Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer


Abstract
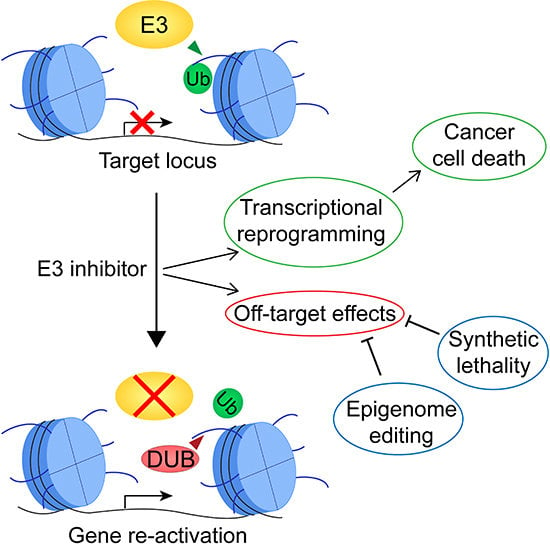
:
1. Introduction
2. Histone Ubiquitination Regulates Gene Transcription and DNA Damage Repair
3. Targeting Aberrant Histone Ubiquitination in Cancer
3.1. Targeting Increased H2AK119ub1 Levels and BMI1 Overexpression in Hematological and Solid Malignancies
3.2. Exploiting Reduced H2BK120ub1 Abundance in Cancer Therapeutics
3.3. Increased H2BK120ub1 Abundance as an Emerging Cancer Therapeutic Target
4. Limitations and Challenges of Therapies Targeting Histone Ubiquitination
5. Harnessing the Full Potential of Therapies Targeting Histone Ubiquitination to Improve Outcomes of Cancer Patients
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Dawson, M.A.; Kouzarides, T. Cancer Epigenetics: From Mechanism to Therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, A.; Helin, K. Chromatin Repressive Complexes in Stem Cells, Development, and Cancer. Cell Stem Cell 2014, 14, 735–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016, 352, aad9780. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.-H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminskas, E.; Farrell, A.T.; Wang, Y.-C.; Sridhara, R.; Pazdur, R. FDA drug approval summary: Azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist 2005, 10, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.J.; Raje, N.S. Panobinostat and Multiple Myeloma in 2018. Oncologist 2018, 23, 516–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Morschhauser, F.; Wilhelm, M.; Pinter-Brown, L.; et al. Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma: Pivotal study update demonstrates durable responses. J. Hematol. Oncol. 2014, 7, 11. [Google Scholar] [CrossRef]
- Bennett, R.L.; Licht, J.D. Targeting Epigenetics in Cancer. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 187–207. [Google Scholar] [CrossRef]
- Morel, D.; Almouzni, G.; Soria, J.-C.; Postel-Vinay, S. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann. Oncol. 2016, 28, mdw552. [Google Scholar] [CrossRef]
- Thompson, L.L.; Guppy, B.J.; Sawchuk, L.; Davie, J.R.; Mcmanus, K.J. Regulation of chromatin structure via histone post-translational modification and the link to carcinogenesis. Cancer Metastasis Rev. 2013, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Takeuchi, M.; Negishi, M.; Oguro, H.; Ichikawa, H.; Iwama, A. Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia 2011, 25, 1335–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdouh, M.; Facchino, S.; Chatoo, W.; Balasingam, V.; Ferreira, J.; Bernier, G. BMI1 Sustains Human Glioblastoma Multiforme Stem Cell Renewal. J. Neurosci. 2009, 29, 8884–8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreso, A.; van Galen, P.; Pedley, N.M.; Lima-Fernandes, E.; Frelin, C.; Davis, T.; Cao, L.; Baiazitov, R.; Du, W.; Sydorenko, N.; et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med. 2014, 20, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Tarcic, O.; Granit, R.Z.; Pateras, I.S.; Masury, H.; Maly, B.; Zwang, Y.; Yarden, Y.; Gorgoulis, V.G.; Pikarsky, E.; Ben-Porath, I.; et al. RNF20 and histone H2B ubiquitylation exert opposing effects in Basal-Like versus luminal breast cancer. Cell Death Differ. 2017, 24, 694–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Huo, D.; Gao, J.; Wu, H.; Ye, Z.; Liu, Z.; Zhang, K.; Shan, L.; Zhou, X.; Wang, Y.; et al. Ubiquitin ligase RNF20/40 facilitates spindle assembly and promotes breast carcinogenesis through stabilizing motor protein Eg5. Nat. Commun. 2016, 7, 12648. [Google Scholar] [CrossRef] [Green Version]
- Shema, E.; Tirosh, I.; Aylon, Y.; Huang, J.; Ye, C.; Moskovits, N.; Raver-Shapira, N.; Minsky, N.; Pirngruber, J.; Tarcic, G.; et al. The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev. 2008, 22, 2664–2676. [Google Scholar] [CrossRef] [Green Version]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Groen, E.J.N.; Gillingwater, T.H. UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration. Trends Mol. Med. 2015, 21, 622–632. [Google Scholar] [CrossRef] [Green Version]
- Tweedie-Cullen, R.Y.; Reck, J.M.; Mansuy, I.M. Comprehensive mapping of post-translational modifications on synaptic, nuclear, and histone proteins in the adult mouse brain. J. Proteome Res. 2009, 8, 4966–4982. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhai, L.; Xu, J.; Joo, H.-Y.; Jackson, S.; Erdjument-Bromage, H.; Tempst, P.; Xiong, Y.; Zhang, Y. Histone H3 and H4 Ubiquitylation by the CUL4-DDB-ROC1 Ubiquitin Ligase Facilitates Cellular Response to DNA Damage. Mol. Cell 2006, 22, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, W.A.; Li, J.; Dou, Y. Effects of histone H2B ubiquitylation on the nucleosome structure and dynamics. Nucleic Acids Res. 2018, 46, 7631–7642. [Google Scholar] [CrossRef]
- McGinty, R.K.; Kim, J.; Chatterjee, C.; Roeder, R.G.; Muir, T.W. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature 2008, 453, 812–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.-Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-Dependent Monoubiquitylation of Histone H2B for Timely Repair of DNA Double-Strand Breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Fierz, B.; Chatterjee, C.; McGinty, R.K.; Bar-Dagan, M.; Raleigh, D.P.; Muir, T.W. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol. 2011, 7, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.M.; Bhattacharyya, A.; Simkus, C.; Vallieres, B.; Veenstra, T.D.; Zhou, M. The RAG1 V(D)J recombinase/ubiquitin ligase promotes ubiquitylation of acetylated, phosphorylated histone 3.3. Immunol. Lett. 2011, 136, 156–162. [Google Scholar] [CrossRef]
- Kalb, R.; Mallery, D.L.; Larkin, C.; Huang, J.T.J.; Hiom, K. BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 2014, 8, 999–1005. [Google Scholar] [CrossRef]
- Goldknopf, I.L.; Taylor, C.W.; Baum, R.M.; Yeoman, L.C.; Olson, M.O.; Prestayko, A.W.; Busch, H. Isolation and characterization of protein A24, a “histone like” non histone chromosomal protein. J. Biol. Chem. 1975, 250, 7182–7187. [Google Scholar] [CrossRef]
- Goldknopf, I.L.; Busch, H. Isopeptide linkage between nonhistone and histone 2A polypeptides of chromosomal conjugate-protein A24. Proc. Natl. Acad. Sci. USA 1977, 74, 864–868. [Google Scholar] [CrossRef]
- West, M.H.; Bonner, W.M. Histone 2B can be modified by the attachment of ubiquitin. Nucleic Acids Res. 1980, 8, 4671–4680. [Google Scholar] [CrossRef] [Green Version]
- Gray, F.; Cho, H.J.; Shukla, S.; He, S.; Harris, A.; Boytsov, B.; Jaremko, Ł.; Jaremko, M.; Demeler, B.; Lawlor, E.R.; Grembecka, J.; et al. BMI1 regulates PRC1 architecture and activity through homo- and hetero-oligomerization. Nat. Commun. 2016, 7, 13343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Cao, R.; Wang, M.; Myers, M.P.; Zhang, Y.; Xu, R.M. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J. Biol. Chem. 2006, 281, 20643–20649. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [Green Version]
- Sahtoe, D.D.; van Dijk, W.J.; Ekkebus, R.; Ovaa, H.; Sixma, T.K. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat. Commun. 2016, 7, 10292. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Jones, A.E.; Yang, W.; Liu, S.; Dai, Q.; Liu, Y.; Swindle, C.S.; Zhou, D.; Zhang, Z.; Ryan, T.M.; et al. The histone H2A deubiquitinase Usp16 regulates hematopoiesis and hematopoietic stem cell function. Proc. Natl. Acad. Sci. USA 2016, 113, E51–E60. [Google Scholar] [CrossRef]
- Nakagawa, T.; Kajitani, T.; Togo, S.; Masuko, N.; Ohdan, H.; Hishikawa, Y.; Koji, T.; Matsuyama, T.; Ikura, T.; Muramatsu, M.; et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 2008, 22, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Chiacchiera, F.; Rossi, A.; Jammula, S.; Piunti, A.; Scelfo, A.; Ordóñez-Morán, P.; Huelsken, J.; Koseki, H.; Pasini, D. Polycomb Complex PRC1 Preserves Intestinal Stem Cell Identity by Sustaining Wnt/β-Catenin Transcriptional Activity. Cell Stem Cell 2016, 18, 91–103. [Google Scholar] [CrossRef]
- Park, I.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef]
- Tian, H.; Biehs, B.; Warming, S.; Leong, K.G.; Rangell, L.; Klein, O.D.; de Sauvage, F.J. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 2011, 478, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguro, H.; Yuan, J.; Ichikawa, H.; Ikawa, T.; Yamazaki, S.; Kawamoto, H.; Nakauchi, H.; Iwama, A. Poised lineage specification in multipotential hematopoietic stem and progenitor cells by the polycomb protein Bmi1. Cell Stem Cell 2010, 6, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Ginjala, V.; Nacerddine, K.; Kulkarni, A.; Oza, J.; Hill, S.J.; Yao, M.; Citterio, E.; van Lohuizen, M.; Ganesan, S. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol. Cell. Biol. 2011, 31, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Ojo, D.; Wei, F.; Wong, N.; Gu, Y.; Tang, D.; Lin, X.; Ojo, D.; Wei, F.; Wong, N.; et al. A Novel Aspect of Tumorigenesis—BMI1 Functions in Regulating DNA Damage Response. Biomolecules 2015, 5, 3396–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chagraoui, J.; Hebert, J.; Girard, S.; Sauvageau, G. An anticlastogenic function for the Polycomb Group gene Bmi1. Proc. Natl. Acad. Sci. USA 2011, 108, 5284–5289. [Google Scholar] [CrossRef] [Green Version]
- Kakarougkas, A.; Ismail, A.; Chambers, A.L.; Riballo, E.; Herbert, A.D.; Künzel, J.; Löbrich, M.; Jeggo, P.A.; Downs, J.A. Requirement for PBAF in Transcriptional Repression and Repair at DNA Breaks in Actively Transcribed Regions of Chromatin. Mol. Cell 2014, 55, 723–732. [Google Scholar] [CrossRef]
- Ochs, F.; Somyajit, K.; Altmeyer, M.; Rask, M.-B.; Lukas, J.; Lukas, C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016, 23, 714–721. [Google Scholar] [CrossRef]
- Mattiroli, F.; Vissers, J.H.A.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 Ubiquitinates K13-15 on H2A/H2AX to Drive DNA Damage Signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Scully, R.; Sobhian, B.; Xie, A.; Shestakova, E.; Livingston, D.M. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011, 25, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Sobhian, B.; Shao, G.; Lilli, D.R.; Culhane, A.C.; Moreau, L.A.; Xia, B.; Livingston, D.M.; Greenberg, R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007, 316, 1198–1202. [Google Scholar] [CrossRef]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1–BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Zhu, Q.; Wani, G.; He, J.; Wang, Q.-E.; Wani, A.A. USP3 counteracts RNF168 via deubiquitinating H2A and γH2AX at lysine 13 and 15. Cell Cycle 2014, 13, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, H.; Liu, J.; Cheruiyot, A.; Lee, J.-H.; Ordog, T.; Lou, Z.; You, Z.; Zhang, Z. USP51 deubiquitylates H2AK13,15ub and regulates DNA damage response. Genes Dev. 2016, 30, 946–959. [Google Scholar] [CrossRef] [Green Version]
- Mosbech, A.; Lukas, C.; Bekker-Jensen, S.; Mailand, N. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J. Biol. Chem. 2013, 288, 16579–16587. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.-M.; Wei, L.; Lan, L.; Huen, M.S.Y. The Lys63-deubiquitylating Enzyme BRCC36 Limits DNA Break Processing and Repair. J. Biol. Chem. 2016, 291, 16197–16207. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Zhu, P.; Wang, J.; Pascual, G.; Ohgi, K.A.; Lozach, J.; Glass, C.K.; Rosenfeld, M.G. Histone H2A Monoubiquitination Represses Transcription by Inhibiting RNA Polymerase II Transcriptional Elongation. Mol. Cell 2008, 29, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Zhou, W.; Wang, J.; Puc, J.; Ohgi, K.A.; Erdjument-Bromage, H.; Tempst, P.; Glass, C.K.; Rosenfeld, M.G. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol. Cell 2007, 27, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Draker, R.; Sarcinella, E.; Cheung, P. USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res. 2011, 39, 3529–3542. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Pfeiffer, H.; Thorne, A.; McMahon, S.B. USP22, an hSAGA subunit and potential cancer stem cell marker, reverses the polycomb-catalyzed ubiquitylation of histone H2A. Cell Cycle 2008, 7, 1522–1524. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Lang, G.; Ito, S.; Bonnet, J.; Metzger, E.; Sawatsubashi, S.; Suzuki, E.; Le Guezennec, X.; Stunnenberg, H.G.; Krasnov, A.; et al. A TFTC/STAGA Module Mediates Histone H2A and H2B Deubiquitination, Coactivates Nuclear Receptors, and Counteracts Heterochromatin Silencing. Mol. Cell 2008, 29, 92–101. [Google Scholar] [CrossRef]
- Zhang, Z.; Jones, A.E.; Wu, W.; Kim, J.; Kang, Y.; Bi, X.; Gu, Y.; Popov, I.K.; Renfrow, M.B.; Vassylyeva, M.N.; et al. Role of remodeling and spacing factor 1 in histone H2A ubiquitination-mediated gene silencing. Proc. Natl. Acad. Sci. USA 2017, 114, E7949–E7958. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.; Dienstbier, M.; Hassan, R.; Schermelleh, L.; Sharif, J.; Blackledge, N.P.; De Marco, V.; Elderkin, S.; Koseki, H.; Klose, R.; et al. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep. 2014, 7, 1456–1470. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.; Grijzenhout, A.; Underwood, E.; Ancelin, K.; Zhang, T.; Nesterova, T.B.; Anil-Kirmizitas, B.; Bassett, A.; Kooistra, S.M.; Agger, K.; et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 2016, 7, 13661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.T.C.; Müller, C.W.; Vermeulen, M.; Müller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Uckelmann, M.; Densham, R.M.; Baas, R.; Winterwerp, H.H.K.; Fish, A.; Sixma, T.K.; Morris, J.R. USP48 restrains resection by site-specific cleavage of the BRCA1 ubiquitin mark from H2A. Nat. Commun. 2018, 9, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zee, B.M.; Wang, Y.; Garcia, B.A.; Dou, Y. The RING Finger Protein MSL2 in the MOF Complex Is an E3 Ubiquitin Ligase for H2B K34 and Is Involved in Crosstalk with H3 K4 and K79 Methylation. Mol. Cell 2011, 43, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zheng, Y.; Pham, A.-D.; Mandal, S.S.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Monoubiquitination of human histone H2B: The factors involved and their roles in HOX gene regulation. Mol. Cell 2005, 20, 601–611. [Google Scholar] [CrossRef]
- Kim, J.; Guermah, M.; McGinty, R.K.; Lee, J.-S.; Tang, Z.; Milne, T.A.; Shilatifard, A.; Muir, T.W.; Roeder, R.G. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell 2009, 137, 459–471. [Google Scholar] [CrossRef]
- Melo-Cardenas, J.; Zhang, Y.; Zhang, D.D.; Fang, D. Ubiquitin-specific peptidase 22 functions and its involvement in disease. Oncotarget 2016, 7, 44848–44856. [Google Scholar] [CrossRef] [Green Version]
- Jeusset, L.; McManus, K. Ubiquitin Specific Peptidase 22 Regulates Histone H2B Mono-Ubiquitination and Exhibits Both Oncogenic and Tumor Suppressor Roles in Cancer. Cancers 2017, 9, 167. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Atanassov, B.S.; Mohan, R.D.; Lan, X.; Kuang, X.; Lu, Y.; Lin, K.; McIvor, E.; Li, W.; Zhang, Y.; Florens, L.; et al. ATXN7L3 and ENY2 Coordinate Activity of Multiple H2B Deubiquitinases Important for Cellular Proliferation and Tumor Growth. Mol. Cell 2016, 62, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, L.; Zhou, B.; Qin, Z.; Dou, Y. H2B Ubiquitylation Promotes RNA Pol II Processivity via PAF1 and pTEFb. Mol. Cell 2014, 54, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kato, A.; Kobayashi, J.; Yanagihara, H.; Sakamoto, S.; Oliveira, D.V.N.P.; Shimada, M.; Tauchi, H.; Suzuki, H.; Tashiro, S.; et al. Regulation of Homologous Recombination by RNF20-Dependent H2B Ubiquitination. Mol. Cell 2011, 41, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Haddad, D.; Li, C.; Le, M.X.; Ling, A.K.; So, C.C.; Nepal, R.M.; Gommerman, J.L.; Yu, K.; Ketela, T.; et al. The SAGA Deubiquitination Module Promotes DNA Repair and Class Switch Recombination through ATM and DNAPK-Mediated γH2AX Formation. Cell Rep. 2016, 15, 1554–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Irrazabal, T.; So, C.C.; Berru, M.; Du, L.; Lam, E.; Ling, A.K.; Gommerman, J.L.; Pan-Hammarström, Q.; Martin, A. The H2B deubiquitinase Usp22 promotes antibody class switch recombination by facilitating non-homologous end joining. Nat. Commun. 2018, 9, 1006. [Google Scholar] [CrossRef] [PubMed]
- Minsky, N.; Oren, M. The RING Domain of Mdm2 Mediates Histone Ubiquitylation and Transcriptional Repression. Mol. Cell 2004, 16, 631–639. [Google Scholar] [CrossRef]
- Li, X.S.; Trojer, P.; Matsumura, T.; Treisman, J.E.; Tanese, N. Mammalian SWI/SNF-A Subunit BAF250/ARID1 Is an E3 Ubiquitin Ligase That Targets Histone H2B. Mol. Cell. Biol. 2010, 30, 1673–1688. [Google Scholar] [CrossRef] [Green Version]
- Nicassio, F.; Corrado, N.; Vissers, J.H.A.; Areces, L.B.; Bergink, S.; Marteijn, J.A.; Geverts, B.; Houtsmuller, A.B.; Vermeulen, W.; Di Fiore, P.P.; et al. Human USP3 Is a Chromatin Modifier Required for S Phase Progression and Genome Stability. Curr. Biol. 2007, 17, 1972–1977. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Varthi, M.; Sykes, S.M.; Phillips, C.; Warzecha, C.; Zhu, W.; Wyce, A.; Thorne, A.W.; Berger, S.L.; McMahon, S.B. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol. Cell 2008, 29, 102–111. [Google Scholar] [CrossRef]
- DeVine, T.; Sears, R.C.; Dai, M.-S. The ubiquitin-specific protease USP36 is a conserved histone H2B deubiquitinase. Biochem. Biophys. Res. Commun. 2018, 495, 2363–2368. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.K.; Vigneron, A.M.; Vousden, K.H. Ubiquitin-specific peptidase 42 (USP42) functions to deubiquitylate histones and regulate transcriptional activity. J. Biol. Chem. 2014, 289, 34862–34870. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Liu, X.; Yang, J.; Li, W.; Liu, S.; Liu, X.; Yang, Z.; Ren, J.; Wang, Y.; Shan, L.; et al. Imbalance of the reciprocally inhibitory loop between the ubiquitin-specific protease USP43 and EGFR/PI3K/AKT drives breast carcinogenesis. Cell Res. 2018, 28, 934–951. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Atanassov, B.S.; Li, W.; Zhang, Y.; Florens, L.; Mohan, R.D.; Galardy, P.J.; Washburn, M.P.; Workman, J.L.; Dent, S.Y.R. USP44 Is an Integral Component of N-CoR that Contributes to Gene Repression by Deubiquitinating Histone H2B. Cell Rep. 2016, 17, 2382–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Jones, A.; Joo, H.-Y.; Zhou, D.; Cao, Y.; Chen, S.; Erdjument-Bromage, H.; Renfrow, M.; He, H.; Tempst, P.; et al. USP49 deubiquitinates histone H2B and regulates cotranscriptional pre-mRNA splicing. Genes Dev. 2013, 27, 1581–1595. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y.; Shema, E.; Moyal, L.; Oren, M. RNF20-RNF40: A ubiquitin-driven link between gene expression and the DNA damage response. FEBS Lett. 2011, 585, 2795–2802. [Google Scholar] [CrossRef] [Green Version]
- Pavri, R.; Zhu, B.; Li, G.; Trojer, P.; Mandal, S.; Shilatifard, A.; Reinberg, D. Histone H2B Monoubiquitination Functions Cooperatively with FACT to Regulate Elongation by RNA Polymerase II. Cell 2006, 125, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Prenzel, T.; Begus-Nahrmann, Y.; Kramer, F.; Hennion, M.; Hsu, C.; Gorsler, T.; Hintermair, C.; Eick, D.; Kremmer, E.; Simons, M.; et al. Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res. 2011, 71, 5739–5753. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, L.; Guo, T.; Wang, Y.; Yang, J. Decreased H2B monoubiquitination and overexpression of ubiquitin-specific protease enzyme 22 in malignant colon carcinoma. Hum. Pathol. 2015, 46, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Urasaki, Y.; Heath, L.; Xu, C.W. Coupling of Glucose Deprivation with Impaired Histone H2B Monoubiquitination in Tumors. PLoS ONE 2012, 7, e36775. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Seshasayee, D.; Noubade, R.; French, D.M.; Liu, J.; Chaurushiya, M.S.; Kirkpatrick, D.S.; Pham, V.C.; Lill, J.R.; Bakalarski, C.E.; et al. Loss of the Tumor Suppressor BAP1 Causes Myeloid Transformation. Science 2012, 337, 1541–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo-Cardenas, J.; Xu, Y.; Wei, J.; Tan, C.; Kong, S.; Gao, B.; Montauti, E.; Kirsammer, G.; Licht, J.D.; Yu, J.; et al. USP22 deficiency leads to myeloid leukemia upon oncogenic Kras activation through a PU.1-dependent mechanism. Blood 2018, 132, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Chroma, K.; Mistrik, M.; Moudry, P.; Gursky, J.; Liptay, M.; Strauss, R.; Skrott, Z.; Vrtel, R.; Bartkova, J.; Kramara, J.; et al. Tumors overexpressing RNF168 show altered DNA repair and responses to genotoxic treatments, genomic instability and resistance to proteotoxic stress. Oncogene 2017, 36, 2405–2422. [Google Scholar] [CrossRef] [PubMed]
- Proctor, E.; Waghray, M.; Lee, C.J.; Heidt, D.G.; Yalamanchili, M.; Li, C.; Bednar, F.; Simeone, D.M. Bmi1 Enhances Tumorigenicity and Cancer Stem Cell Function in Pancreatic Adenocarcinoma. PLoS ONE 2013, 8, e55820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Sui, L.; Xin, T. Correlations of BMI-1 expression and telomerase activity in ovarian cancer tissues. Exp. Oncol. 2008, 30, 70–74. [Google Scholar] [PubMed]
- Senthil Kumar, S.; Sengupta, S.; Lee, K.; Hura, N.; Fuller, C.; DeWire, M.; Stevenson, C.B.; Fouladi, M.; Drissi, R. BMI-1 is a Potential Therapeutic Target in Diffuse Intrinsic Pontine Glioma. Oncotarget 2017, 8, 62962–62975. [Google Scholar] [CrossRef]
- Rizo, A.; Olthof, S.; Han, L.; Vellenga, E.; de Haan, G.; Schuringa, J.J. Repression of BMI1 in normal and leukemic human CD34+ cells impairs self-renewal and induces apoptosis. Blood 2009, 114, 1498–1505. [Google Scholar] [CrossRef] [Green Version]
- Lessard, J.; Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003, 423, 255–260. [Google Scholar] [CrossRef]
- Nishida, Y.; Maeda, A.; Kim, M.J.; Cao, L.; Kubota, Y.; Ishizawa, J.; AlRawi, A.; Kato, Y.; Iwama, A.; Fujisawa, M.; et al. The novel BMI-1 inhibitor PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis in acute myeloid leukemia progenitor cells. Blood Cancer J. 2017, 7, e527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña-Llopis, S.; Vega-Rubín-de-Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFave, L.M.; Béguelin, W.; Koche, R.; Teater, M.; Spitzer, B.; Chramiec, A.; Papalexi, E.; Keller, M.D.; Hricik, T.; Konstantinoff, K.; et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 2015, 21, 1344–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Greenberg, R.A. Deciphering the BRCA1 Tumor Suppressor Network. J. Biol. Chem. 2015, 290, 17724–17732. [Google Scholar] [CrossRef] [Green Version]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [Green Version]
- Chernikova, S.B.; Razorenova, O.V.; Higgins, J.P.; Sishc, B.J.; Nicolau, M.; Dorth, J.A.; Chernikova, D.A.; Kwok, S.; Brooks, J.D.; Bailey, S.M.; et al. Deficiency in mammalian histone H2B ubiquitin ligase Bre1 (Rnf20/Rnf40) leads to replication stress and chromosomal instability. Cancer Res. 2012, 72, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Tarcic, O.; Pateras, I.S.; Cooks, T.; Shema, E.; Kanterman, J.; Ashkenazi, H.; Boocholez, H.; Hubert, A.; Rotkopf, R.; Baniyash, M.; et al. RNF20 Links Histone H2B Ubiquitylation with Inflammation and Inflammation-Associated Cancer. Cell Rep. 2016, 14, 1462–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinsky, G.V.; Berezovska, O.; Glinskii, A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Investig. 2005, 115, 1503–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.L.; Yang, Y.M.; Xu, H.; Dong, X.S. Increased expression of ubiquitin-specific protease 22 can promote cancer progression and predict therapy failure in human colorectal cancer. J. Gastroenterol. Hepatol. 2010, 25, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-D.; Cui, B.-B.; Sun, L.; Zheng, H.; Huang, Q.; Tong, J.-X.; Zhang, Q.-F. The Co-expression of USP22 and BMI-1 May Promote Cancer Progression and Predict Therapy Failure in Gastric Carcinoma. Cell Biochem. Biophys. 2011, 61, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Ning, Z.; Gao, W.; Ling, J.; Wang, A.; Luo, H.; Liang, Y.; Yan, Q.; Wang, Z. Ubiquitin-specific protease 22-induced autophagy is correlated with poor prognosis of pancreatic cancer. Oncol. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Z.; Li, Y. USP22 nuclear expression is significantly associated with progression and unfavorable clinical outcome in human esophageal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2012, 138, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, L.; Zhang, X.; Ji, H.; Wang, L.; Sun, S.; Pang, D. Elevated expression of USP22 in correlation with poor prognosis in patients with invasive breast cancer. J. Cancer Res. Clin. Oncol. 2011, 137, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Shi, H.; Xie, Y.; Zhao, Z.; Li, S.; Chang, C.; Cheng, X.; Li, Y. Ubiquitin specific protease 22 promotes cell proliferation and tumor growth of epithelial ovarian cancer through synergy with transforming growth factor beta1. Oncol. Rep. 2015, 33, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Guppy, B.J.; McManus, K.J. Synthetic lethal targeting of RNF20 through PARP1 silencing and inhibition. Cell. Oncol. 2017, 40, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Kawaoka, S.; Yu, M.; Shi, J.; Ni, T.; Yang, W.; Zhu, J.; Roeder, R.G.; Vakoc, C.R. Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 3901–3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudy, N.S.; Fawzy, I.M.; Azmy, E.; Goda, E.F.; Eneen, A.; Abdul Salam, E.M. BMI1 gene expression in myeloid leukemias and its impact on prognosis. Blood Cells Mol. Dis. 2014, 53, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Xiong, X.; Crim, A.; Dwivedi, S.K.D.; Mustafi, S.B.; Mukherjee, P.; Cao, L.; Sydorenko, N.; Baiazitov, R.; Moon, Y.-C.; et al. Evaluating the Mechanism and Therapeutic Potential of PTC-028, a Novel Inhibitor of BMI-1 Function in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Bedard, P.L.; Shapiro, G.; Bauer, T.M.; Prawira, A.; Laskin, O.; Weetall, M.; Baird, J.; Branstrom, A.; Mara, E.O.; et al. Phase 1 results of PTC596, a novel small molecule targeting cancer stem cells (CSCs) by reducing levels of BMI1 protein. ASCO Annu. Meet. Abstr. 2017, 35, 2574. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.X.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.M.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.; Jeusset, L.; Lepage, C.; McManus, K. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef]
- Schrecengost, R.S.; Dean, J.L.; Goodwin, J.F.; Schiewer, M.J.; Urban, M.W.; Stanek, T.J.; Sussman, R.T.; Hicks, J.L.; Birbe, R.C.; Draganova-Tacheva, R.A.; et al. USP22 regulates oncogenic signaling pathways to drive lethal cancer progression. Cancer Res. 2014, 74, 272–286. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2017, 17, 57–78. [Google Scholar] [CrossRef]
- Landré, V.; Rotblat, B.; Melino, S.; Bernassola, F.; Melino, G. Screening for E3-ubiquitin ligase inhibitors: Challenges and opportunities. Oncotarget 2014, 5, 7988–8013. [Google Scholar] [CrossRef]
- Ritorto, M.S.; Ewan, R.; Perez-Oliva, A.B.; Knebel, A.; Buhrlage, S.J.; Wightman, M.; Kelly, S.M.; Wood, N.T.; Virdee, S.; Gray, N.S.; et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat. Commun. 2014, 5, 4763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrigley, J.D.; Eckersley, K.; Hardern, I.M.; Millard, L.; Walters, M.; Peters, S.W.; Mott, R.; Nowak, T.; Ward, R.A.; Simpson, P.B.; et al. Enzymatic Characterisation of USP7 Deubiquitinating activity and Inhibition. Cell Biochem. Biophys. 2011, 60, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Orcutt, S.J.; Wu, J.; Eddins, M.J.; Leach, C.A.; Strickler, J.E. Bioluminescence assay platform for selective and sensitive detection of Ub/Ubl proteases. Biochim. Biophys. Acta 2012, 1823, 2079–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, A.; Merkx, R.; Berlin, I.; Rodenko, B.; Wijdeven, R.H.M.; El Atmioui, D.; Yalçin, Z.; Robson, C.N.; Neefjes, J.J.; Ovaa, H. Ubiquitin-based probes prepared by total synthesis to profile the activity of deubiquitinating enzymes. Chembiochem 2012, 13, 2251–2258. [Google Scholar] [CrossRef]
- Harrigan, J.; Jacq, X. Monitoring Target Engagement of Deubiquitylating Enzymes Using Activity Probes: Past, Present, and Future; Humana Press: New York, NY, USA, 2016; pp. 395–410. [Google Scholar]
- Herman, A.G.; Hayano, M.; Poyurovsky, M.V.; Shimada, K.; Skouta, R.; Prives, C.; Stockwell, B.R. Discovery of Mdm2-MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discov. 2011, 1, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Kungulovski, G.; Jeltsch, A. Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet. 2016, 32, 101–113. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Ramachandran, S.; Ient, J.; Göttgens, E.-L.; Krieg, A.J.; Hammond, E.M. Epigenetic Therapy for Solid Tumors: Highlighting the Impact of Tumor Hypoxia. Genes 2015, 6, 935–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Writers 1 | Erasers | Readers and Function |
|---|---|---|
| H2AK15ub1 | ||
| RNF168 (recruited by RNF8-mediated H1 polyubiquitination) [27,48] | -USP3 [52] -USP51 [53] May also be directly or indirectly regulated by: -USP44 [54] | DNA damage repair: In the vicinity of a DNA double-strand break (DSB), H2AK15ub1 mediates recruitment of 53BP1 (in conjunction with H4K20 di-methylation). 53BP1 acts as a protein scaffold recruiting DNA repair proteins, which modulate DNA end resection and promote error-free repair [47]. |
| H2AK15 polyubiquitination (K63-linked ubiquitin chains) | ||
| Initiated by RNF168, elongated by RNF8 [48] | BRCC36 [55] | DNA damage repair: Following DSB, H2AK15 polyubiquitination recruits the BRCA1-A complex (including E3 ubiquitin ligase BRCA1/BARD1), which inhibits DNA end resection [49,50]. |
| H2AK119ub1 | ||
| -RING1A/RING1B (catalytic subunit of PRC1 complexes) activated by BMI1 [33,34,35] -DZIP3 [56] | -BAP1, activated by ASXL1, ASLX2 or ASLX3 [36] -MYSM1 [57] -USP3 (DNA damage repair) [52] -USP10 (H2A.Z variant) [58] -USP16 (regulation of hematopoiesis) [37] -USP21 [38] -USP22 [59,60] | Transcriptional repression: -Prevents recruitment of the FACT (facilitates chromatin transcription) complex, which blocks transcription elongation [56] -Prevents methylation of gene-activating marks H3K4me2-3 [38] -Recruits chromatin remodeling protein RSF1 [61] -Recruits PRC2 polycomb repressive complexes, which catalyze the repressive mark H3K27me3 [62,63,64] -Deubiquitination by MYSM1 promotes androgen receptor-dependent gene activation [57] DNA damage response: -Represses transcription of DSB-flanking regions [46] -Promotes rapid DSB repair [46] |
| H2AK127ub1 and H2AK129ub1 | ||
| BRCA1/BARD1 [29] | USP48 [65] | DNA damage response: Recruits SMARCAD1, which promotes DNA end resection and homologous recombination [51] |
| Writers 1 | Erasers | Readers and Function |
|---|---|---|
| H2BK34ub1 | ||
| MSL1/MSL2 [66,73] | Unknown | Transcription activation: -Promotes methylation of transcription-activating marks H3K4me3 and H3K79me2 [66] -Promotes conversion of nucleosomes into hexasomes via H2A/H2B dimer eviction [23] |
| H2BK120ub1 | ||
| -RNF20/RNF40 [67,68] -MDM2 (H2BK120 activity may be restricted to p53 target genes) [77] -BAF250/Elongin-C/Cullin-2/RBX1 [78] | -USP3 (DNA damage repair) [79] -USP22 (transcription regulation; DSB repair) [60,80] -USP27X (transcription regulation) [72] -USP36 (transcriptional repression of target genes) [81] -USP42 (transcriptional activation of target genes) [82] -USP43 (transcriptional repression of NuRD target genes) [83] -USP44 (transcriptional repression of N-CoR target genes) [84] -USP49 (control of mRNA splicing) [85] -USP51 (transcription regulation) [72] | Transcription activation: -Promotes accessible chromatin conformation [26,86] -Promotes transcription elongation by RNA polymerase II in cooperation with FACT [87] -Promotes methylation of transcription-activating marks H3K4me2-3 (by SET1 complex) [68] and H3K79me1-2 (by DOT1L) [24] Transcriptional repression: -MDM2-mediated H2BK120ub1 may promote transcription repression [77] DNA damage repair: -Recruits effectors of the two major DSB repair pathways, non-homologous end-joining and homologous recombination [25,74] -Removal required for class-switch recombination repair, non-homologous end-joining and homologous recombination [75,76] |
| Alteration of Writers or Erasers in Cancer | Clinical Development |
|---|---|
| H2AK15ub1 and H2AK15 poly-ub | |
| Proteotoxic stress (endogenous or treatment-induced) depletes ubiquitin pools available for DNA damage signaling. Overexpression of RNF168 and subsequent alteration of DSB repair processes promotes resistance to proteotoxic stress in cancer cells [95]. | Not applicable |
| H2AK119ub1 | |
| BMI1, the activator of the RING1A/RING1B E3 ubiquitin ligase, is overexpressed and promotes cancer cell self-renewal in multiple cancer types, including pancreatic cancer, glioblastoma multiforme, pediatric diffuse intrinsic pontine glioma, colorectal cancer, epithelial ovarian cancer, and acute myeloid leukemia [12,13,14,96,97,98,99,100]. BMI1 promotes self-renewal of leukemic cells in part via H2AK119ub1-mediated repression of the INK4A/ARF locus [12]. In glioblastoma and colorectal cancer, the effect of BMI1 overexpression on cancer cell self-renewal is independent of the INK4A/ARF locus and involves repression of distinct genes [13,14]. | BMI1 inhibitor PTC-596: -Preclinical studies for acute myeloid leukemia [101] -Phase I for advanced solid tumors (NCT02404480 complete) -Phase Ib in combination with carboplatin/paclitaxel for ovarian cancer patients (NCT03206645) -Phase Ib in combination with radiation for pediatric diffuse intrinsic pontine glioma or high-grade glioma (NCT03605550) |
| Reduced expression of BAP1 (DUB) occurs frequently in metastatic uveal melanoma, pleural mesothelioma, and clear-cell renal cell carcinoma [92,102,103,104]. BAP1 germline mutations are associated with a familial syndrome of predisposition to mesothelioma and uveal and cutaneous melanoma [92,105]. Relevance of aberrant H2AK119ub1 in BAP1-deficient cancers is not established. | BAP1-deficient cells may be targeted with an EZH2 inhibitor via a synthetic lethal strategy [106]. EZH2 inhibitor tazemetostat is in phase II clinical trial for patients with BAP1-deficient malignant mesothelioma (NCT02860286). |
| H2AK127ub1 and H2AK129ub1 | |
| BRCA1, which is frequently deficient in breast and ovarian cancer, is a well-established tumor suppressor protein maintaining genome integrity via its multiple roles in DNA damage repair [107]. Recent identification of BRCA1 as the E3 ubiquitin ligase for H2AK127/129 indicates that misregulation of H2AK127/129ub1 may contribute to genome instability in cancer [29]. | BRCA1 deficiency sensitizes cancer cells to synthetic lethal targeting with PARP1 inhibitors [108,109]. Advanced BRCA1-deficient ovarian cancers are treated with PARP1 inhibitors such as olaparib [110]. |
| H2BK120ub1 | |
| Global loss of H2BK120ub1 is observed in ~70% of breast and colorectal cancers [88,89]. RNF20 promoter is hypermethylated in breast cancer and RNF20/RNF40 expression is reduced in seminoma, basal-like breast cancer, and colorectal cancer [15,17,111,112]. USP22 overexpression is part of the “death from cancer” signature [113] and observed in multiple cancer types, including breast cancer and colorectal cancer [89,114,115,116,117,118,119] | Preclinical study indicates that RNF20-depleted cells can be selectively targeted with PARP1 inhibitor via a synthetic lethal strategy [120] |
| RNF20 promotes breast luminal tumor growth and RNF20-mediated H2BK120ub1 promotes expression of estrogen receptor-α target genes in luminal breast cancer cells [15]. RNF20 expression is required for proliferation of MLL rearrangement-driven leukemia [121] | Not applicable |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeusset, L.M.-P.; McManus, K.J. Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer. Cells 2019, 8, 165. https://doi.org/10.3390/cells8020165
Jeusset LM-P, McManus KJ. Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer. Cells. 2019; 8(2):165. https://doi.org/10.3390/cells8020165
Chicago/Turabian StyleJeusset, Lucile M-P, and Kirk J McManus. 2019. "Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer" Cells 8, no. 2: 165. https://doi.org/10.3390/cells8020165
APA StyleJeusset, L. M. -P., & McManus, K. J. (2019). Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer. Cells, 8(2), 165. https://doi.org/10.3390/cells8020165