathways. Front. Pharm. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Simó, A.; Just-Borràs, L.; Cilleros-Mañé, V.; Hurtado, E.; Nadal, L.; Tomàs, M.; Garcia, N.; Lanuza, M.A.; Tomàs, J. BDNF-TrkB Signaling Coupled to nPKCε and cPKCβI Modulate the Phosphorylation of the Exocytotic Protein Munc18-1 During Synaptic Activity at the Neuromuscular Junction. Front. Mol. Neurosci. 2018, 11, 207–227. [Google Scholar] [CrossRef] [Green Version]
- Simó, A.; Cilleros-Mañé, V.; Just-Borràs, L.; Hurtado, E.; Nadal, L.; Tomàs, M.; Garcia, N.; Lanuza, M.A.; Tomàs, J. nPKCε Mediates SNAP-25 Phosphorylation of Ser-187 in Basal Conditions and After Synaptic Activity at the Neuromuscular Junction. Mol. Neurobiol. 2019, 56, 5346–5364. [Google Scholar] [CrossRef]
- Snider, W.D. How do you feel? Neurotrophins and mechanotransduction. Nat. Neurosci. 1998, 1, 5–6. [Google Scholar] [CrossRef]
- Mantilla, C.B.; Stowe, J.M.; Sieck, D.C.; Ermilov, L.G.; Greising, S.M.; Zhang, C.; Shokat, K.M.; Sieck, G.C. TrkB kinase activity maintains synaptic function and structural integrity at adult neuromuscular junctions. J. Appl. Physiol. 2014, 117, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk Receptors: Roles in Neuronal Signal Transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, B. BDNF and Activity-Dependent Synaptic Modulation. Learn. Mem. 2003, 10, 86–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Je, H.-S.; Ji, Y.; Nagappan, G.; Hempstead, B.; Lu, B. Pro-BDNF-induced synaptic depression and retraction at developing neuromuscular synapses. J. Cell Biol. 2009, 185, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Hempstead, B.L. Dissecting the diverse actions of pro- and mature neurotrophins. Curr. Alzheimer Res. 2006, 3, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. B 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middlemas, D.S.; Lindberg, R.A.; Hunter, T. trkB, a neural receptor protein-tyrosine kinase: Evidence for a full-length and two truncated receptors. Mol. Cell. Biol. 1991, 11, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Baxter, G.T.; Radeke, M.J.; Kuo, R.C.; Makrides, V.; Hinkle, B.; Hoang, R.; Medina-Selby, A.; Coit, D.; Valenzuela, P.; Feinstein, S.C. Signal transduction mediated by the truncated trkB receptor isoforms, trkB.T1 and trkB.T2. J. Neurosci. 1997, 17, 2683–2690. [Google Scholar] [CrossRef]
- Dorsey, S.G.; Lovering, R.M.; Renn, C.L.; Leitch, C.C.; Liu, X.; Tallon, L.J.; Sadzewicz, L.D.; Pratap, A.; Ott, S.; Sengamalay, N.; et al. Genetic deletion of trkB.T1 increases neuromuscular function. Am. J. Physiol. 2012, 302, 141–153. [Google Scholar] [CrossRef] [Green Version]
- Eide, F.F.; Vining, E.R.; Eide, B.L.; Zang, K.; Wang, X.Y.; Reichardt, L.F. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J. Neurosci. 1996, 16, 3123–3129. [Google Scholar] [CrossRef]
- Rose, C.R.; Blum, R.; Pichler, B.; Lepier, A.; Kafitz, K.W.; Konnerth, A. Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature 2003, 426, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Garner, B. Evidence that truncated TrkB isoform, TrkB-Shc can regulate phosphorylated TrkB protein levels. Biochem. Biophys. Res. Commun. 2012, 420, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.; Ruggiero, F.P.; Chang, Q.; Shi, Y.J.; Rich, M.M.; Kraner, S.; Balice-Gordon, R.J. Disruption of Trkb-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron 1999, 24, 567–583. [Google Scholar] [CrossRef] [Green Version]
- Haapasalo, A.; Koponen, E.; Hoppe, E.; Wong, G.; Castrén, E. Truncated trkB.T1 Is Dominant Negative Inhibitor of trkB.TK+-Mediated Cell Survival. Biochem. Biophys. Res. Commun. 2001, 280, 1352–1358. [Google Scholar] [CrossRef]
- Cuppini, R.; Sartini, S.; Agostini, D.; Guescini, M.; Ambrogini, P.; Betti, M.; Bertini, L.; Vallasciani, M.; Stocchi, V. Bdnf expression in rat skeletal muscle after acute or repeated exercise. Arch. Ital. Biol. 2007, 145, 99–110. [Google Scholar]
- Gómez-Pinilla, F.; Ying, Z.; Opazo, P.; Roy, R.R.; Edgerton, V.R. Differential regulation by exercise of BDNF and NT-3 in rat spinal cord and skeletal muscle. Eur. J. Neurosci. 2001, 13, 1078–1084. [Google Scholar] [CrossRef]
- Gómez-Pinilla, F.; Ying, Z.; Roy, R.R.; Molteni, R.; Edgerton, V.R. Voluntary Exercise Induces a BDNF-Mediated Mechanism That Promotes Neuroplasticity. J. Neurophysiol. 2002, 88, 2187–2195. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Pinilla, F.; Ying, Z.; Zhuang, Y. Brain and Spinal Cord Interaction: Protective Effects of Exercise Prior to Spinal Cord Injury. PLoS ONE 2012, 7, 32298. [Google Scholar] [CrossRef] [Green Version]
- Zoladz, J.A.; Pilc, A. The effect of physical activity on the brain derived neurotrophic factor: From animal to human studies. J. Physiol. Pharm. 2010, 61, 533–541. [Google Scholar]
- Adlard, P.A.; Perreau, V.M.; Engesser-Cesar, C.; Cotman, C.W. The timecourse of induction of brain-derived neurotrophic factor mRNA and protein in the rat hippocampus following voluntary exercise. Neurosci. Lett. 2004, 363, 43–48. [Google Scholar] [CrossRef]
- Liem, R.; Brouwer, N.; Copray, J. Ultrastructural localisation of intramuscular expression of BDNF mRNA by silver-gold intensified non-radioactive in situ hybridisation. Histochem. Cell Biol. 2001, 116, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. Physical activity and muscle–brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Franke, A.; Wilkinson, G.A.; Kruttgen, A.; Hu, M.; Munro, E.; Hanson, M.G.; Reichardt, L.F.; Barres, B.A. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 1998, 21, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Aloyz, R.; Fawcett, J.P.; Kaplan, D.R.; Murphy, R.A.; Miller, F.D. Activity-dependent activation of TrkB neurotrophin receptors in the adult CNS. Learn. Mem. 1999, 6, 216–231. [Google Scholar]
- Patterson, S.L.; Pittenger, C.; Morozov, A.; Martin, K.C.; Scanlin, H.; Drake, C.; Kandel, E.R. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron 2001, 32, 123–140. [Google Scholar] [CrossRef] [Green Version]
- Skup, M.; Dwornik, A.; Macias, M.; Sulejczak, D.; Wiater, M.; Czarkowska-Bauch, J. Long-term locomotor training up-regulates TrkB(FL) receptor-like proteins, brain-derived neurotrophic factor, and neurotrophin 4 with different topographies of expression in oligodendroglia and neurons in the spinal cord. Exp. Neurol. 2002, 176, 289–307. [Google Scholar] [CrossRef]
- Guiton, M.; Gunn-Moore, F.J.; Stitt, T.N.; Yancopoulos, G.D.; Tavaré, J.M. Identification of in vivo brain-derived neurotrophic factor-stimulated autophosphorylation sites on the TrkB receptor tyrosine kinase by site-directed mutagenesis. J. Biol. Chem. 1994, 269, 30370–30377. [Google Scholar]
- Cunningham, M.E.; Stephens, R.M.; Kaplan, D.R.; Greene, L.A. Autophosphorylation of activation loop tyrosines regulates signaling by the TRK nerve growth factor receptor. J. Biol. Chem. 1997, 272, 10957–10967. [Google Scholar] [CrossRef] [Green Version]
- Friedman, W.J.; Greene, L.A. Neurotrophin Signaling via Trks and p75. Exp. Cell Res. 1999, 253, 131–142. [Google Scholar] [CrossRef]
- Middlemas, D.S.; Meisenhelder, J.; Hunter, T. Identification of TrkB autophosphorylation sites and evidence that phospholipase C-gamma1 is a substrate of the TrkB receptor. J. Biol. Chem. 1994, 269, 5458–5466. [Google Scholar]
- Segal, R.A.; Bhattacharyya, A.; Rua, L.A.; Alberta, J.A.; Stephens, R.M.; Kaplan, D.R.; Stiles, C.D. Differential utilization of Trk autophosphorylation sites. J. Biol. Chem. 1996, 271, 20175–20181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, G.; Ji, Q. Phospholipase C-γ as a Signal-Transducing Element. Exp. Cell Res. 1999, 253, 15–24. [Google Scholar] [CrossRef]
- Kleiman, R.J.; Tian, N.; Krizaj, D.; Hwang, T.N.; Copenhagen, D.R.; Reichardt, L.F. BDNF-Induced potentiation of spontaneous twitching in innervated myocytes requires calcium release from intracellular stores. J. Neurophysiol. 2000, 84, 472–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colón-González, F.; Kazanietz, M.G. C1 domains exposed: From diacylglycerol binding to protein-protein interactions. Biochim. Biophys. Acta 2006, 1761, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef]
- Balendran, A.; Hare, G.R.; Kieloch, A.; Williams, M.R.; Alessi, D.R. Further evidence that 3-phosphoinositide-dependent protein kinase-1 (PDK1) is required for the stability and phosphorylation of protein kinase C (PKC) isoforms. FEBS Lett. 2000, 484, 217–223. [Google Scholar] [CrossRef]
- Chou, M.M.; Hou, W.; Johnson, J.; Graham, L.K.; Lee, M.H.; Chen, C.-S.; Newton, A.C.; Schaffhausen, B.S.; Toker, A. Regulation of protein kinase C ζ by PI 3-kinase and PDK-1. Curr. Biol. 1998, 8, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Keranen, L.M.; Dutil, E.M.; Newton, A.C. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr. Biol. 1995, 5, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Le Good, J.A.; Ziegler, W.H.; Parekh, D.B.; Alessi, D.R.; Cohen, P.; Parker, P.J. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 1998, 281, 2042–2045. [Google Scholar] [CrossRef]
- Sonnenburg, E.D.; Gao, T.; Newton, A.C. The phosphoinositide-dependent kinase, PDK-1, phosphorylates conventional protein kinase C isozymes by a mechanism that is independent of phosphoinositide 3-kinase. J. Biol. Chem. 2001, 276, 45289–45297. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, E.; Cilleros, V.; Just, L.; Simó, A.; Nadal, L.; Tomàs, M.; Garcia, N.; Lanuza, M.A.; Tomàs, J. Synaptic Activity and Muscle Contraction Increases PDK1 and PKCβI Phosphorylation in the Presynaptic Membrane of the Neuromuscular Junction. Front. Mol. Neurosci. 2017, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanuza, M.A.; Santafe, M.M.; Garcia, N.; Besalduch, N.; Tomàs, M.; Obis, T.; Priego, M.; Nelson, P.G.; Tomàs, J. Protein kinase C isoforms at the neuromuscular junction: Localization and specific roles in neurotransmission and development. J. Anat. 2014, 224, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Besalduch, N.; Tomàs, M.; Santafé, M.M.; Garcia, N.; Tomàs, J.; Lanuza, M.A. Synaptic activity-related classical protein kinase C isoform localization in the adult rat neuromuscular synapse. J. Comp. Neurol. 2010, 518, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Obis, T.; Besalduch, N.; Hurtado, E.; Nadal, L.; Santafe, M.M.; Garcia, N.; Tomàs, M.; Priego, M.; Lanuza, M.A.; Tomàs, J. The novel protein kinase C epsilon isoform at the adult neuromuscular synapse: Location, regulation by synaptic activity-dependent muscle contraction through TrkB signaling and coupling to ACh release. Mol. Brain 2015, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obis, T.; Hurtado, E.; Nadal, L.; Tomàs, M.; Priego, M.; Simon, A.; Garcia, N.; Santafe, M.M.; Lanuza, M.A.; Tomàs, J. The novel protein kinase C epsilon isoform modulates acetylcholine release in the rat neuromuscular junction. Mol. Brain 2015, 8, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santafé, M.M.; Lanuza, M.A.; Garcia, N.; Tomàs, J. Muscarinic autoreceptors modulate transmitter release through protein kinase C and protein kinase A in the rat motor nerve terminal. Eur. J. Neurosci. 2006, 23, 2048–2056. [Google Scholar] [CrossRef]
- Santafé, M.M.; Lanuza, M.A.; Garcia, N.; Tomàs, M.; Tomàs, J.M. Coupling of presynaptic muscarinic autoreceptors to serine kinases in low and high release conditions on the rat motor nerve terminal. Neuroscience 2007, 148, 432–440. [Google Scholar] [CrossRef]
- Mantilla, C.B.; Zhan, W.-Z.; Sieck, G.C. Neurotrophins improve neuromuscular transmission in the adult rat diaphragm. Muscle Nerve 2004, 29, 381–386. [Google Scholar] [CrossRef]
- Garcia, N.; Tomàs, M.; Santafé, M.M.; Besalduch, N.; Lanuza, M.A.; Tomàs, J. The interaction between tropomyosin-related kinase B receptors and presynaptic muscarinic receptors modulates transmitter release in adult rodent motor nerve terminals. J. Neurosci. 2010, 30, 16514–16522. [Google Scholar] [CrossRef] [Green Version]
- Santafé, M.M.; Garcia, N.; Tomàs, M.; Obis, T.; Lanuza, M.A.; Besalduch, N.; Tomàs, J. The interaction between tropomyosin-related kinase B receptors and serine kinases modulates acetylcholine release in adult neuromuscular junctions. Neurosci. Lett. 2014, 561, 171–175. [Google Scholar] [CrossRef]
- Pousinha, P.A.; Diogenes, M.J.; Ribeiro, J.A.; Sebastião, A.M. Triggering of BDNF facilitatory action on neuromuscular transmission by adenosine A2A receptors. Neurosci. Lett. 2006, 404, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Tomàs, J.; Santafé, M.M.; Garcia, N.; Lanuza, M.A.; Tomàs, M.; Besalduch, N.; Obis, T.; Priego, M.; Hurtado, E. Presynaptic membrane receptors in acetylcholine release modulation in the neuromuscular synapse. J. Neurosci. Res. 2014, 92, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, J.; Noakes, P.G.; Bellingham, M.C. The Role of Altered BDNF/TrkB Signaling in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2019, 13, 1–16. [Google Scholar] [CrossRef]
- Just-Borràs, L.; Hurtado, E.; Cilleros-Mañé, V.; Biondi, O.; Charbonnier, F.; Tomàs, M.; Garcia, N.; Lanuza, M.A.; Tomàs, J. Overview of Impaired BDNF Signaling, Their Coupled Downstream Serine-Threonine Kinases and SNARE/SM Complex in the Neuromuscular Junction of the Amyotrophic Lateral Sclerosis Model SOD1-G93A Mice. Mol. Neurobiol. 2019, 56, 6856–6872. [Google Scholar] [CrossRef] [PubMed]
- Harandi, V.M.; Gaied, A.R.N.; Brännström, T.; Pedrosa Domellöf, F.; Liu, J.-X. Unchanged Neurotrophic Factors and Their Receptors Correlate With Sparing in Extraocular Muscles in Amyotrophic Lateral Sclerosis. Investig. Opthalmology Vis. Sci. 2016, 57, 6831–6842. [Google Scholar] [CrossRef] [Green Version]
- Nijssen, J.; Aguila, J.; Hoogstraaten, R.; Kee, N.; Hedlund, E. Axon-Seq Decodes the Motor Axon Transcriptome and Its Modulation in Response to ALS. Stem Cell Rep. 2018, 11, 1565–1578. [Google Scholar] [CrossRef] [Green Version]
- Deforges, S.; Branchu, J.; Biondi, O.; Grondard, C.; Pariset, C.; Lécolle, S.; Lopes, P.; Vidal, P.-P.; Chanoine, C.; Charbonnier, F. Motoneuron survival is promoted by specific exercise in a mouse model of amyotrophic lateral sclerosis. J. Physiol. 2009, 587, 3561–3571. [Google Scholar] [CrossRef]
- Hegedus, J.; Putman, C.T.; Tyreman, N.; Gordon, T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Physiol. 2008, 586, 3337–3351. [Google Scholar] [CrossRef]
- Küst, B.M.; Copray, J.C.V.M.; Brouwer, N.; Troost, D.; Boddeke, H.W.G.M. Elevated Levels of Neurotrophins in Human Biceps Brachii Tissue of Amyotrophic Lateral Sclerosis. Exp. Neurol. 2002, 177, 419–427. [Google Scholar] [CrossRef]
- Mutoh, T.; Sobue, G.; Hamano, T.; Kuriyama, M.; Hirayama, M.; Yamamoto, M.; Mitsuma, T. Decreased Phosphorylation Levels of TrkB Neurotrophin Receptor in the Spinal Cords from Patients with Amyotrophic Lateral Sclerosis. Neurochem. Res. 2000, 25, 239–245. [Google Scholar] [CrossRef]
- Funakoshi, H.; Belluardo, N.; Arenas, E.; Yamamoto, Y.; Casabona, A.; Persson, H.; Ibanez, C. Muscle-derived neurotrophin-4 as an activity-dependent trophic signal for adult motor neurons. Science 1995, 268, 1495–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campanari, M.-L.; García-Ayllón, M.-S.; Ciura, S.; Sáez-Valero, J.; Kabashi, E. Neuromuscular Junction Impairment in Amyotrophic Lateral Sclerosis: Reassessing the Role of Acetylcholinesterase. Front. Mol. Neurosci. 2016, 9, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanpallewar, S.U.; Barrick, C.A.; Buckley, H.; Becker, J.; Tessarollo, L. Deletion of the BDNF Truncated Receptor TrkB.T1 Delays Disease Onset in a Mouse Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, 1–7. [Google Scholar] [CrossRef]
- Delezie, J.; Weihrauch, M.; Maier, G.; Tejero, R.; Ham, D.J.; Gill, J.F.; Karrer-Cardel, B.; Rüegg, M.A.; Tabares, L.; Handschin, C. BDNF is a mediator of glycolytic fiber-type specification in mouse skeletal muscle. Proc. Natl. Acad. Sci. USA 2019, 116, 16111–16120. [Google Scholar] [CrossRef] [Green Version]
- Chevrel, G.; Hohlfeld, R.; Sendtner, M. The role of neurotrophins in muscle under physiological and pathological conditions. Muscle Nerve. 2006, 33, 462–476. [Google Scholar] [CrossRef]
- Kulakowski, S.A.; Parker, S.D.; Personius, K.E. Reduced TrkB expression results in precocious age-like changes in neuromuscular structure, neurotransmission, and muscle function. J. Appl. Physiol. 2011, 111, 844–852. [Google Scholar] [CrossRef] [Green Version]
- Ochs, G.; Penn, R.D.; York, M.; Giess, R.; Beck, M.; Tonn, J.; Haigh, J.; Malta, E.; Traub, M.; Sendtner, M.; et al. A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 201–206. [Google Scholar] [CrossRef]
- Beck, M.; Flachenecker, P.; Magnus, T.; Giess, R.; Reiners, K.; Toyka, K.V.; Naumann, M. Autonomic dysfunction in ALS: A preliminary study on the effects of intrathecal BDNF. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2005, 6, 100–103. [Google Scholar] [CrossRef]
- Lee, F.S.; Chao, M. V Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc. Natl. Acad. Sci. USA 2001, 98, 3555–3560. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.; Nanduri, V.; Jing, S.; Lamballe, F.; Tapley, P.; Bryant, S.; Cordon-Cardo, C.; Jones, K.R.; Reichardt, L.F.; Barbacid, M. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell 1991, 66, 395–403. [Google Scholar] [CrossRef]
- Barker, P.A. p75NTR is positively promiscuous: Novel partners and new insights. Neuron 2004, 42, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibel, M.; Hoppe, E.; Barde, Y.A. Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. Embo. J. 1999, 18, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykjaer, A.; Willnow, T.E.; Petersen, C.M. p75NTR—live or let die. Curr. Opin. Neurobiol. 2005, 15, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lee, X.; Shao, Z.; Apicco, D.; Huang, G.; Gong, B.J.; Pepinsky, R.B.; Mi, S. A DR6/p75(NTR) complex is responsible for β-amyloid-induced cortical neuron death. Cell Death Dis. 2013, 4, e579. [Google Scholar] [CrossRef]
- Lowry, K.S.; Murray, S.S.; McLean, C.A.; Talman, P.; Mathers, S.; Lopes, E.C.; Cheema, S.S. A potential role for the p75 low-affinity neurotrophin receptor in spinal motor neuron degeneration in murine and human amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2001, 2, 127–134. [Google Scholar] [CrossRef]
- Dupuis, L.; Pehar, M.; Cassina, P.; Rene, F.; Castellanos, R.; Rouaux, C.; Gandelman, M.; Dimou, L.; Schwab, M.E.; Loeffler, J.P.; et al. Nogo receptor antagonizes p75NTR-dependent motor neuron death. Proc. Natl. Acad. Sci. USA 2008, 105, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Hempstead, B.L. The many faces of p75NTR. Curr. Opin. Neurobiol. 2002, 12, 260–267. [Google Scholar] [CrossRef]
- Turner, B.J.; Cheah, I.K.; Macfarlane, K.J.; Lopes, E.C.; Petratos, S.; Langford, S.J.; Cheema, S.S. Antisense peptide nucleic acid-mediated knockdown of the p75 neurotrophin receptor delays motor neuron disease in mutant SOD1 transgenic mice. J. Neurochem. 2003, 87, 752–763. [Google Scholar] [CrossRef]
- Shepheard, S.R.; Chataway, T.; Schultz, D.W.; Rush, R.A.; Rogers, M.-L. The extracellular domain of neurotrophin receptor p75 as a candidate biomarker for amyotrophic lateral sclerosis. PLoS ONE 2014, 9, e87398. [Google Scholar] [CrossRef] [Green Version]
- Küst, B.M.; Brouwer, N.; Mantingh, I.J.; Boddeke, H.W.G.M.; Copray, J.C.V.M. Reduced p75NTR expression delays disease onset only in female mice of a transgenic model of familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2003, 4, 100–105. [Google Scholar] [CrossRef]
- Peng, H.B.; Yang, J.-F.; Dai, Z.; Lee, C.W.; Hung, H.W.; Feng, Z.H.; Ko, C.-P. Differential effects of neurotrophins and schwann cell-derived signals on neuronal survival/growth and synaptogenesis. J. Neurosci. 2003, 23, 5050–5060. [Google Scholar] [CrossRef] [PubMed]
- Mantilla, C.B.; Gransee, H.M.; Zhan, W.-Z.; Sieck, G.C. Motoneuron BDNF/TrkB signaling enhances functional recovery after cervical spinal cord injury. Exp. Neurol. 2013, 247, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahara, A.H.; Tuszynski, M.H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2011, 10, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Nichols, N.L.; Satriotomo, I.; Allen, L.L.; Grebe, A.M.; Mitchell, G.S. Mechanisms of enhanced phrenic long-term facilitation in SOD1 G93a rats. J. Neurosci. 2017, 37, 5834–5845. [Google Scholar] [CrossRef] [Green Version]
- Patapoutian, A.; Reichardt, L.F. Trk receptors: Mediators of neurotrophin action. Curr. Opin. Neurobiol. 2001, 11, 272–280. [Google Scholar] [CrossRef]
- Nadal, L.; Garcia, N.; Hurtado, E.; Simó, A.; Tomàs, M.; Lanuza, M.A. Presynaptic Muscarinic Acetylcholine Receptors and TrkB Receptor Cooperate in the Elimination of Redundant Motor Nerve Terminals during Development. Front. Aging Neurosci. 2017, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Garcia, N.; Priego, M.; Obis, T.; Santafe, M.M.; Tomàs, M.; Besalduch, N.; Lanuza, Ma.; Tomàs, J. Adenosine A1 and A2A receptor-mediated modulation of acetylcholine release in the mice neuromuscular junction. Eur. J. Neurosci. 2013, 38, 2229–2241. [Google Scholar] [CrossRef]
- Tomàs, J.; Garcia, N.; Lanuza, M.A.; Santafé, M.M.; Tomàs, M.; Nadal, L.; Hurtado, E.; Simó, A.; Cilleros, V. Presynaptic Membrane Receptors Modulate ACh Release, Axonal Competition and Synapse Elimination during Neuromuscular Junction Development. Front. Mol. Neurosci. 2017, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, M.-X.; Jia, M.; Yang, L.-X.; Jiang, H.; Lanuza, M.A.; Gonzalez, C.M.; Nelson, P.G. The Role of the Theta Isoform of Protein Kinase C (PKC) in Activity-Dependent Synapse Elimination: Evidence from the PKC Theta Knock-Out Mouse In Vivo and In Vitro. J. Neurosci. 2004, 24, 3762–3769. [Google Scholar] [CrossRef] [Green Version]
- Lanuza, M.A.; Besalduch, N.; González, C.; Santafé, M.M.; Garcia, N.; Tomàs, M.; Nelson, P.G.; Tomàs, J. Decreased phosphorylation of δ and ε subunits of the acetylcholine receptor coincides with delayed postsynaptic maturation in PKC θ deficient mouse. Exp. Neurol. 2010, 225, 183–195. [Google Scholar] [CrossRef]
- Camerino, G.M.; Fonzino, A.; Conte, E.; De Bellis, M.; Mele, A.; Liantonio, A.; Tricarico, D.; Tarantino, N.; Dobrowolny, G.; Musarò, A.; et al. Elucidating the Contribution of Skeletal Muscle Ion Channels to Amyotrophic Lateral Sclerosis in search of new therapeutic options. Sci. Rep. 2019, 9, 3185. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolny, G.; Martini, M.; Scicchitano, B.M.; Romanello, V.; Boncompagni, S.; Nicoletti, C.; Pietrangelo, L.; De Panfilis, S.; Catizone, A.; Bouchè, M.; et al. Muscle Expression of SOD1 G93A Triggers the Dismantlement of Neuromuscular Junction via PKC-Theta. Antioxid. Redox Signal 2018, 28, 1105–1119. [Google Scholar] [CrossRef]
- Nagao, M.; Kato, S.; Oda, M.; Hirai, S. Decrease of protein kinase C in the spinal motor neurons of amyotrophic lateral sclerosis. Acta. Neuropathol. 1998, 96, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Plomp, J.J.; Vergouwe, M.N.; Van den Maagdenberg, A.M.; Ferrari, M.D.; Frants, R.R.; Molenaar, P.C. Abnormal transmitter release at neuromuscular junctions of mice carrying the tottering alpha1A Ca2+ channel mutation. Brain 2000, 123, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Felipo, V.; Miñana, M.D.; Grisolía, S. Inhibitors of protein kinase C prevent the toxicity of glutamate in primary neuronal cultures. Brain Res. 1993, 604, 192–196. [Google Scholar] [CrossRef]
- Krieger, C.; Lanius, R.A.; Pelech, S.L.; Shaw, C. a Amyotrophic lateral sclerosis: The involvement of intracellular Ca2+ and protein kinase C. Trends Pharm. Sci 1996, 17, 114–120. [Google Scholar] [CrossRef]
- Mondola, P.; Damiano, S.; Sasso, A.; Santillo, M. The Cu, Zn Superoxide Dismutase: Not Only a Dismutase Enzyme. Front. Physiol. 2016, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Krieger, C.; Hu, J.H.; Pelech, S. Aberrant protein kinases and phosphoproteins in amyotrophic lateral sclerosis. Trends Pharm. Sci. 2003, 24, 535–541. [Google Scholar] [CrossRef]
- Eisen, A. Clinical Electrophysiology of the Upper and Lower Motor Neuron in Amyotrophic Lateral Sclerosis. Semin. Neurol. 2001, 21, 141–154. [Google Scholar] [CrossRef]
- Rocha, M.C.; Pousinha, P.A.; Correia, A.M.; Sebastião, A.M.; Ribeiro, J.A. Early Changes of Neuromuscular Transmission in the SOD1(G93A) Mice Model of ALS Start Long before Motor Symptoms Onset. PLoS ONE 2013, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wood, S.J.; Slater, C.R. The contribution of postsynaptic folds to the safety factor for neuromuscular transmission in rat fast- and slow-twitch muscles. J. Physiol. 1997, 500, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Andonian, M.H.; Fahim, M.A. Effects of endurance exercise on the morphology of mouse neuromuscular junctions during ageing. J. Neurocytol. 1987, 16, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Tomas, J.; Batlle, J.; Fenoll, M.R.; Santafé, M.; Lanuza, M.A. Activity-dependent plastic changes in the motor nerve terminals of the adult rat. Biol. Cell 1993, 79, 133–137. [Google Scholar] [CrossRef]
- Tomas, J.; Santafé, M.; Lanuza, M.A.; Fenoll-Brunet, M.R. Physiological activity-dependent ultrastructural plasticity in normal adult rat neuromuscular junctions. Biol. Cell 1997, 89, 19–28. [Google Scholar] [CrossRef]
- Deschenes, M.R.; Maresh, C.M.; Crivello, J.F.; Armstrong, L.E.; Kraemer, W.J.; Covault, J. The effects of exercise training of different intensities on neuromuscular junction morphology. J. Neurocytol. 1993, 22, 603–615. [Google Scholar] [CrossRef]
- Dorlöchter, M.; Irintchev, A.; Brinkers, M.; Wernig, A. Effects of enhanced activity on synaptic transmission in mouse extensor digitorum longus muscle. J. Physiol. 1991, 436, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Husain, K.; Somani, S.M. Response of cardiac antioxidant system to alcohol and exercise training in the rat. Alcohol 1997, 14, 301–307. [Google Scholar] [CrossRef]
- Miyazaki, H.; Oh-ishi, S.; Ookawara, T.; Kizaki, T.; Toshinai, K.; Ha, S.; Haga, S.; Ji, L.L.; Ohno, H. Strenuous endurance training in humans reduces oxidative stress following exhausting exercise. Eur. J. Appl. Physiol. 2001, 84, 1–6. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Oscai, L.B.; Don, I.J.; Molé, P.A. Mitochondrial citric acid cycle and related enzymes: Adaptive response to exercise. Biochem. Biophys. Res. Commun. 1970, 40, 1368–1373. [Google Scholar] [CrossRef]
- Acsadi, G.; Anguelov, R.A.; Yang, H.; Toth, G.; Thomas, R.; Jani, A.; Wang, Y.; Ianakova, E.; Mohammad, S.; Lewis, R.A.; et al. Increased Survival and Function of SOD1 Mice After Glial Cell-Derived Neurotrophic Factor Gene Therapy. Hum. Gene 2002, 13, 1047–1059. [Google Scholar] [CrossRef]
- Manabe, Y.; Nagano, I.; Gazi, M.S.A.; Murakami, T.; Shiote, M.; Shoji, M.; Kitagawa, H.; Setoguchi, Y.; Abe, K. Adenovirus-mediated gene transfer of glial cell line-derived neurotrophic factor prevents motor neuron loss of transgenic model mice for amyotrophic lateral sclerosis. Apoptosis 2002, 7, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Funakoshi, H.; Nakamura, T. Overexpression of HGF retards disease progression and prolongs life span in a transgenic mouse model of ALS. J. Neurosci. 2002, 22, 6537–6548. [Google Scholar] [CrossRef]
- Drory, V.E.; Goltsman, E.; Reznik, J.G.; Mosek, A.; Korczyn, A.D. The value of muscle exercise in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2001, 191, 133–137. [Google Scholar] [CrossRef]
- Pinto, A.C.; Alves, M.; Nogueira, A.; Evangelista, T.; Carvalho, J.; Coelho, A.; de Carvalho, M.; Sales-Luís, M.L. Can amyotrophic lateral sclerosis patients with respiratory insufficiency exercise? J. Neurol. Sci. 1999, 169, 69–75. [Google Scholar] [CrossRef]
- Gordon, T.; Tyreman, N.; Li, S.; Putman, C.T.; Hegedus, J. Functional over-load saves motor units in the SOD1-G93A transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2010, 37, 412–422. [Google Scholar] [CrossRef]
- Bello-Haas, V.D.; Florence, J.M.; Kloos, A.D.; Scheirbecker, J.; Lopate, G.; Hayes, S.M.; Pioro, E.P.; Mitsumoto, H. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology 2007, 68, 2003–2007. [Google Scholar] [CrossRef]
- Lunetta, C.; Lizio, A.; Sansone, V.A.; Cellotto, N.M.; Maestri, E.; Bettinelli, M.; Gatti, V.; Melazzini, M.G.; Meola, G.; Corbo, M. Strictly monitored exercise programs reduce motor deterioration in ALS: Preliminary results of a randomized controlled trial. J. Neurol. 2016, 263, 52–60. [Google Scholar] [CrossRef]
- Meyer, R.; Spittel, S.; Steinfurth, L.; Funke, A.; Kettemann, D.; Münch, C.; Meyer, T.; Maier, A. Patient-Reported Outcome of Physical Therapy in Amyotrophic Lateral Sclerosis: Observational Online Study. JMIR Rehabil. Assist. Technol. 2018, 5, e10099. [Google Scholar] [CrossRef]
- Merico, A.; Cavinato, M.; Gregorio, C.; Lacatena, A.; Gioia, E.; Piccione, F.; Angelini, C. Effects of combined endurance and resistance training in Amyotrophic Lateral Sclerosis: A pilot, randomized, controlled study. Eur. J. Transl. Myol. 2018, 28, 72–78. [Google Scholar]
- Kaspar, B.K.; Frost, L.M.; Christian, L.; Umapathi, P.; Gage, F.H. Synergy of insulin-like growth factor-1 and exercise in amyotrophic lateral sclerosis. Ann. Neurol. 2005, 57, 649–655. [Google Scholar] [CrossRef]
- Kirkinezos, I.G.; Hernandez, D.; Bradley, W.G.; Moraes, C.T. Regular exercise is beneficial to a mouse model of amyotrophic lateral sclerosis. Ann. Neurol 2003, 53, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Veldink, J.H.; Bär, P.R.; Joosten, E.A.J.; Otten, M.; Wokke, J.H.J.; Van Den Berg, L.H. Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscul. Disord. 2003, 13, 737–743. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Rodriguez, C.; Devries, M.; Yasuda, N.; Tarnopolsky, M.A. Effects of high-intensity endurance exercise training in the G93A mouse model of amyotrophic lateral sclerosis. Muscle Nerve 2004, 29, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Carreras, I.; Yuruker, S.; Aytan, N.; Hossain, L.; Choi, J.-K.; Jenkins, B.G.; Kowall, N.W.; Dedeoglu, A. Moderate exercise delays the motor performance decline in a transgenic model of ALS. Brain Res. 2010, 1313, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Just-Borràs, L.; Hurtado, E.; Cilleros-Mañé, V.; Biondi, O.; Charbonnier, F.; Tomàs, M.; Garcia, N.; Tomàs, J.; Lanuza, M.A. Running and swimming prevent the deregulation of the BDNF/TrkB neurotrophic signalling at the neuromuscular junction in mice with amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Henriques, A.; Pitzer, C.; Schneider, A. Henriques Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: Where do we stand? Front. Neurosci. 2010, 4, 1–14. [Google Scholar]
- Nagy, G.; Matti, U.; Nehring, R.B.; Binz, T.; Rettig, J.; Neher, E.; Sørensen, J.B. Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J. Neurosci. 2002, 22, 9278–9286. [Google Scholar] [CrossRef] [Green Version]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [Green Version]
- de Majo, M.; Topp, S.D.; Smith, B.N.; Nishimura, A.L.; Chen, H.-J.; Gkazi, A.S.; Miller, J.; Wong, C.H.; Vance, C.; Baas, F.; et al. ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol. Aging 2018, 71, 1–10. [Google Scholar] [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radtke, A.L.; Delbridge, L.M.; Balachandran, S.; Barber, G.N.; O’Riordan, M.X.D. TBK1 protects vacuolar integrity during intracellular bacterial infection. PLoS Pathog. 2007, 3, e29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korac, J.; Schaeffer, V.; Kovacevic, I.; Clement, A.M.; Jungblut, B.; Behl, C.; Terzic, J.; Dikic, I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci. 2013, 126, 580–592. [Google Scholar] [CrossRef] [Green Version]
- N’Diaye, E.-N.; Debnath, J.; Brown, E.J. Ubiquilins accelerate autophagosome maturation and promote cell survival during nutrient starvation. Autophagy 2009, 5, 573–575. [Google Scholar] [CrossRef] [Green Version]
- Rothenberg, C.; Srinivasan, D.; Mah, L.; Kaushik, S.; Peterhoff, C.M.; Ugolino, J.; Fang, S.; Cuervo, A.M.; Nixon, R.A.; Monteiro, M.J. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum. Mol. Genet. 2010, 19, 3219–3232. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, 4439–4448. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Morton, S.; Hesson, L.; Peggie, M.; Cohen, P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 2008, 582, 997–1002. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Řehořová, M.; Vargová, I.; Forostyak, S.; Vacková, I.; Turnovcová, K.; Kupcová Skalníková, H.; Vodička, P.; Kubinová, Š.; Syková, E.; Jendelová, P. A Combination of Intrathecal and Intramuscular Application of Human Mesenchymal Stem Cells Partly Reduces the Activation of Necroptosis in the Spinal Cord of SOD1 G93A Rats. Stem Cells Transl. Med. 2019, 8, 535–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, C.B.; Pluchon, P.; Harley, P.; Rigby, M.; Gonzalez Sabater, V.; Stevenson, D.C.; Hynes, S.; Lowe, A.; Burrone, J.; Viasnoff, V.; et al. In Vitro Modelling of Nerve-Muscle Connectivity in a Compartmentalised Tissue Culture Device. Adv. Biosyst. 2019, 3. [Google Scholar] [CrossRef] [Green Version]
- Burden, S.J.; Yumoto, N.; Zhang, W. The Role of MuSK in Synapse Formation and Neuromuscular Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a009167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilmont, V.; Cadot, B.; Vezin, E.; Le Grand, F.; Gomes, E.R. Dynein disruption perturbs post-synaptic components and contributes to impaired MuSK clustering at the NMJ: Implication in ALS. Sci. Rep. 2016, 6, 27804. [Google Scholar] [CrossRef] [Green Version]
- Sengupta-Ghosh, A.; Dominguez, S.L.; Xie, L.; Barck, K.H.; Jiang, Z.; Earr, T.; Imperio, J.; Phu, L.; Budayeva, H.G.; Kirkpatrick, D.S.; et al. Muscle specific kinase (MuSK) activation preserves neuromuscular junctions in the diaphragm but is not sufficient to provide a functional benefit in the SOD1 G93A mouse model of ALS. Neurobiol. Dis. 2019, 124, 340–352. [Google Scholar] [CrossRef]
- Cantor, S.; Zhang, W.; Delestrée, N.; Remédio, L.; Mentis, G.Z.; Burden, S.J. Preserving neuromuscular synapses in ALS by stimulating MuSK with a therapeutic agonist antibody. eLife 2018, 7, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, S.; Tezuka, T.; Arimura, S.; Tomono, T.; Okada, T.; Yamanashi, Y. DOK7 gene therapy enhances motor activity and life span in ALS model mice. EMBO Mol. Med. 2017, 9, 880–889. [Google Scholar] [CrossRef]
- Perez-Garcia, M.J.; Burden, S.J. Increasing MuSK Activity Delays Denervation and Improves Motor Function in ALS Mice. Cell Rep. 2012, 2, 497. [Google Scholar] [CrossRef] [Green Version]
- Gorlewicz, A.; Wlodarczyk, J.; Wilczek, E.; Gawlak, M.; Cabaj, A.; Majczynski, H.; Nestorowicz, K.; Herbik, M.A.; Grieb, P.; Slawinska, U.; et al. CD44 is expressed in non-myelinating Schwann cells of the adult rat, and may play a role in neurodegeneration-induced glial plasticity at the neuromuscular junction. Neurobiol. Dis. 2009, 34, 245–258. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fukuda, Y.; Yoshimura, J.; Toyoda, A.; Kurppa, K.; Moritoyo, H.; Belzil, V.V.; Dion, P.A.; Higasa, K.; Doi, K.; et al. ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am. J. Hum. Genet. 2013, 93, 900–905. [Google Scholar] [CrossRef] [Green Version]
- Dols-Icardo, O.; García-Redondo, A.; Rojas-García, R.; Borrego-Hernández, D.; Illán-Gala, I.; Muñoz-Blanco, J.L.; Rábano, A.; Cervera-Carles, L.; Juárez-Rufián, A.; Spataro, N.; et al. Analysis of known amyotrophic lateral sclerosis and frontotemporal dementia genes reveals a substantial genetic burden in patients manifesting both diseases not carrying the C9orf72 expansion mutation. J. Neurol. Neurosurg. Psychiatry 2018, 89, 162–168. [Google Scholar] [CrossRef]
- Le Pichon, C.E.; Dominguez, S.L.; Solanoy, H.; Ngu, H.; Lewin-Koh, N.; Chen, M.; Eastham-Anderson, J.; Watts, R.; Scearce-Levie, K. EGFR Inhibitor Erlotinib Delays Disease Progression but Does Not Extend Survival in the SOD1 Mouse Model of ALS. PLoS ONE 2013, 8, e62342. [Google Scholar]
- Chico, L.; Modena, M.; Lo Gerfo, A.; Ricci, G.; Caldarazzo Ienco, E.; Ryskalin, L.; Fornai, F.; Siciliano, G. Cross-talk between pathogenic mechanisms in neurodegeneration: The role of oxidative stress in Amyotrophic Lateral Sclerosis. Arch. Ital. Biol. 2017, 155, 131–141. [Google Scholar]
- Boillée, S.; Cleveland, D.W. Revisiting oxidative damage in ALS: Microglia, Nox, and mutant SOD1. J. Clin. Investig. 2008, 118, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Estevez, A.O.; Morgan, K.L.; Szewczyk, N.J.; Gems, D.; Estevez, M. The neurodegenerative effects of selenium are inhibited by FOXO and PINK1/PTEN regulation of insulin/insulin-like growth factor signaling in Caenorhabditis elegans. Neurotoxicology 2014, 41, 28–43. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.C.; Tresse, E.; Kolaitis, R.-M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P.; et al. VCP Is Essential for Mitochondrial Quality Control by PINK1/Parkin and this Function Is Impaired by VCP Mutations. Neuron 2013, 78, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L.F. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 2015, 11, 422–434. [Google Scholar] [CrossRef] [Green Version]
- Chouhan, A.K.; Zhang, J.; Zinsmaier, K.E.; Macleod, G.T. Presynaptic Mitochondria in Functionally Different Motor Neurons Exhibit Similar Affinities for Ca2+ But Exert Little Influence as Ca2+ Buffers at Nerve Firing Rates In Situ. J. Neurosci. 2010, 30, 1869–1881. [Google Scholar] [CrossRef]
- Recabarren-Leiva, D.; Alarcón, M. New insights into the gene expression associated to amyotrophic lateral sclerosis. Life Sci. 2018, 193, 110–123. [Google Scholar] [CrossRef]
- Léger, B.; Vergani, L.; Sorarù, G.; Hespel, P.; Derave, W.; Gobelet, C.; D’Ascenzio, C.; Angelini, C.; Russell, A.P. Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J. 2006, 20, 583–585. [Google Scholar] [CrossRef]
- Moreno-Igoa, M.; Calvo, A.C.; Penas, C.; Manzano, R.; Oliván, S.; Muñoz, M.J.; Mancuso, R.; Zaragoza, P.; Aguilera, J.; Navarro, X.; et al. Fragment C of tetanus toxin, more than a carrier. Novel perspectives in non-viral ALS gene therapy. J. Mol. Med. 2010, 88, 297–308. [Google Scholar] [CrossRef]
- Kirby, J.; Ning, K.; Ferraiuolo, L.; Heath, P.R.; Ismail, A.; Kuo, S.-W.; Valori, C.F.; Cox, L.; Sharrack, B.; Wharton, S.B.; et al. Phosphatase and tensin homologue/protein kinase B pathway linked to motor neuron survival in human superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 2011, 134, 506–517. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanuza, M.A.; Just-Borràs, L.; Hurtado, E.; Cilleros-Mañé, V.; Tomàs, M.; Garcia, N.; Tomàs, J. The Impact of Kinases in Amyotrophic Lateral Sclerosis at the Neuromuscular Synapse: Insights into BDNF/TrkB and PKC Signaling. Cells 2019, 8, 1578. https://doi.org/10.3390/cells8121578
Lanuza MA, Just-Borràs L, Hurtado E, Cilleros-Mañé V, Tomàs M, Garcia N, Tomàs J. The Impact of Kinases in Amyotrophic Lateral Sclerosis at the Neuromuscular Synapse: Insights into BDNF/TrkB and PKC Signaling. Cells. 2019; 8(12):1578. https://doi.org/10.3390/cells8121578
Chicago/Turabian StyleLanuza, Maria A., Laia Just-Borràs, Erica Hurtado, Víctor Cilleros-Mañé, Marta Tomàs, Neus Garcia, and Josep Tomàs. 2019. "The Impact of Kinases in Amyotrophic Lateral Sclerosis at the Neuromuscular Synapse: Insights into BDNF/TrkB and PKC Signaling" Cells 8, no. 12: 1578. https://doi.org/10.3390/cells8121578
APA StyleLanuza, M. A., Just-Borràs, L., Hurtado, E., Cilleros-Mañé, V., Tomàs, M., Garcia, N., & Tomàs, J. (2019). The Impact of Kinases in Amyotrophic Lateral Sclerosis at the Neuromuscular Synapse: Insights into BDNF/TrkB and PKC Signaling. Cells, 8(12), 1578. https://doi.org/10.3390/cells8121578