Antiglycative Activity and RAGE Expression in Rett Syndrome

 ,
,  ,
,  ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Study Approval
2.3. Blood Sampling
2.4. Human Fibroblasts Culture
2.5. Cell Extract Preparation for Enzymatic Activity Assessments
2.6. Glyoxalase 1 (GLO1) Activity
2.7. Glyoxalase 2 (GLO2) Activity
2.8. Western Immunoblot Analysis
2.9. RNA Extraction and Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) Analysis
2.10. Detection of MG–Protein Adducts by ELISA Assay
2.11. Exogenous MG-Related Cytotoxicity
2.12. Statistics
3. Results
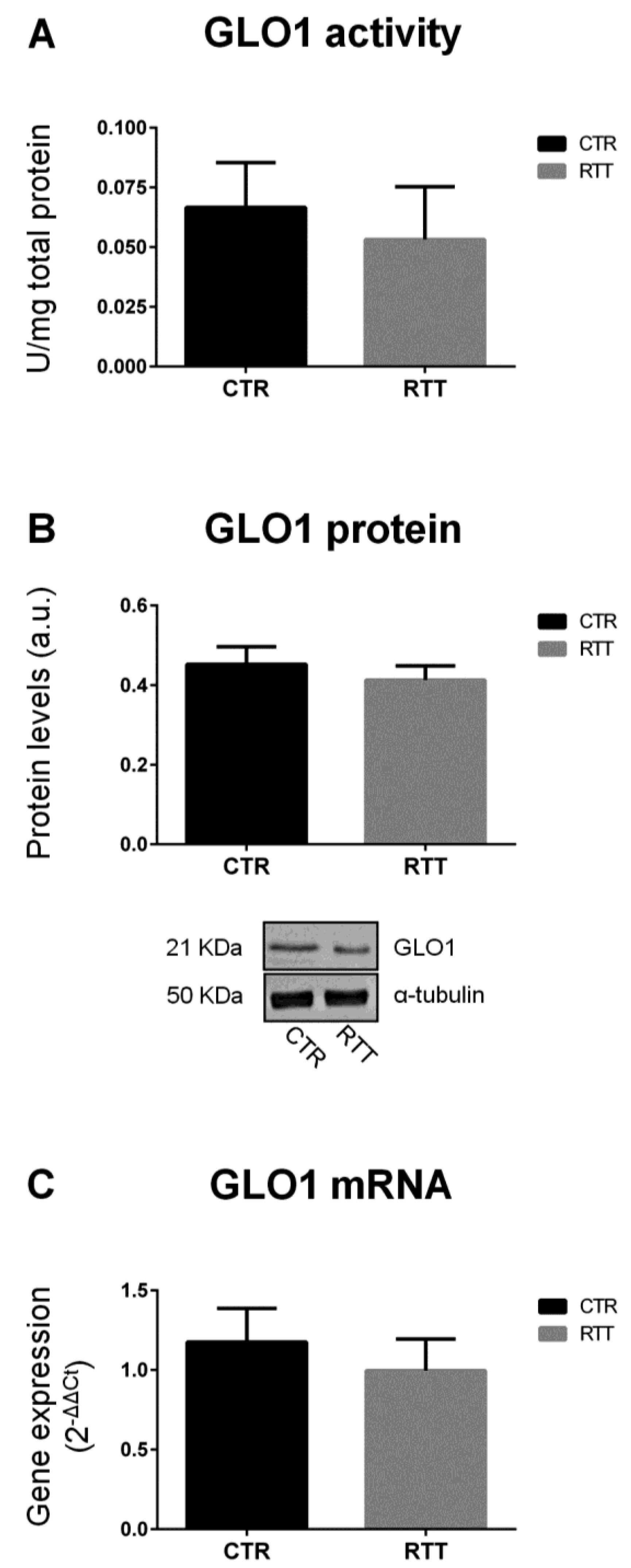
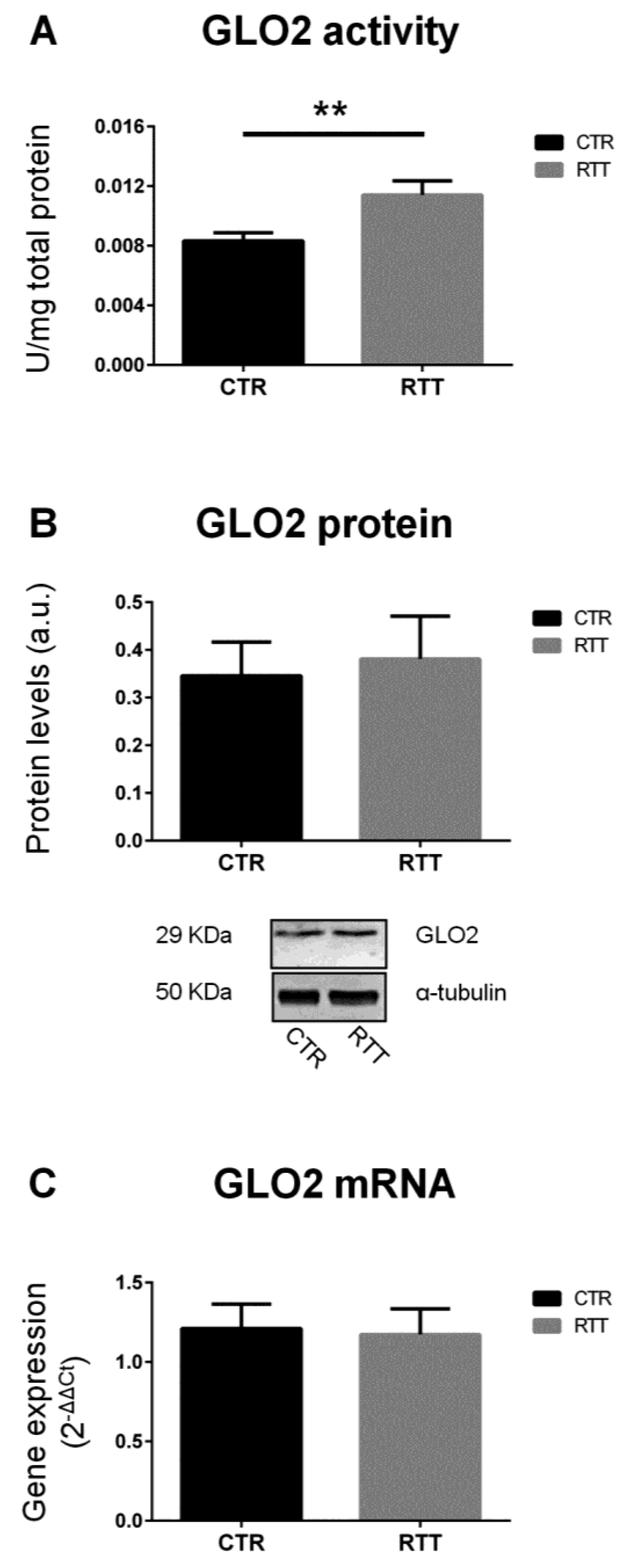
3.1. Evaluation of Glyoxalase (GLO1 and GLO2) Expression and Activity in RTT Cells
3.2. Effect of MG on Cellular Viability
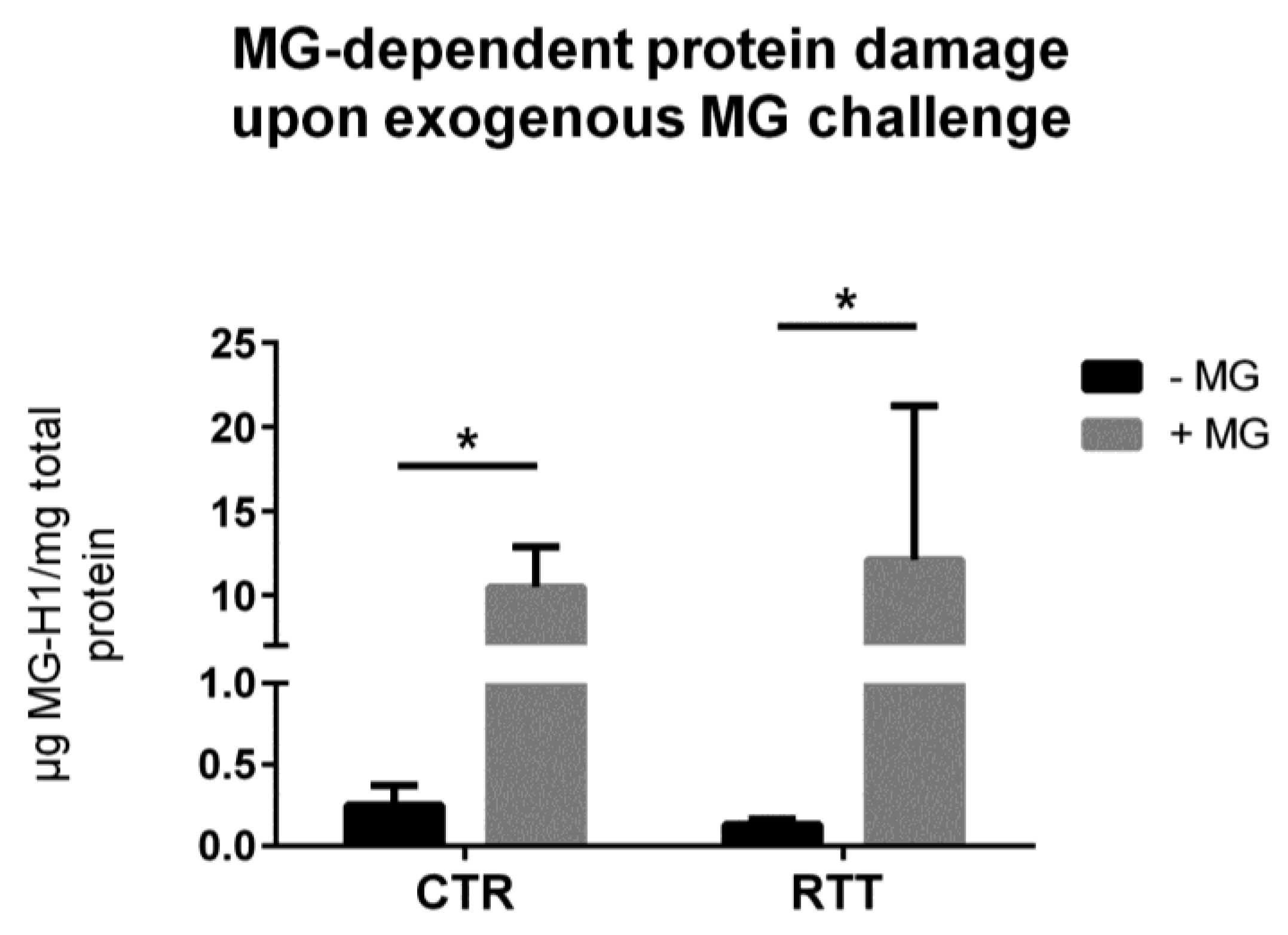
3.3. MG-Dependent Protein Damage in RTT Cells
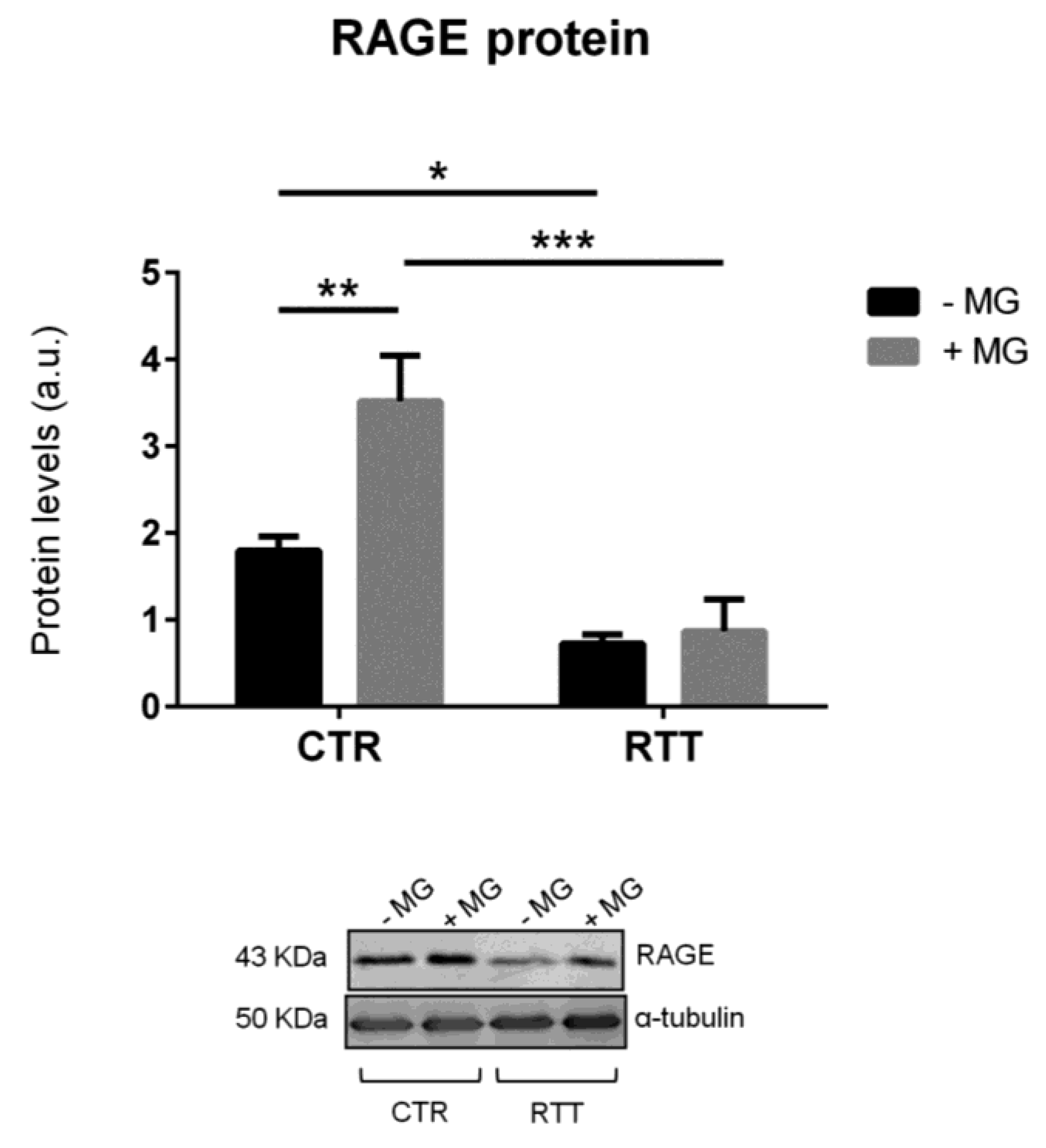
3.4. Evaluation of Cellular RAGE Levels
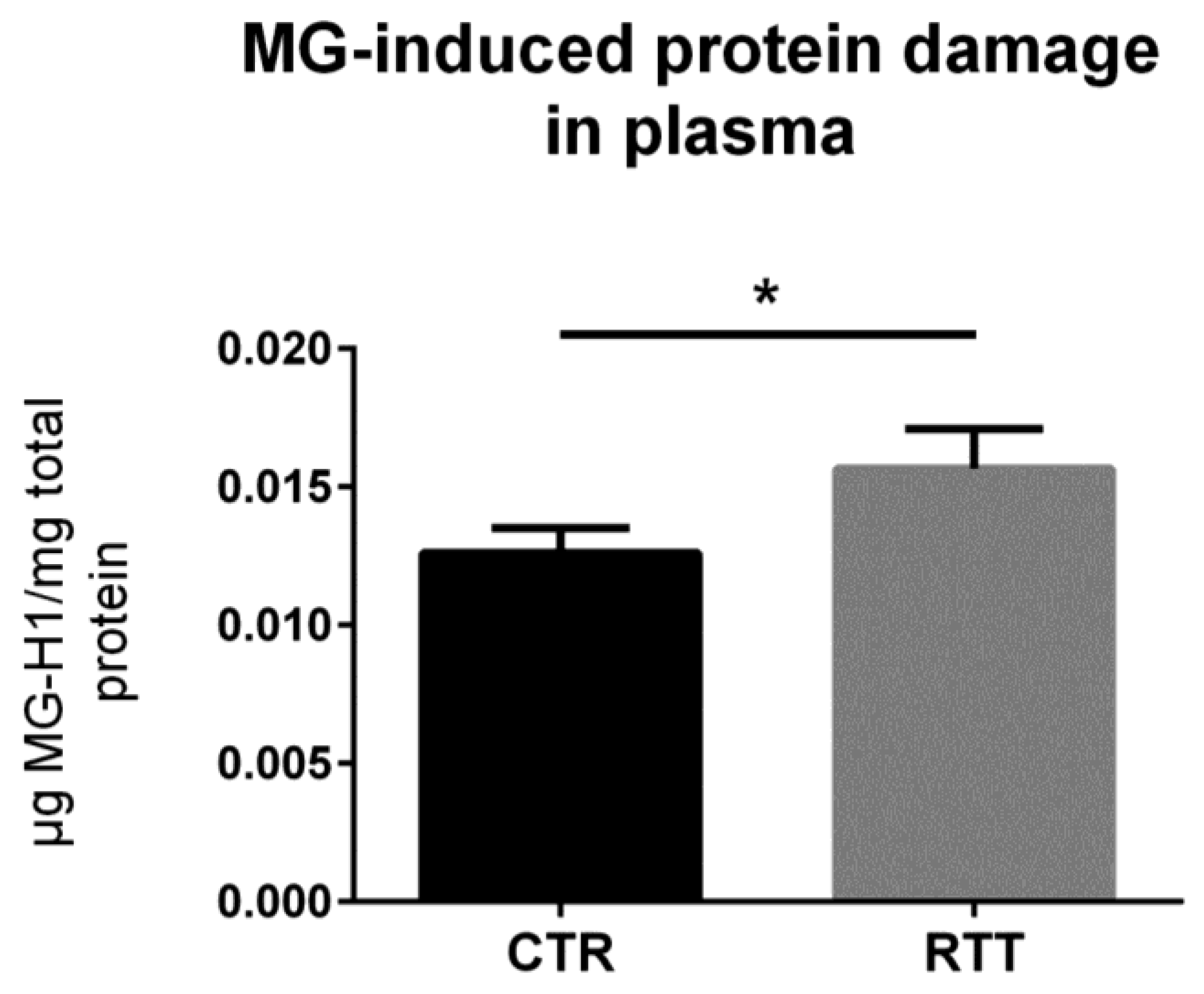
3.5. MG-Dependent Dicarbonyl Damage in Plasma
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Witt-Engerström, I.; Gillberg, C. Rett syndrome in Sweden. J. Autism. Dev. Disord. 1987, 17, 149–150. [Google Scholar] [CrossRef] [PubMed]
- Leonard, H.; Bower, C.; English, D. The prevalence and incidence of Rett syndrome in Australia. Eur. Child Adolesc. Psychiatry 1997, 6, 8–10. [Google Scholar] [PubMed]
- Bienvenu, T.; Philippe, C.; De Roux, N.; Raynaud, M.; Bonnefond, J.P.; Pasquier, L.; Lesca, G.; Mancini, J.; Jonveaux, P.; Moncla, A.; et al. The incidence of Rett syndrome in France. Pediatr. Neurol. 2006, 34, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M.; et al. RettSearch Consortium. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Lyst, M.J.; Bird, A. Rett syndrome: A complex disorder with simple roots. Nat. Rev. Genet. 2015, 16, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Cortelazzo, A.; De Felice, C.; Guerranti, R.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Zollo, G.; Landi, C.; Valacchi, G.; Ciccoli, L.; et al. Subclinical inflammatory status in Rett syndrome. Mediators Inflamm. 2014, 2014, 480980. [Google Scholar] [CrossRef] [PubMed]
- Pecorelli, A.; Cervellati, C.; Hayek, J.; Valacchi, G. OxInflammation in Rett syndrome. Int. J. Biochem. Cell Biol. 2016, 81, 246–253. [Google Scholar] [CrossRef]
- Pecorelli, A.; Ciccoli, L.; Signorini, C.; Leoncini, S.; Giardini, A.; D’Esposito, M.; Filosa, S.; Hayek, J.; De Felice, C.; Valacchi, G. Increased levels of 4HNE-protein plasma adducts in Rett syndrome. Clin. Biochem. 2011, 44, 368–371. [Google Scholar] [CrossRef]
- Leoncini, S.; De Felice, C.; Signorini, C.; Zollo, G.; Cortelazzo, A.; Durand, T.; Galano, J.M.; Guerranti, R.; Rossi, M.; Ciccoli, L.; et al. Cytokine Dysregulation in MECP2- and CDKL5-Related Rett Syndrome: Relationships with Aberrant Redox Homeostasis, Inflammation, and ω-3 PUFAs. Oxid. Med. Cell. Longev. 2015, 2015, 421624. [Google Scholar] [CrossRef] [PubMed]
- Valacchi, G.; Virgili, F.; Cervellati, C.; Pecorelli, A. OxInflammation: From Subclinical Condition to Pathological Biomarker. Front. Physiol. 2018, 9, 858. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, P.; Somanathan, R. Cell signaling and receptors in toxicity of advanced glycation end products (AGEs): α-dicarbonyls, radicals, oxidative stress and antioxidants. J. Recept. Signal Transduct. Res. 2011, 31, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.J.; Hsia, S.M.; Lee, W.H.; Wu, C.H. Polyphenols with antiglycation activity and mechanisms of action: A review of recent findings. J. Food Drug Anal. 2017, 25, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinucci, L.; Balloni, S.; Fettucciari, K.; Bodo, M.; Talesa, V.N.; Antognelli, C. Nicotine induces apoptosis in human osteoblasts via a novel mechanism driven by H(2)O(2) and entailing Glyoxalase 1-dependent MG-H1 accumulation leading to TG2-mediated NF-kB desensitization: Implication for smokers-related osteoporosis. Free Radic. Biol. Med. 2018, 117, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Amicarelli, F.; Colafarina, S.; Cattani, F.; Cimini, A.; Di Ilio, C.; Ceru, M.P.; Miranda, M. Scavenging system efficiency is crucial for cell resistance to ROS-mediated methylglyoxal injury. Free Radic. Biol. Med. 2003, 35, 856–871. [Google Scholar] [CrossRef]
- Kalapos, M.P. The tandem of free radicals and methylglyoxal. Chem. Biol. Interact. 2008, 171, 251–271. [Google Scholar] [CrossRef]
- Antognelli, C.; Gambelunghe, A.; Talesa, V.N.; Muzi, G. Reactive oxygen species induce apoptosis in bronchial epithelial BEAS-2B cells by inhibiting the antiglycation glyoxalase I defence: Involvement of superoxide anion, hydrogen peroxide and NF-κB. Apoptosis 2014, 19, 102–116. [Google Scholar] [CrossRef]
- Di Loreto, S.; Caracciolo, V.; Colafarina, S.; Sebastiani, P.; Gasbarri, A.; Amicarelli, F. Methylglyoxal induces oxidative stress-dependent cell injury and up-regulation of interleukin-1beta and nerve growth factor in cultured hippocampal neuronal cells. Brain Res. 2004, 1006, 157–167. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; Koh, Y.H.; Takahashi, M.; Miyamoto, Y.; Suzuki, K.; Dohmae, N.; Takio, K.; Honke, K.; Taniguchi, N. Identification of the binding site of methylglyoxal on glutathione peroxidase: methylglyoxal inhibits glutathione peroxidase activity via binding to glutathione binding sites Arg 184 and 185. Free Radic. Res. 2003, 37, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, J.; Wu, L. Methylglyoxal-induced mitochondrial dysfunction in vascular smooth muscle cells. Biochem. Pharmacol. 2009, 77, 1709–1716. [Google Scholar] [CrossRef] [PubMed]
- Morcos, M.; Du, X.; Pfisterer, F.; Hutter, H.; Sayed, A.A.; Thornalley, P.; Ahmed, N.; Baynes, J.; Thorpe, S.; Kukudov, G.; et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell 2008, 7, 260–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, K.; Ki, S.H.; Shin, S.M. Methylglyoxal induces mitochondrial dysfunction and cell death in liver. Toxicol. Res. 2014, 30, 193–198. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.H.; Lv, B.L.; Xie, J.Z.; Liu, J.; Zhou, X.W.; Wang, J.Z. AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol. Aging 2012, 33, 1400–1410. [Google Scholar] [CrossRef]
- Salahuddin, P.; Rabbani, G.; Khan, R.H. The role of advanced glycation end products in various types of neurodegenerative disease: A therapeutic approach. Cell. Mol. Biol. Lett. 2014, 19, 407–437. [Google Scholar] [CrossRef]
- Maher, P. Methylglyoxal, advanced glycation end products and autism: Is there a connection? Med. Hypotheses 2012, 78, 548–552. [Google Scholar] [CrossRef]
- Vicente Miranda, H.; Outeiro, T.F. The sour side of neurodegenerative disorders: The effects of protein glycation. J. Pathol. 2010, 221, 13–25. [Google Scholar] [CrossRef]
- Li, X.H.; Du, L.L.; Cheng, X.S.; Jiang, X.; Zhang, Y.; Lv, B.L.; Liu, R.; Wang, J.Z.; Zhou, X.W. Glycation exacerbates the neuronal toxicity of β-amyloid. Cell Death Dis. 2013, 4, e673. [Google Scholar] [CrossRef] [PubMed]
- More, S.S.; Vartak, A.P.; Vince, R. Restoration of glyoxalase enzyme activity precludes cognitive dysfunction in a mouse model of Alzheimer’s disease. ACS Chem. Neurosci. 2013, 4, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Desai, K.M.; Chang, T.; Wang, H.; Banigesh, A.; Dhar, A.; Liu, J.; Untereiner, A.; Wu, L. Oxidative stress and aging: Is methylglyoxal the hidden enemy? Can. J. Physiol. Pharmacol. 2010, 88, 273–284. [Google Scholar] [CrossRef]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Lue, L.F.; Yan, S.; Xu, H.; Luddy, J.S.; Chen, D.; Walker, D.G.; Stern, D.M.; Yan, S.; Schmidt, A.M.; et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010, 24, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Abdelsalam, R.M.; Safar, M.M. Neuroprotective effects of vildagliptin in rat rotenone Parkinson’s disease model: Role of RAGE-NFκB and Nrf2-antioxidant signaling pathways. J. Neurochem. 2015, 133, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Juranek, J.K.; Rai, V. RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55. [Google Scholar] [CrossRef]
- Gasparotto, J.; Girardi, C.S.; Somensi, N.; Ribeiro, C.T.; Moreira, J.C.F.; Michels, M.; Sonai, B.; Rocha, M.; Steckert, A.V.; Barichello, T.; et al. Receptor for advanced glycation end products mediates sepsis-triggered amyloid-ß accumulation, Tau phosphorylation, and cognitive impairment. J. Biol. Chem. 2018, 293, 226–244. [Google Scholar] [CrossRef]
- Cervellati, C.; Sticozzi, C.; Romani, A.; Belmonte, G.; De Rasmo, D.; Signorile, A.; Cervellati, F.; Milanese, C.; Mastroberardino, P.G.; Pecorelli, A.; et al. Impaired enzymatic defensive activity, mitochondrial dysfunction and proteasome activation are involved in RTT cell oxidative damage. Biochim. Biophys. Acta 2015, 1852, 2066–2074. [Google Scholar] [CrossRef] [Green Version]
- Van der Vaart, M.; Svoboda, O.; Weijts, B.G.; Espín-Palazón, R.; Sapp, V.; Pietri, T.; Bagnat, M.; Muotri, A.R.; Traver, D. Mecp2 regulates tnfa during zebrafish embryonic development and acute inflammation. Dis. Model. Mech. 2017, 10, 1439–1451. [Google Scholar] [CrossRef]
- Auburger, G.; Klinkenberg, M.; Drost, J.; Marcus, K.; Morales-Gordo, B.; Kunz, W.S.; Brandt, U.; Broccoli, V.; Reichmann, H.; Gispert, S.; et al. Primary skin fibroblasts as a model of Parkinson’s disease. Mol. Neurobiol. 2012, 46, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Monzio Compagnoni, G.; Kleiner, G.; Bordoni, A.; Fortunato, F.; Ronchi, D.; Salani, S.; Guida, M.; Corti, C.; Pichler, I.; Bergamini, C.; Fato, R.; et al. Mitochondrial dysfunction in fibroblasts of Multiple System Atrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3588–3597. [Google Scholar] [CrossRef]
- Sticozzi, C.; Belmonte, G.; Pecorelli, A.; Cervellati, F.; Leoncini, S.; Signorini, C.; Ciccoli, L.; De Felice, C.; Hayek, J.; Valacchi, G. Scavenger receptor B1 post-translational modifications in Rett syndrome. FEBS Lett. 2013, 587, 2199–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Mannervik, B.; Aronsson, A.C.; Marmstål, E.; Tibbelin, G. Glyoxalase I (rat liver). Methods Enzymol. 1981, 77, 297–301. [Google Scholar] [PubMed]
- Guha, M.K.; Vander Jagt, D.L.; Creighton, D.J. Diffusion-dependent rates for the hydrolysis reaction catalyzed by glyoxalase II from rat erythrocytes. Biochemistry 1988, 27, 8818–8822. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Falone, S.; Santini, S., Jr.; di Loreto, S.; Cordone, V.; Grannonico, M.; Cesare, P.; Cacchio, M.; Amicarelli, F. Improved Mitochondrial and Methylglyoxal-Related Metabolisms Support Hyperproliferation Induced by 50 Hz Magnetic Field in Neuroblastoma Cells. J. Cell. Physiol. 2016, 231, 2014–2025. [Google Scholar] [CrossRef] [PubMed]
- Cavicchio, C.; Benedusi, M.; Pambianchi, E.; Pecorelli, A.; Cervellati, F.; Savelli, V.; Calamandrei, D.; Maellaro, E.; Rispoli, G.; Maioli, E.; Valacchi, G. Potassium Ascorbate with Ribose: Promising Therapeutic Approach for Melanoma Treatment. Oxid. Med. Cell. Longev. 2017, 2017, 4256519. [Google Scholar] [CrossRef] [PubMed]
- Masterjohn, C.; Park, Y.; Lee, J.; Noh, S.K.; Koo, S.I.; Bruno, R.S. Dietary fructose feeding increases adipose methylglyoxal accumulation in rats in association with low expression and activity of glyoxalase-2. Nutrients 2013, 5, 3311–3328. [Google Scholar] [CrossRef] [PubMed]
- Sousa Silva, M.; Gomes, R.A.; Ferreira, A.E.; Ponces Freire, A.; Cordeiro, C. The glyoxalase pathway: The first hundred years... and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Thornalley, P.J.; Dawczynski, J.; Franke, S.; Strobel, J.; Stein, G.; Haik, G.M. Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins. Invest. Ophthalmol. Vis. Sci. 2003, 44, 5287–5292. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Ray, R.; Singer, D.; Böhme, D.; Burz, D.S.; Rai, V.; Hoffmann, R.; Shekhtman, A. The receptor for advanced glycation end products (RAGE) specifically recognizes methylglyoxal-derived AGEs. Biochemistry 2014, 53, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Signorini, C.; Leoncini, S.; De Felice, C.; Pecorelli, A.; Meloni, I.; Ariani, F.; Mari, F.; Amabile, S.; Paccagnini, E.; Gentile, M.; et al. Redox imbalance and morphological changes in skin fibroblasts in typical Rett syndrome. Oxid. Med. Cell. Longev. 2014, 2014, 195935. [Google Scholar] [CrossRef] [PubMed]
- Ercolani, L.; Scirè, A.; Galeazzi, R.; Massaccesi, L.; Cianfruglia, L.; Amici, A.; Piva, F.; Urbanelli, L.; Emiliani, C.; Principato, G.; Armeni, T. A possible S-glutathionylation of specific proteins by glyoxalase II: An in vitro and in silico study. Cell Biochem. Funct. 2016, 34, 620–627. [Google Scholar] [CrossRef]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef] [PubMed]
- Birkenmeier, G.; Stegemann, C.; Hoffmann, R.; Günther, R.; Huse, K.; Birkemeyer, C. Posttranslational modification of human glyoxalase 1 indicates redox-dependent regulation. PLoS ONE. 2010, 5, e10399. [Google Scholar] [CrossRef]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Pecorelli, A.; Cervellati, C.; Cortelazzo, A.; Cervellati, F.; Sticozzi, C.; Mirasole, C.; Guerranti, R.; Trentini, A.; Zolla, L.; Savelli, V.; et al. Proteomic analysis of 4-hydroxynonenal and nitrotyrosine modified proteins in RTT fibroblasts. Int. J. Biochem. Cell Biol. 2016, 81, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef] [Green Version]
- Esterbauer, H.; Cheeseman, K.H.; Dianzani, M.U.; Poli, G.; Slater, T.F. Separation and characterization of the aldehydic products of lipid peroxidation stimulated by ADP-Fe2+ in rat liver microsomes. Biochem. J. 1982, 208, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Hoffbuhr, K.; Devaney, J.M.; LaFleur, B.; Sirianni, N.; Scacheri, C.; Giron, J.; Schuette, J.; Innis, J.; Marino, M.; Philippart, M.; et al. MeCP2 mutations in children with and without the phenotype of Rett syndrome. Neurology 2001, 56, 1486–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffbuhr, K.C.; Moses, L.M.; Jerdonek, M.A.; Naidu, S.; Hoffman, E.P. Associations between MeCP2 mutations, X-chromosome inactivation, and phenotype. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, D.; Simcox, T.; Samaco, R.C.; LaSalle, J.M. X-Chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2-/+ mouse brain. Hum. Mol. Genet. 2004, 13, 1275–1286. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.Y.; Horvath, L.M.; Carrel, L.; Ellis, J. X-chromosome inactivation in rett syndrome human induced pluripotent stem cells. Front. Psychiatry 2012, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Kyle, S.M.; Vashi, N.; Justice, M.J. Rett syndrome: A neurological disorder with metabolic components. Open Biol. 2018, 8, 170216. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Mishra, M. AGE-RAGE Stress, Stressors, and Antistressors in Health and Disease. Int. J. Angiol. 2018, 27, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Ott, C.; Hörlacher, M.; Weber, D.; Höhn, A.; Grune, T. Advanced-glycation-end-product-induced formation of immunoproteasomes: Involvement of RAGE and Jak2/STAT1. Biochem. J. 2012, 448, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortelazzo, A.; De Felice, C.; De Filippis, B.; Ricceri, L.; Laviola, G.; Leoncini, S.; Signorini, C.; Pescaglini, M.; Guerranti, R.; Timperio, A.M.; et al. Persistent Unresolved Inflammation in the Mecp2-308 Female Mutated Mouse Model of Rett Syndrome. Mediators Inflamm. 2017, 2017, 9467819. [Google Scholar] [CrossRef] [PubMed]
- Pecorelli, A.; Cervellati, F.; Belmonte, G.; Montagner, G.; Waldon, P.; Hayek, J.; Gambari, R.; Valacchi, G. Cytokines profile and peripheral blood mononuclear cells morphology in Rett and autistic patients. Cytokine 2016, 77, 180–188. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Mutation Type | AA Change | Age (years) | CSS Total Score |
|---|---|---|---|---|
| 1 | Early Truncating | R168X | 6 | 35 |
| 2 | Early Truncating | K144fs | 22 | 37 |
| 3 | Deletion | c.806delG | 11 | 15 |
| 4 | Missense | R133C | 30 | 9 |
| 5 | Missense | D156E | 8 | 26 |
| 6 | Early Truncating | R270X | 6 | 25 |
| 7 | Early Truncating | R255X | 29 | 12 |
| 8 | Missense | T158M | 12 | 14 |
| 9 | Missense | T158M | 24 | 31 |
| 10 | Large Deletion | 22 | 33 | |
| 11 | Early Truncating | R270X | 14 | 22 |
| 12 | Missense | R106C | 9 | 19 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordone, V.; Pecorelli, A.; Benedusi, M.; Santini, S., Jr.; Falone, S.; Hayek, J.; Amicarelli, F.; Valacchi, G. Antiglycative Activity and RAGE Expression in Rett Syndrome. Cells 2019, 8, 161. https://doi.org/10.3390/cells8020161
Cordone V, Pecorelli A, Benedusi M, Santini S Jr., Falone S, Hayek J, Amicarelli F, Valacchi G. Antiglycative Activity and RAGE Expression in Rett Syndrome. Cells. 2019; 8(2):161. https://doi.org/10.3390/cells8020161
Chicago/Turabian StyleCordone, Valeria, Alessandra Pecorelli, Mascia Benedusi, Silvano Santini, Jr., Stefano Falone, Joussef Hayek, Fernanda Amicarelli, and Giuseppe Valacchi. 2019. "Antiglycative Activity and RAGE Expression in Rett Syndrome" Cells 8, no. 2: 161. https://doi.org/10.3390/cells8020161
APA StyleCordone, V., Pecorelli, A., Benedusi, M., Santini, S., Jr., Falone, S., Hayek, J., Amicarelli, F., & Valacchi, G. (2019). Antiglycative Activity and RAGE Expression in Rett Syndrome. Cells, 8(2), 161. https://doi.org/10.3390/cells8020161