The Catalase Gene Family in Cotton: Genome-Wide Characterization and Bioinformatics Analysis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Databases
2.2. Sequence Identification and Functional Annotation Analysis
2.3. Phylogenetic and Synteny Analysis
2.4. Transcription Factor Binding Sites Prediction
2.5. Potential Alternative Splicing Events Analysis
2.6. Putative microRNA Target Sites Analysis
2.7. Plant Growth Conditions and Fungal Pathogen Infection Assays
2.8. RNA Isolation and Expression Profiling Analysis
2.9. Gene Cloning, Vector Construction, Genetic Transformation
3. Results
3.1. Identification of CAT Genes in Upland Cotton and Sea-Island Cotton
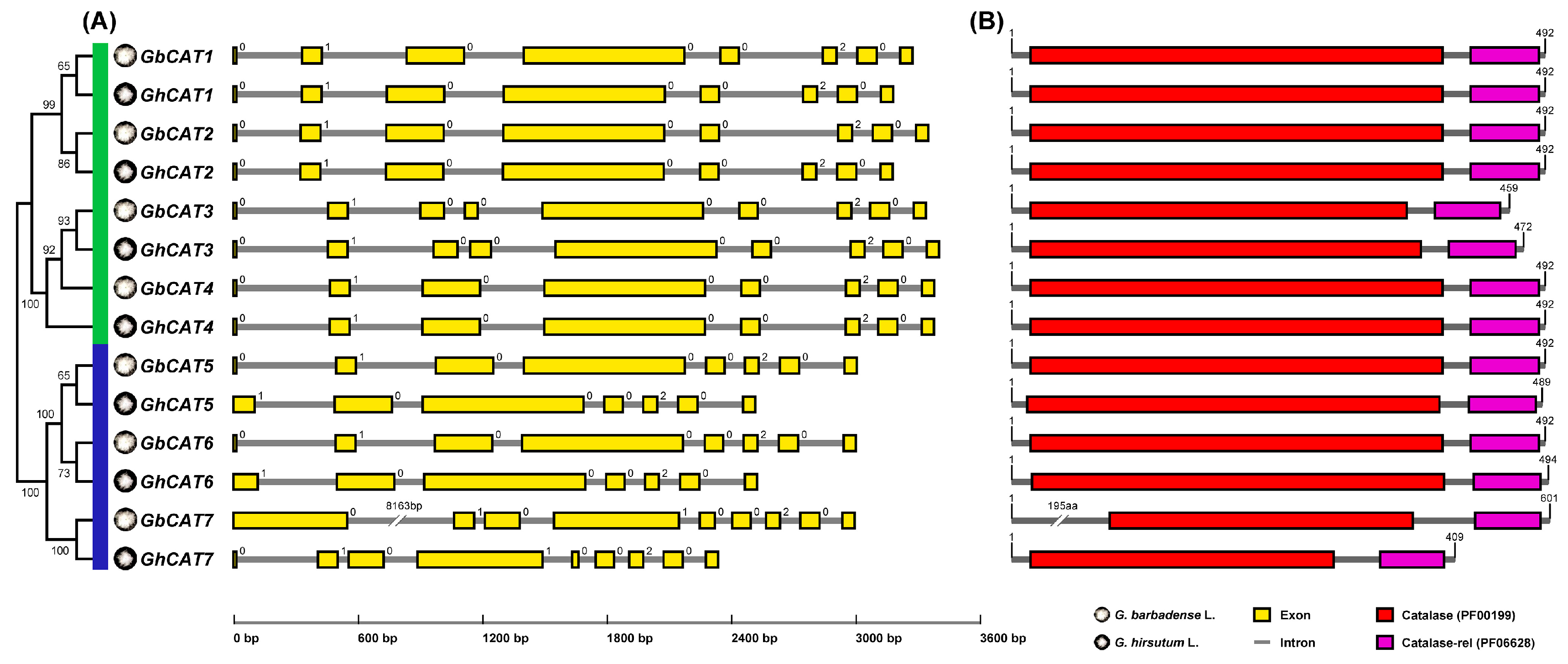
3.2. Sequence Analysis of GhCATs and GbCATs
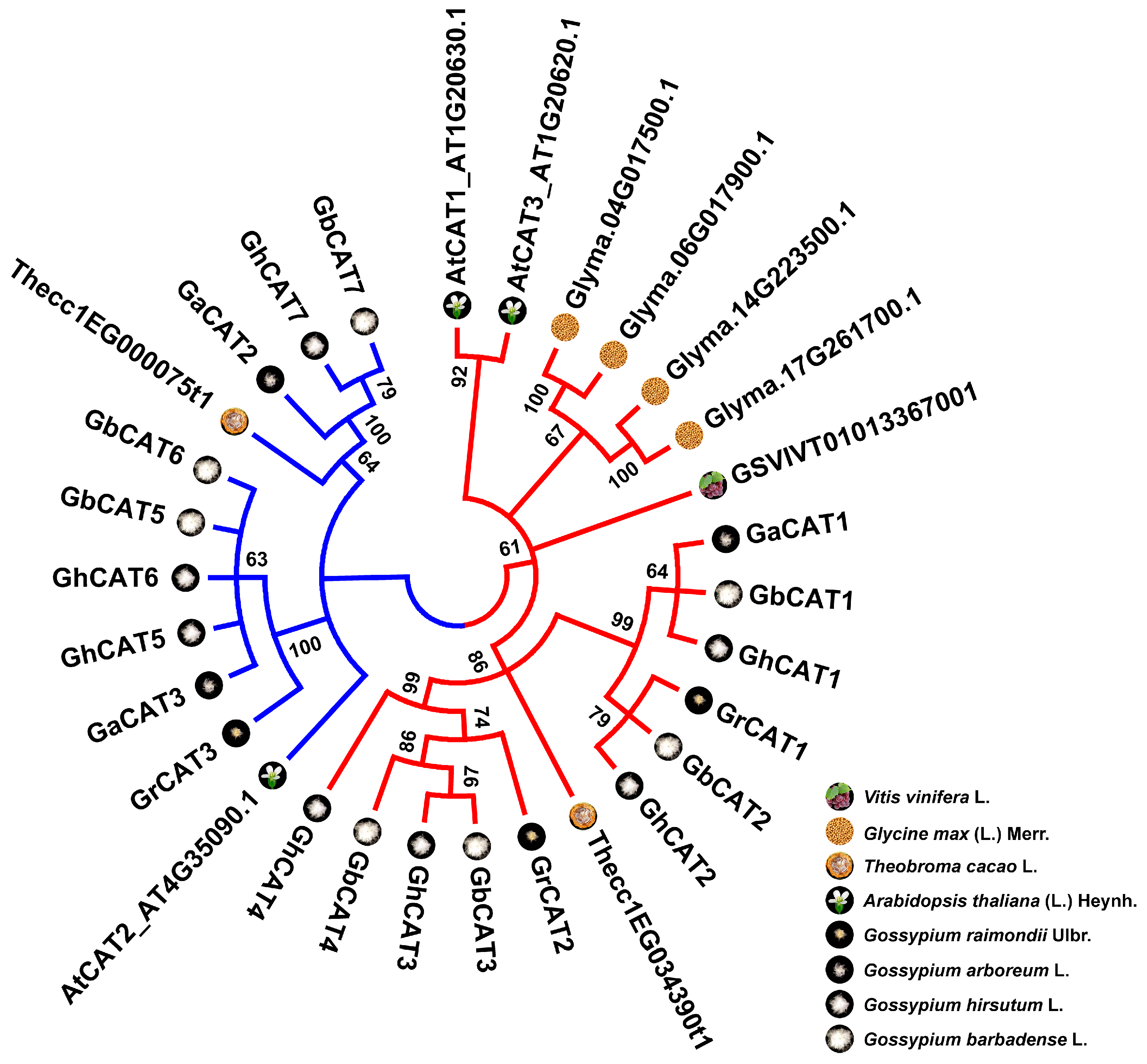
3.3. Phylogenetic Analysis of CAT Gene Family
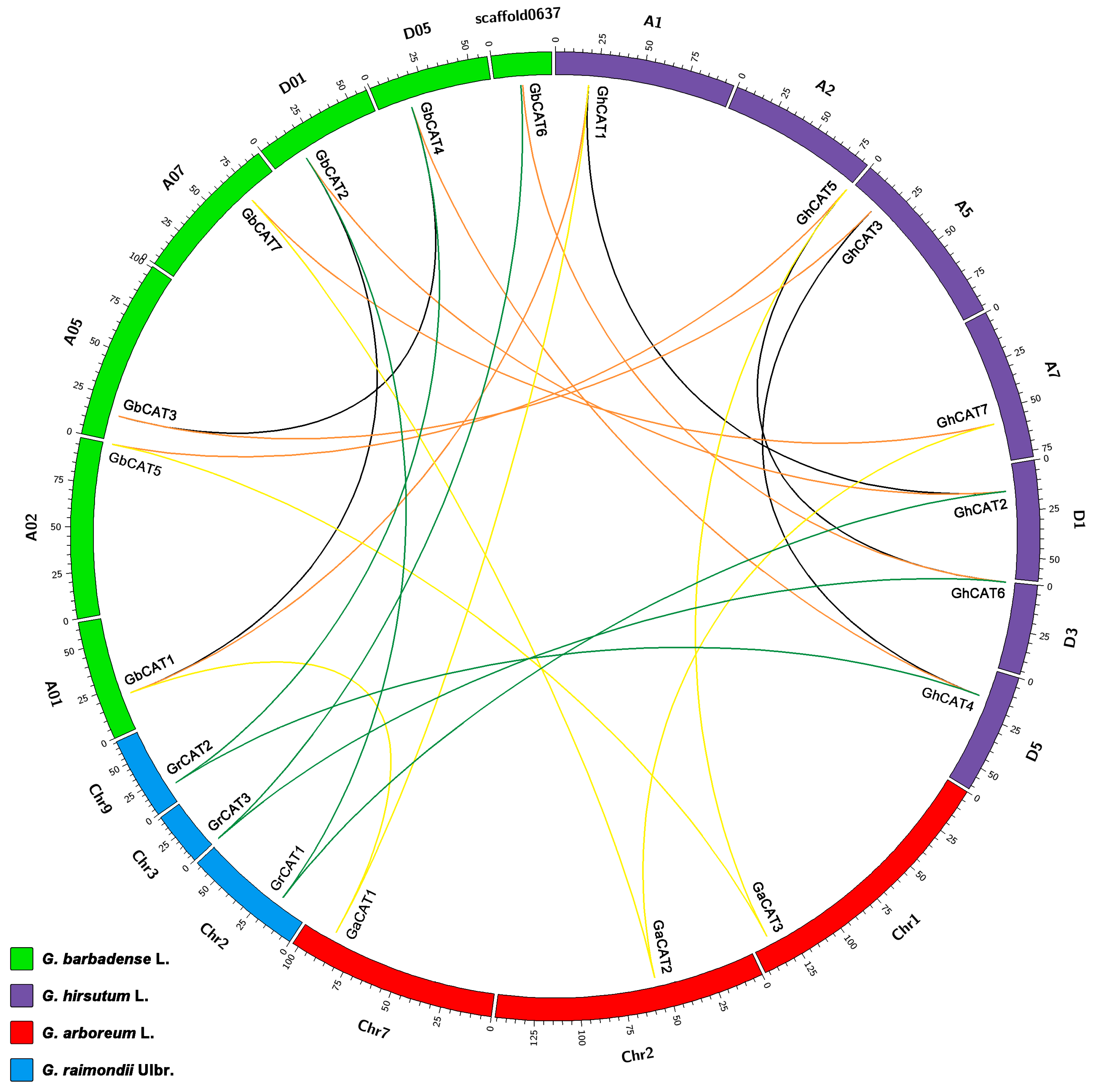
3.4. Synteny Analysis of CAT Genes between Gossypium
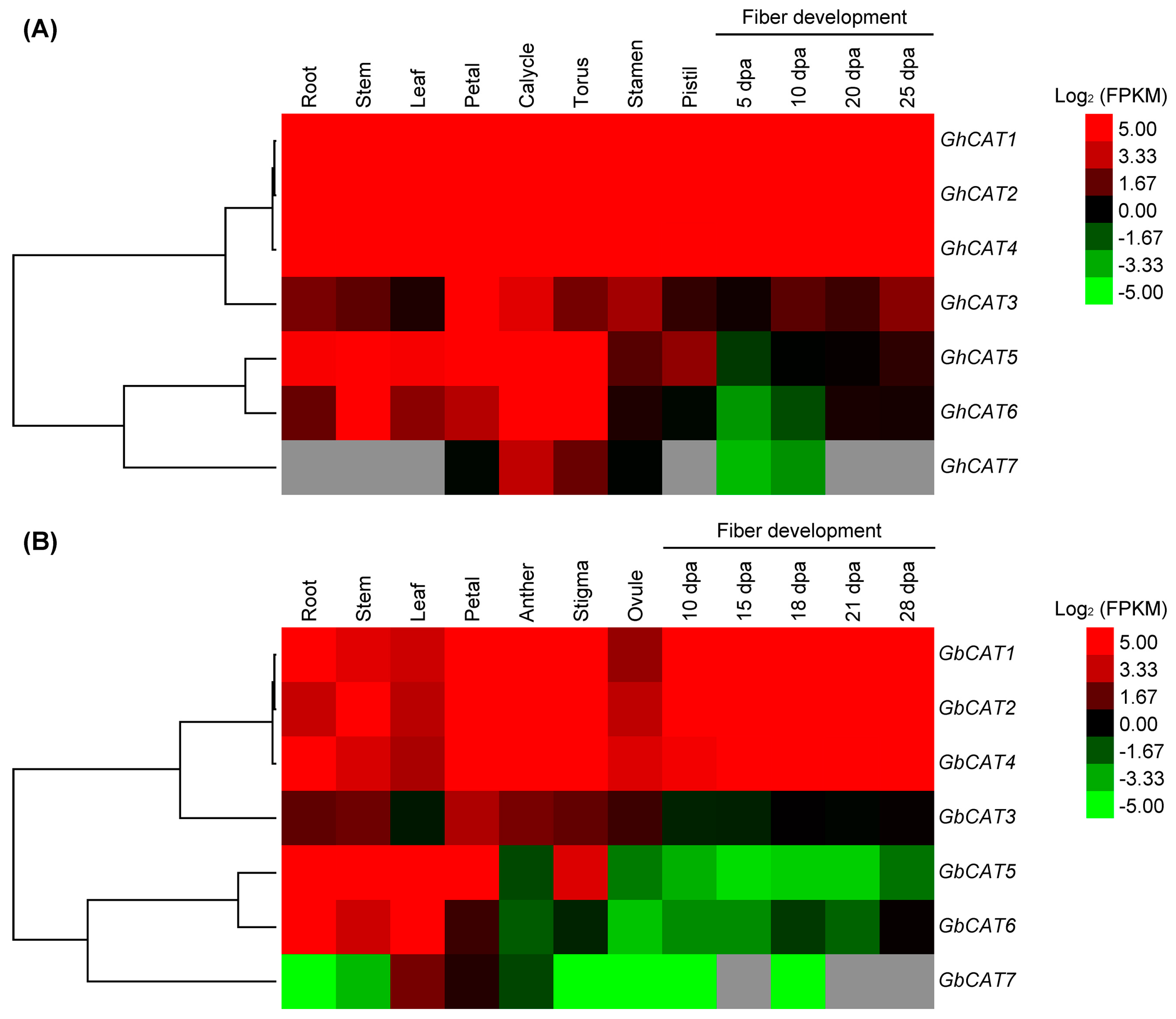
3.5. Expression Analysis of GhCATs and GbCATs in Different Tissues/Organs and Fiber Development Stages
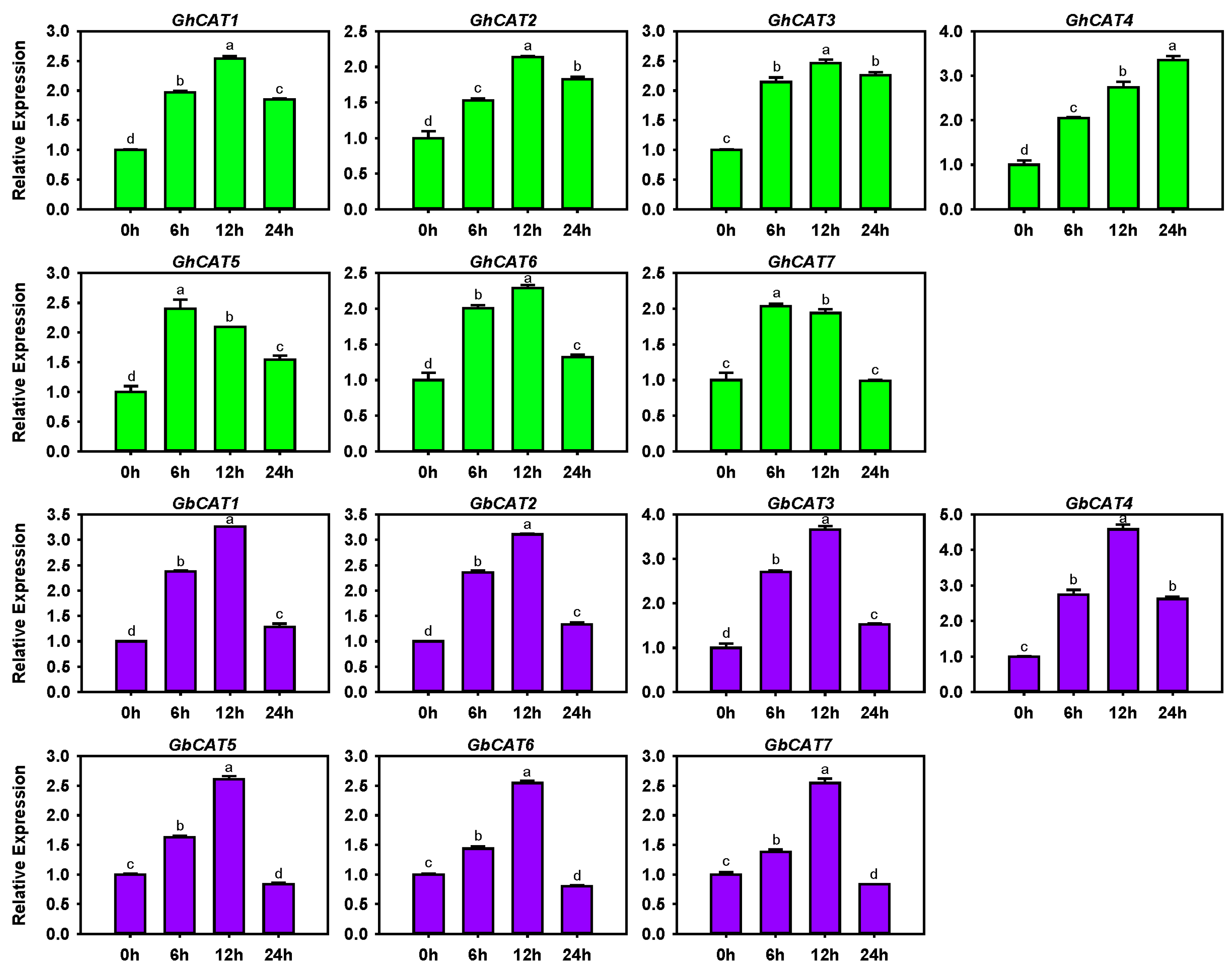
3.6. Transcription Levels of GhCATs and GbCATs under V. dahliae Treatment
3.7. Predictions of Putative Molecular Regulatory Mechanisms of Cotton CATs
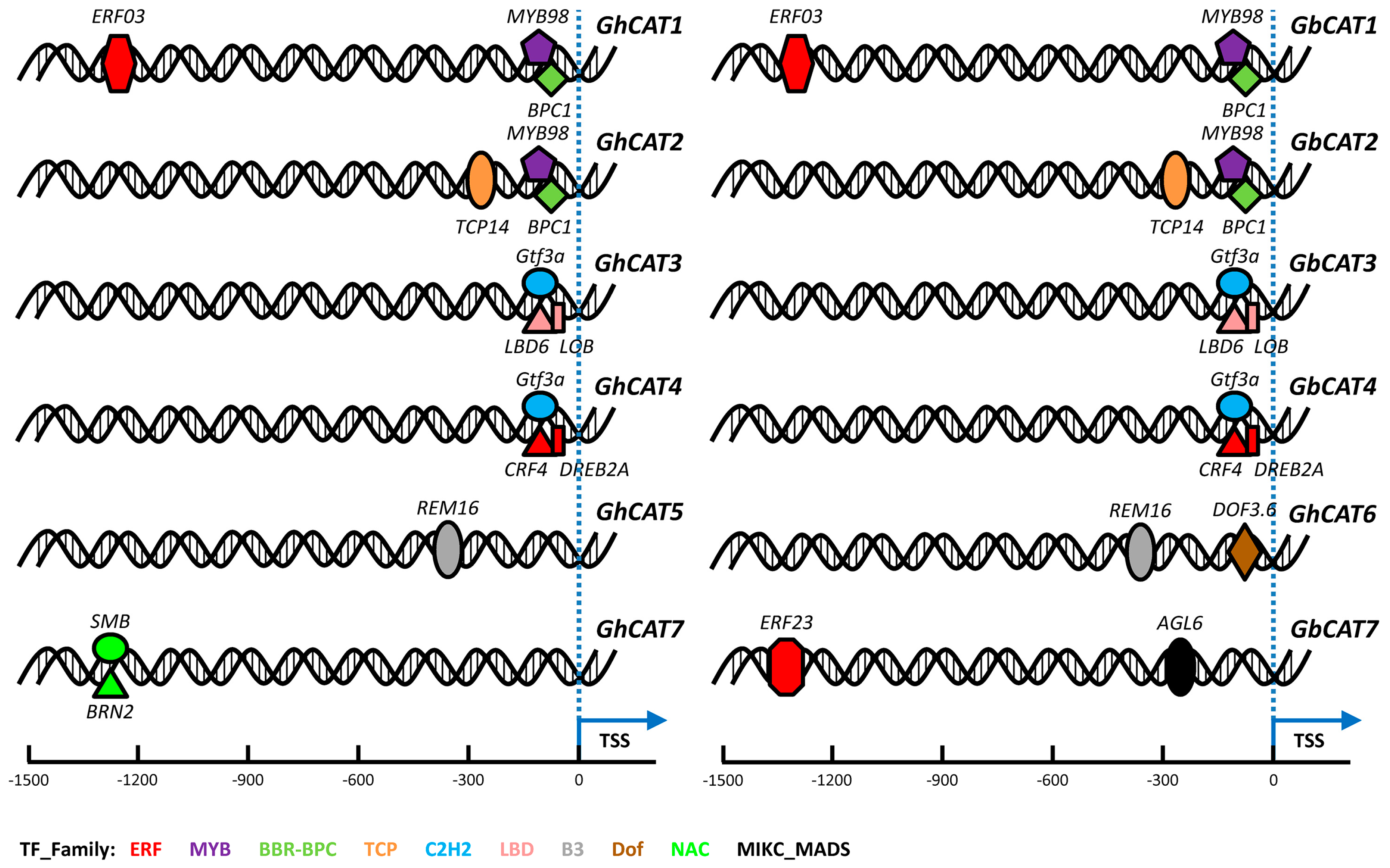
3.7.1. Prediction of Transcription Factor Binding Sites
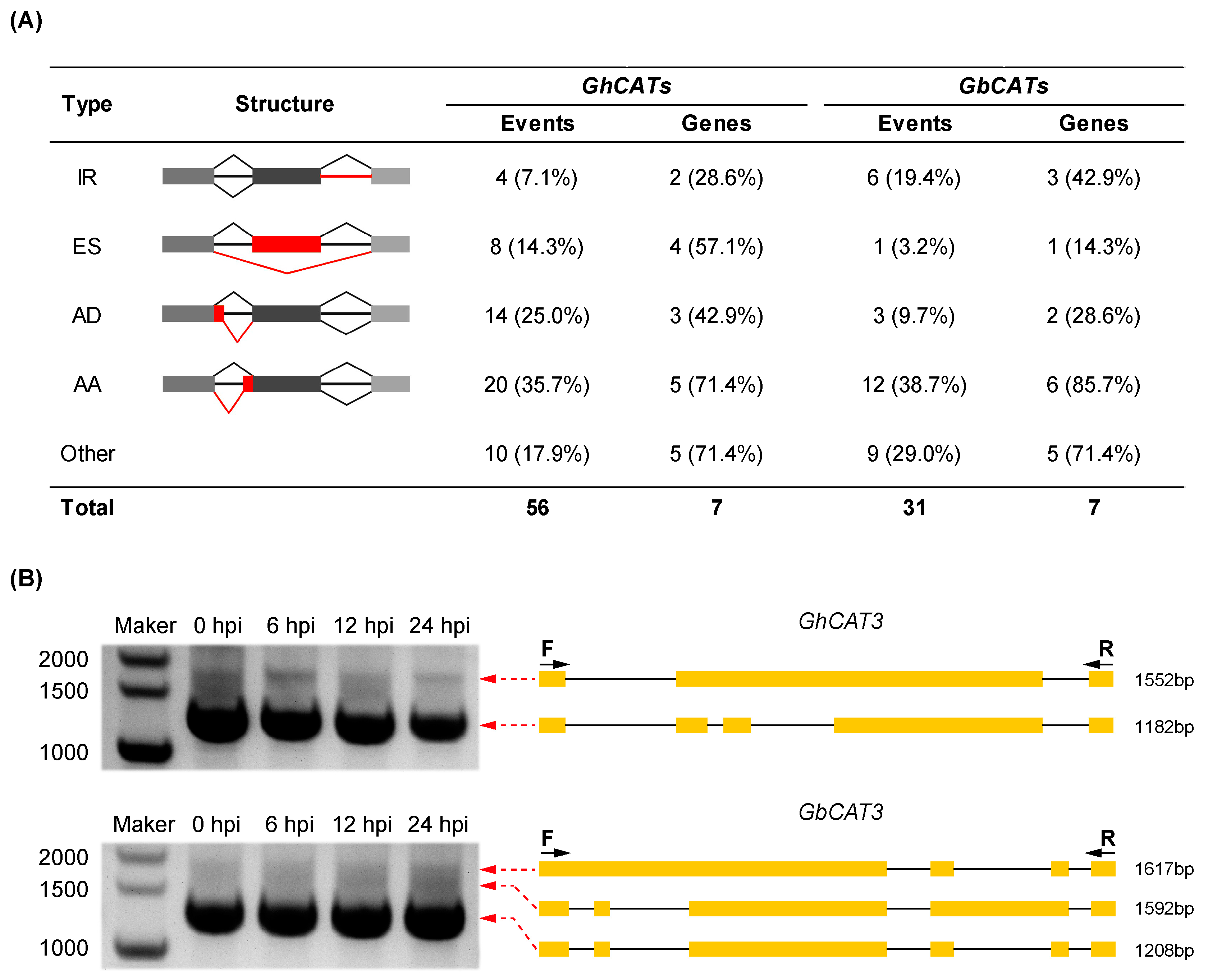
3.7.2. Survey of Alternative Splicing
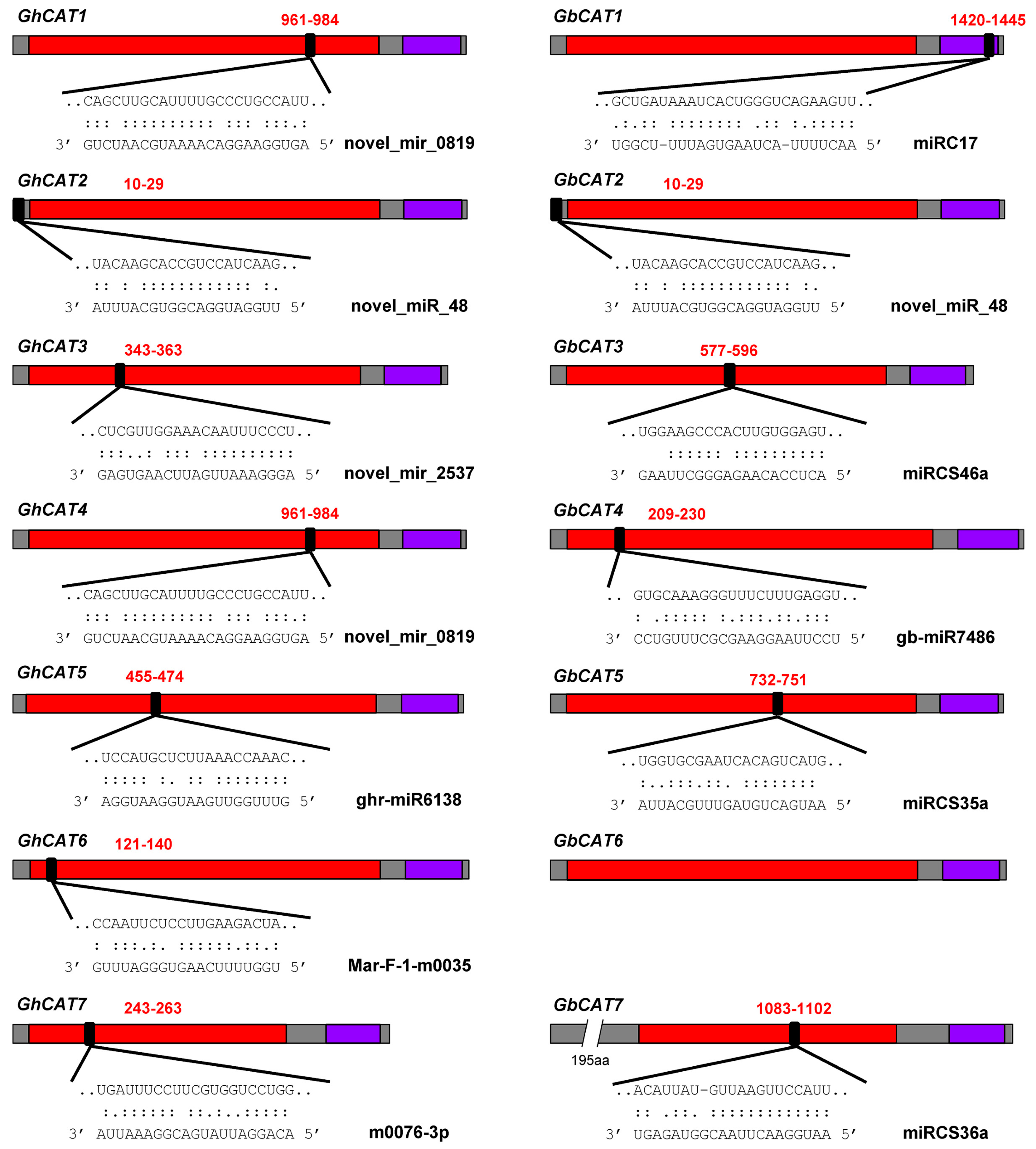
3.7.3. Prediction of miRNA Target Sites
3.7.4. Regulation of Isoforms by miRNAs
4. Discussion
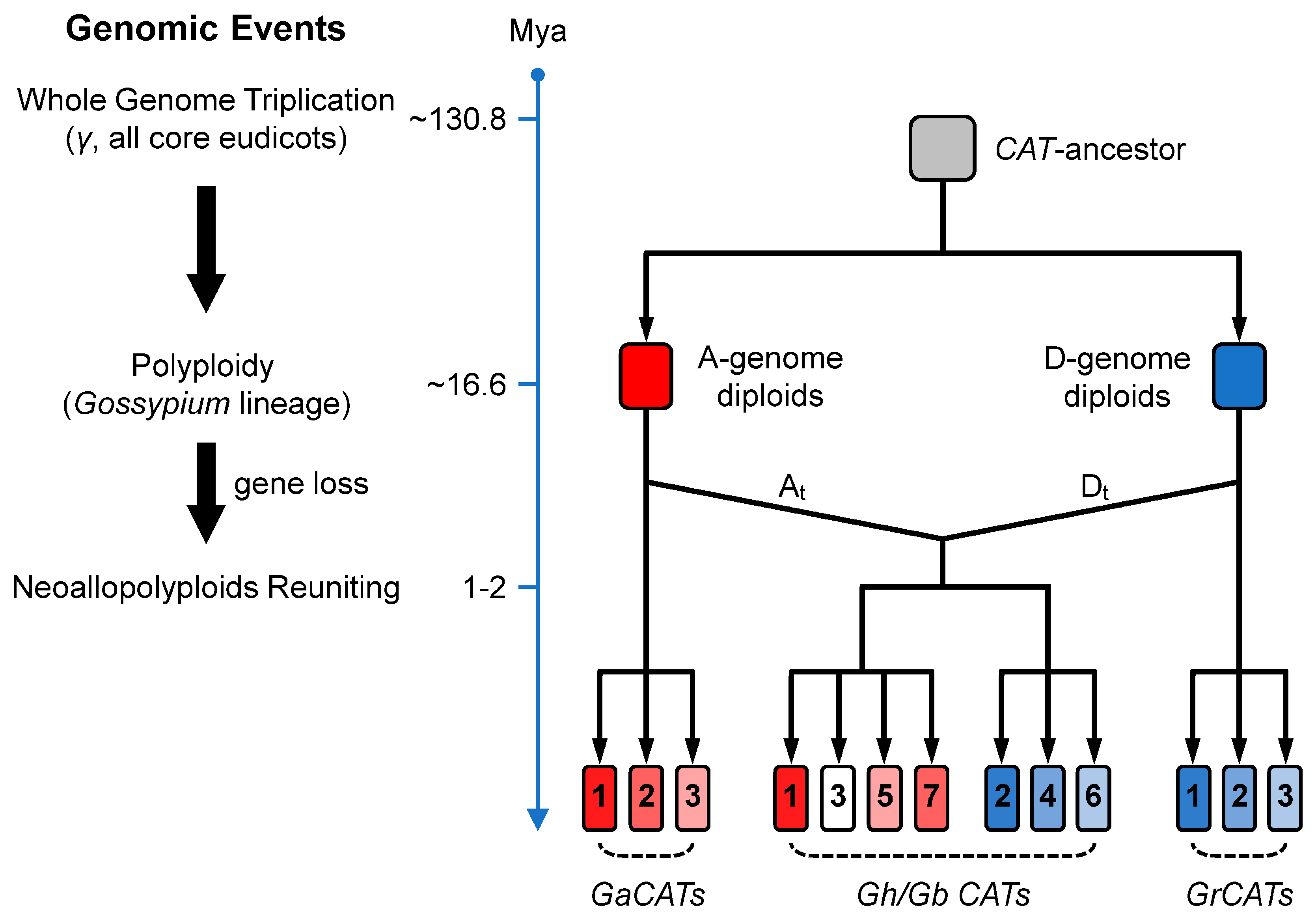
4.1. Evolutionary History of the CAT Gene Family in Gossypium Spp.
4.2. Cotton CAT Genes Probably Function in Defense against Biotic Stress by Mediating ROS Metabolism
4.3. The Potential Regulatory Mechanisms of Cotton CAT Gene Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Choudhury, F.K.; Rivero, R.M.; Blumwald, E.; Mittler, R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 2017, 90, 856–867. [Google Scholar] [CrossRef]
- Mittler, R. Ros are good. Trends Plant Sci. 2016, 22, 11–19. [Google Scholar] [CrossRef]
- Ma, H.; Xiang, G.; Li, Z.; Wang, Y.; Dou, M.; Su, L.; Yin, X.; Liu, R.; Wang, Y.; Xu, Y. Grapevine vppr10.1 functions in resistance to plasmopara viticola through triggering a cell death-like defence response by interacting with vpvdac3. Plant Biotechnol. J. 2018, 16, 1488–1501. [Google Scholar] [CrossRef]
- Fan, J.; Bai, P.; Ning, Y.; Wang, J.; Shi, X.; Xiong, Y.; Zhang, K.; He, F.; Zhang, C.; Wang, R.; et al. The monocot-specific receptor-like kinase SDS2 controls cell death and immunity in rice. Cell Host Microbe 2018, 23, 498–510. [Google Scholar] [CrossRef]
- Del Pozo, J.C. Reactive oxygen species: From harmful molecules to fine-tuning regulators of stem cell niche maintenance. PLoS Genet. 2016, 12, e1006251. [Google Scholar] [CrossRef]
- Wang, W.; Xia, M.X.; Chen, J.; Yuan, R.; Deng, F.N.; Shen, F.F. Gene expression characteristics and regulation mechanisms of superoxide dismutase and its physiological roles in plants under stress. Biochem. Moscow 2016, 81, 465–480. [Google Scholar]
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, X.; Deng, F.; Yuan, R.; Shen, F. Genome-wide characterization and expression analyses of superoxide dismutase (sod) genes in gossypium hirsutum. BMC Genom. 2017, 18, 376. [Google Scholar] [CrossRef]
- Zhou, Y.-B.; Liu, C.; Tang, D.-Y.; Yan, L.; Wang, D.; Yang, Y.-Z.; Gui, J.-S.; Zhao, X.-Y.; Li, L.-G.; Tang, X.-D.; et al. The receptor-like cytoplasmic kinase strk1 phosphorylates and activates catc, thereby regulating h2o2 homeostasis and improving salt tolerance in rice. Plant Cell 2018, 30, 1100–1118. [Google Scholar] [CrossRef]
- Mhamdi, A.; Queval, G.; Chaouch, S.; Vanderauwera, S.; Van Breusegem, F.; Noctor, G. Catalase function in plants: A focus on arabidopsis mutants as stress-mimic models. J. Exp. Bot. 2010, 61, 4197–4220. [Google Scholar] [CrossRef]
- Du, Y.-Y.; Wang, P.-C.; Chen, J.; Song, C.-P. Comprehensive functional analysis of the catalase gene family in arabidopsis thaliana. J. Integr. Plant Biol. 2008, 50, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Willekens, H.; Villarroel, R.; Van Montagu, M.; Inzé, D.; Van Camp, W. Molecular identification of catalases from nicotiana plumbaginifolia (L.). FEBS Lett. 1994, 352, 79–83. [Google Scholar] [CrossRef]
- Guan, L.; Scandalios, J.G. Developmentally related responses of maize catalase genes to salicylic acid. Proc. Natl. Acad. Sci. USA 1995, 92, 5930–5934. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.; Lee, Y.H.; Song, S.I. Rice CatA, CatB, and CatC are involved in environmental stress response, root growth, and photorespiration, respectively. J. Plant Biol. 2014, 57, 375–382. [Google Scholar] [CrossRef]
- Esaka, M.; Yamada, N.; Kitabayashi, M.; Setoguchi, Y.; Tsugeki, R.; Kondo, M.; Nishimura, M. cDNA cloning and differential gene expression of three catalases in pumpkin. Plant Mol. Biol. 1997, 33, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Yang, Y.; Jiang, L.; Liu, S. The Catalase gene family in cucumber: Genome-wide identification and organization. Genet. Mol. Biol. 2016, 39, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Skadsen, R.W.; Schulze-Lefert, P.; Herbst, J.M. Molecular cloning, characterization and expression analysis of two catalase isozyme genes in barley. Plant Mol. Biol. 1995, 29, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-J.; Wu, S.-D.; Huang, G.-J.; Shen, C.-Y.; Afiyanti, M.; Li, W.-J.; Lin, Y.-H. Expression of a cloned sweet potato catalase spcat1 alleviates ethephon-mediated leaf senescence and h2o2 elevation. J. Plant Physiol. 2012, 169, 86–97. [Google Scholar] [CrossRef] [PubMed]
- González, E. The c-terminal domain of plant catalases implications for a glyoxysomal targeting sequence. Eur. J. Biochem. 1991, 199, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Drory, A.; Woodson, W.R. Molecular cloning and nucleotide sequence of a cDNA encoding catalase from tomato. Plant Physiol. 1992, 100, 1605–1606. [Google Scholar] [CrossRef]
- Inaba, J.-I.; Kim, B.M.; Shimura, H.; Masuta, C. Virus-induced necrosis is a consequence of direct protein-protein interaction between a viral RNA-silencing suppressor and a host catalase. Plant Physiol. 2011, 156, 2026–2036. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, T.; Juul, T.; Auzina, A.; Gwiżdż, S.; Małolepszy, A.; Van Der Kelen, K.; Dam, S.; Bressendorff, S.; Lorentzen, A.; Roepstorff, P.; et al. Catalase and NO CATALASE ACTIVITY1 promote autophagy-dependent cell death in Arabidopsis. Plant Cell 2013, 25, 4616–4626. [Google Scholar] [CrossRef]
- Singh, S.; Braus-Stromeyer, S.A.; Timpner, C.; Valerius, O.; von Tiedemann, A.; Karlovsky, P.; Druebert, C.; Polle, A.; Braus, G.H. The plant host Brassica napus induces in the pathogen Verticillium longisporum the expression of functional catalase peroxidase which is required for the late phase of disease. Mol. Plant Microbe Interact. 2011, 25, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Queval, G.; Issakidis-Bourguet, E.; Hoeberichts, F.A.; Vandorpe, M.; Gakière, B.; Vanacker, H.; Miginiac-Maslow, M.; Van Breusegem, F.; Noctor, G. Conditional oxidative stress responses in the arabidopsis photorespiratory mutant cat2 demonstrate that redox state is a key modulator of daylength-dependent gene expression, and define photoperiod as a crucial factor in the regulation of h2o2-induced cell death. Plant J. 2007, 52, 640–657. [Google Scholar]
- Contento, A.L.; Bassham, D.C. Increase in catalase-3 activity as a response to use of alternative catabolic substrates during sucrose starvation. Plant Physiol. Biochem. 2010, 48, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomska, E.; Polzien, L.; Desel, C.; Rozpadek, P.; Miszalski, Z.; Krupinska, K. Spatial patterns of senescence and development-dependent distribution of reactive oxygen species in tobacco (Nicotiana tabacum) leaves. J. Plant Physiol. 2009, 166, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Polidoros, A.N.; Mylona, P.V.; Scandalios, J.G. Transgenic tobacco plants expressing the maize Cat2 gene have altered catalase levels that affect plant-pathogen interactions and resistance to oxidative stress. Transgenic Res. 2001, 10, 555–569. [Google Scholar] [CrossRef]
- Chen, Z.J.; Scheffler, B.E.; Dennis, E.; Triplett, B.A.; Zhang, T.; Guo, W.; Chen, X.; Stelly, D.M.; Rabinowicz, P.D.; Town, C.D.; et al. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 2007, 145, 1303–1310. [Google Scholar] [CrossRef]
- Rathore, K.; Campbell, L.; Sherwood, S.; Nunes, E. Cotton (Gossypium hirsutum L.). In Agrobacterium Protocols; Wang, K., Ed.; Springer: New York, NY, USA, 2015; Volume 1224, pp. 11–23. [Google Scholar]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef]
- Berlanger, I.; Powelson, M.L. Verticillium wilt. Plant Health Instr. 2000, 151, 109–110. [Google Scholar] [CrossRef]
- Li, C.; He, X.; Luo, X.; Xu, L.; Liu, L.; Min, L.; Jin, L.; Zhu, L.; Zhang, X. Cotton wrky1 mediates the plant defense-to-development transition during infection of cotton by Verticillium dahliae by activating Jasmonate zim-domain1 expression. Plant Physiol. 2014, 166, 2179–2194. [Google Scholar] [CrossRef]
- Li, Y.-B.; Han, L.-B.; Wang, H.-Y.; Zhang, J.; Sun, S.-T.; Feng, D.-Q.; Yang, C.-L.; Sun, Y.-D.; Zhong, N.-Q.; Xia, G.-X. The thioredoxin gbnrx1 plays a crucial role in homeostasis of Apoplastic reactive oxygen species in response to Verticillium dahliae infection in cotton. Plant Physiol. 2016, 170, 2392–2406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Rong, W.; Yang, J.; Li, Z.; Wu, L.; Zhang, G.; Ma, Z. Histochemical analyses reveal that stronger intrinsic defenses in Gossypium barbadense than in G. Hirsutum are associated with resistance to Verticillium dahliae. Mol. Plant Microbe Interact. 2017, 30, 984–996. [Google Scholar] [CrossRef]
- Sun, L.; Zhu, L.; Xu, L.; Yuan, D.; Min, L.; Zhang, X. Cotton cytochrome P450 CYP82D regulates systemic cell death by modulating the octadecanoid pathway. Nat. Commun. 2014, 5, 5372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.-L.; Liang, S.; Wang, H.-Y.; Han, L.-B.; Wang, F.-X.; Cheng, H.-Q.; Wu, X.-M.; Qu, Z.-L.; Wu, J.-H.; Xia, G.-X. Cotton major latex protein 28 functions as a positive regulator of the ethylene responsive factor 6 in defense against Verticillium dahliae. Mol. Plant 2014, 8, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. Acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, B.; Zheng, H.-J.; Hu, Y.; Lu, G.; Yang, C.-Q.; Chen, J.-D.; Chen, J.-J.; Chen, D.-Y.; Zhang, L.; et al. Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci. Rep. 2015, 5, 14139. [Google Scholar] [CrossRef]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. CottonFGD: An integrated functional genomics database for cotton. BMC Plant Biol. 2017, 17, 101. [Google Scholar] [CrossRef]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. Mega: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Van Bel, M.; Diels, T.; Vancaester, E.; Kreft, L.; Botzki, A.; Van de Peer, Y.; Coppens, F.; Vandepoele, K. Plaza 4.0: An integrative resource for functional, evolutionary and comparative plant genomics. NAR 2018, 46, D1190–D1196. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, L.; Yuan, D.; Lindsey, K.; Zhang, X. Small RNA and degradome sequencing reveal complex mirna regulation during cotton somatic embryogenesis. J. Exp. Bot. 2013, 64, 1521–1536. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Li, Y.; Han, X.; Shen, F. Genome-wide profiling of miRNAs and other small non-coding RNAs in the Verticillium dahlia-inoculated cotton roots. PLoS ONE 2012, 7, e35765. [Google Scholar]
- Yin, Z.; Li, Y.; Yu, J.; Liu, Y.; Li, C.; Han, X.; Shen, F. Difference in miRNA expression profiles between two cotton cultivars with distinct salt sensitivity. Mol. Biol. Rep. 2012, 39, 4961–4970. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.M.; Xue, W.; Dong, C.J.; Jin, L.G.; Bian, S.M.; Wang, C.; Wu, X.Y.; Liu, J.Y. A comparative mirnaome analysis reveals seven fiber initiation-related and 36 novel mirnas in developing cotton ovules. Mol. Plant 2012, 5, 889–900. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W.; Chen, J.; Liu, J.; Xia, M.; Shen, F. Identification of miRNAs and their targets in cotton inoculated with Verticillium dahliae by high-throughput sequencing and degradome analysis. Int. J. Mol. Sci. 2015, 16, 14749–14768. [Google Scholar] [CrossRef]
- Xie, F.; Wang, Q.; Sun, R.; Zhang, B. Deep sequencing reveals important roles of micrornas in response to drought and salinity stress in cotton. J. Exp. Bot. 2015, 66, 789–804. [Google Scholar] [CrossRef]
- Hu, H.; Yu, D.; Liu, H. Bioinformatics analysis of small RNAs in pima (Gossypium barbadense L.). PLoS ONE 2015, 10, e0116826. [Google Scholar] [CrossRef]
- Liu, N.; Tu, L.; Tang, W.; Gao, W.; Lindsey, K.; Zhang, X. Small RNA and degradome profiling reveals a role for miRNAs and their targets in the developing fibers of Gossypium barbadense. Plant J. 2014, 80, 331–344. [Google Scholar] [CrossRef]
- Maojun, W.; Pengcheng, W.; Fan, L.; Zhengxiu, Y.; Jianying, L.; Chao, S.; Liuling, P.; Feng, W.; Jiang, H.; Lili, T.; et al. A global survey of alternative splicing in allopolyploid cotton: Landscape, complexity and regulation. New Phytol. 2018, 217, 163–178. [Google Scholar]
- Lu, K.; Li, T.; He, J.; Chang, W.; Zhang, R.; Liu, M.; Yu, M.; Fan, Y.; Ma, J.; Sun, W.; et al. qPrimerDB: A thermodynamics-based gene-specific qPCR primer database for 147 organisms. Nucleic Acids Res. 2018, 46, D1229–D1236. [Google Scholar] [CrossRef] [PubMed]
- Sang, J.; Wang, Z.; Li, M.; Cao, J.; Niu, G.; Xia, L.; Zou, D.; Wang, F.; Xu, X.; Han, X.; et al. ICG: A wiki-driven knowledgebase of internal control genes for RT-qPCR normalization. Nucleic Acids Res. 2018, 46, D121–D126. [Google Scholar] [CrossRef] [PubMed]
- Klotz, M.G.; Klassen, G.R.; Loewen, P.C. Phylogenetic relationships among prokaryotic and eukaryotic catalases. Mol. Biol. Evol. 1997, 14, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Bowers, J.E.; Wang, X.; Ming, R.; Alam, M.; Paterson, A.H. Synteny and collinearity in plant genomes. Science 2008, 320, 486–488. [Google Scholar] [CrossRef]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and Angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Ma, J.; Liu, F.; Wang, Q.; Wang, K.; Jones, D.C.; Zhang, B. Comprehensive analysis of TCP transcription factors and their expression during cotton (Gossypium arboreum) fiber early development. Sci. Rep. 2016, 6, 21535. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, H.; Qin, G.; He, C.; Li, B.; Tian, S. The mads-box transcription factor bcmads1 is required for growth, sclerotia production and pathogenicity of Botrytis cinerea. Sci. Rep. 2016, 6, 33901. [Google Scholar] [CrossRef]
- Cosio, C.; Ranocha, P.; Francoz, E.; Burlat, V.; Zheng, Y.; Perry, S.E.; Ripoll, J.J.; Yanofsky, M.; Dunand, C. The class III peroxidase PRX17 is a direct target of the mads-box transcription factor AGAMOUS-LIKE15 (AGL15) and participates in lignified tissue formation. New Phytol. 2017, 213, 250–263. [Google Scholar] [CrossRef]
- Liu, J.; Osbourn, A.; Ma, P. MYB transcription factors as regulators of phenylpropanoid metabolism in plants. Mol. Plant 2015, 8, 689–708. [Google Scholar] [CrossRef]
- Kim, H.J.; Nam, H.G.; Lim, P.O. Regulatory network of NAC transcription factors in leaf senescence. Curr. Opin. Plant Biol. 2016, 33, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-Y.; Guo, X.-J.; Chen, Z.-X.; Chen, W.-Y.; Liu, D.-C.; Zheng, Y.-L.; Liu, Y.-X.; Wei, Y.-M.; Wang, J.-R. Genome-wide characterization of developmental stage- and tissue-specific transcription factors in wheat. BMC Genom. 2015, 16, 125. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413. [Google Scholar] [CrossRef] [PubMed]
- Laloum, T.; Martín, G.; Duque, P. Alternative splicing control of abiotic stress responses. Trends Plant Sci. 2018, 23, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.M. Studies of catalase in plants under abiotic stress. In Antioxidants and Antioxidant Enzymes in Higher Plants; Gupta, D.K., Palma, J.M., Corpas, F.J., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 27–39. [Google Scholar]
- Guo, P.; Li, Z.; Huang, P.; Li, B.; Fang, S.; Chu, J.; Guo, H. A tripartite amplification loop involving the transcription factor WRKY75, salicylic acid, and reactive oxygen species accelerates leaf senescence. Plant Cell 2017, 29, 2854–2870. [Google Scholar] [CrossRef]
- Xing, Y.; Jia, W.; Zhang, J. Atmek1 mediates stress-induced gene expression of cat1 catalase by triggering H2O2 production in Arabidopsis. J. Exp. Bot. 2007, 58, 2969–2981. [Google Scholar] [CrossRef] [PubMed]
- Corpas, F.J.; Barroso, J.B. Lead-induced stress, which triggers the production of nitric oxide (no) and superoxide anion (O2·−) in arabidopsis peroxisomes, affects catalase activity. Nitric Oxide 2017, 68, 103–110. [Google Scholar] [CrossRef]
- Zou, J.-J.; Li, X.-D.; Ratnasekera, D.; Wang, C.; Liu, W.-X.; Song, L.-F.; Zhang, W.-Z.; Wu, W.-H. Arabidopsis CALCIUM-DEPENDENT PROTEIN KINASE8 and CATALASE3 function in abscisic acid-mediated signaling and H2O2 homeostasis in stomatal guard cells under drought stress. Plant Cell 2015, 27, 1445–1460. [Google Scholar] [CrossRef]
- Vighi, I.L.; Benitez, L.C.; do Amaral, M.N.; Auler, P.A.; Moraes, G.P.; Rodrigues, G.S.; da Maia, L.C.; Pinto, L.S.; Braga, E.J. Changes in gene expression and catalase activity in Oryza sativa L. Under abiotic stress. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, S.; Yang, Z.; Yang, Y.; Jiang, L.; Hu, L. Cscat3, a catalase gene from cucumis sativus, confers resistance to a variety of stresses to Escherichia coli. Biotechnol. Biotechnol. Equip. 2017, 31, 886–896. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, X.; Yao, Y.; Xu, L.; Xing, S. Isolation and expression analysis of catalase genes in Erianthus arundinaceus and sugarcane. SugarTech 2016, 18, 468–477. [Google Scholar] [CrossRef]
- Yong, B.; Wang, X.; Xu, P.; Zheng, H.; Fei, X.; Hong, Z.; Ma, Q.; Miao, Y.; Yuan, X.; Jiang, Y.; et al. Isolation and abiotic stress resistance analyses of a catalase gene from Ipomoea batatas (L.) lam. BioMed Res. Int. 2017, 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Ye-Qin, H.; Sheng, L.; Hong-Mei, Y.; Jing, L.; Da-Wei, Y.; Jian-Feng, Z.; Ying-Tang, L. Functional comparison of catalase genes in the elimination of photorespiratory h2o2 using promoter- and 3′-untranslated region exchange experiments in the Arabidopsis Cat2 photorespiratory mutant. Plant Cell Environ. 2010, 33, 1656–1670. [Google Scholar]
- Yamasaki, K.; Kigawa, T.; Seki, M.; Shinozaki, K.; Yokoyama, S. DNA-binding domains of plant-specific transcription factors: Structure, function, and evolution. Trends Plant Sci. 2013, 18, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, Z.; Chern, M.; Yin, J.; Yang, C.; Ran, L.; Cheng, M.; He, M.; Wang, K.; Wang, J.; et al. A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell 2017, 170, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Pang, C.; Fan, S.; Song, M.; Wei, H.; Yu, S. Global analysis of the Gossypium hirsutum L. Transcriptome during leaf senescence by RNA-Seq. BMC Plant Biol. 2015, 15, 43. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, J.; Du, M.-L.; Li, L.; Wang, X.-L.; Li, X.-B. A cotton gene encoding myb-like transcription factor is specifically expressed in pollen and is involved in regulation of late anther/pollen development. Plant Cell Physiol. 2013, 54, 893–906. [Google Scholar] [CrossRef]
- Sun, H.; Hu, M.; Li, J.; Chen, L.; Li, M.; Zhang, S.; Zhang, X.; Yang, X. Comprehensive analysis of NAC transcription factors uncovers their roles during fiber development and stress response in cotton. BMC Plant Biol. 2018, 18, 150. [Google Scholar] [CrossRef]
- Chen, T.; Li, W.; Hu, X.; Guo, J.; Liu, A.; Zhang, B. A cotton MYB transcription factor, GbMYB5, is positively involved in plant adaptive response to drought stress. Plant Cell Physiol. 2015, 56, 917–929. [Google Scholar] [CrossRef]
- Xu, F.-C.; Liu, H.-L.; Xu, Y.-Y.; Zhao, J.-R.; Guo, Y.-W.; Long, L.; Gao, W.; Song, C.-P. Heterogeneous expression of the cotton R2R3-MYB transcription factor GbMYB60 increases salt sensitivity in transgenic Arabidopsis. Plant Cell 2018, 133, 15–25. [Google Scholar] [CrossRef]
- He, X.; Zhu, L.; Xu, L.; Guo, W.; Zhang, X. GhATAF1, a NAC transcription factor, confers abiotic and biotic stress responses by regulating phytohormonal signaling networks. Plant Cell Rep. 2016, 35, 2167–2179. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.N.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Green, R.E.; Brenner, S.E. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mrna decay in humans. Proc. Natl. Acad. Sci. USA 2003, 100, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.; Zhao, J.-H.; Guo, H.-S. Trans-kingdom RNA silencing in plant–fungal pathogen interactions. Mol. Plant 2018, 11, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, D.; Zhang, X.; Chen, D.; Cheng, Y.; Shen, F. Plant microRNAs in cross-kingdom regulation of gene expression. Int. J. Mol. Sci. 2018, 19, 2007. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Burd, S.; Lers, A. Mir408 is involved in abiotic stress responses in Arabidopsis. Plant J. 2015, 84, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, G.; Saini, A.; Sunkar, R. Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta 2009, 229, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Weiberg, A.; Wang, M.; Lin, F.-M.; Zhao, H.; Zhang, Z.; Kaloshian, I.; Huang, H.-D.; Jin, H. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 2013, 342, 118–123. [Google Scholar] [CrossRef]
- Wang, B.; Sun, Y.; Song, N.; Zhao, M.; Liu, R.; Feng, H.; Wang, X.; Kang, Z. Puccinia striiformis f. sp. Tritici microRNA-like RNA 1 (Pst-milR1), an important pathogenicity factor of Pst, impairs wheat resistance to Pst by suppressing the wheat pathogenesis-related 2 gene. New Phytol. 2017, 215, 338–350. [Google Scholar] [CrossRef]
- Wang, M.; Weiberg, A.; Lin, F.-M.; Thomma, B.P.H.J.; Huang, H.-D.; Jin, H. Bidirectional cross-kingdom rnai and fungal uptake of external rnas confer plant protection. Nat. Plants 2016, 2, 16151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, Y.-L.; Zhao, J.-H.; Wang, S.; Jin, Y.; Chen, Z.-Q.; Fang, Y.-Y.; Hua, C.-L.; Ding, S.-W.; Guo, H.-S. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2016, 2, 16153. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Locus ID | Gene Features | Transcript Features | Protein Statistics | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genomic Position | Length (bp) | CDS Length (bp) | CDS GC Content (%) | Exon Number | Protein Length (aa) | Molecular Weight (kDa) | Isoelectric Point (pI) | GRAVY | Predicted Subcellular Localization | ||
| GhCAT1 | Gh_A01G0845 | A01:19440294-19443475 − | 3182 | 1479 | 46.9 | 8 | 492 | 56.829 | 7.173 | −0.533 | Peroxisome |
| GhCAT2 | Gh_D01G0873 | D01:14348913-14352092 − | 3180 | 1479 | 47.0 | 8 | 492 | 56.817 | 7.173 | −0.538 | Peroxisome |
| GhCAT3 | Gh_A05G1539 | A05:15642154-15645557 − | 3404 | 1419 | 46.1 | 9 | 472 | 54.706 | 7.068 | −0.561 | Peroxisome |
| GhCAT4 | Gh_D05G1710 | D05:15409889-15413267 − | 3379 | 1479 | 46.5 | 8 | 492 | 56.957 | 7.413 | −0.587 | Peroxisome |
| GhCAT5 | Gh_A02G1698 | A02:83178810-83181326 + | 2517 | 1470 | 45.4 | 7 | 489 | 56.528 | 7.454 | −0.546 | Peroxisome |
| GhCAT6 | Gh_D03G0021 | D03:171438-173963 − | 2526 | 1485 | 45.5 | 7 | 494 | 57.285 | 7.310 | −0.543 | Peroxisome |
| GhCAT7 | Gh_A07G1556 | A07:57094046-57096383 + | 2338 | 1230 | 46.0 | 9 | 409 | 46.962 | 6.869 | −0.498 | Peroxisome |
| GbCAT1 | GOBAR_AA22711 | A01:19685673-19688948 − | 3276 | 1479 | 47.0 | 8 | 492 | 56.845 | 7.173 | −0.539 | Peroxisome |
| GbCAT2 | GOBAR_DD08701 | D01:16738967-16742318 + | 3352 | 1479 | 47.0 | 8 | 492 | 56.817 | 7.173 | −0.538 | Peroxisome |
| GbCAT3 | GOBAR_AA35088 | A05:14744651-14749782 − | 5132 | 1380 | 46.7 | 9 | 459 | 53.290 | 7.251 | −0.651 | Peroxisome |
| GbCAT4 | GOBAR_DD30012 | D05:16498614-16501993 + | 3380 | 1479 | 46.6 | 8 | 492 | 56.924 | 7.294 | −0.585 | Peroxisome |
| GbCAT5 | GOBAR_AA31934 | A02:98649356-98652362 + | 3007 | 1479 | 45.4 | 8 | 492 | 56.903 | 7.453 | −0.557 | Peroxisome |
| GbCAT6 | GOBAR_AA38517 | scaffold_0637.UKA:19604-22605 + | 3002 | 1479 | 45.6 | 8 | 492 | 56.939 | 7.453 | −0.567 | Peroxisome |
| GbCAT7 | GOBAR_AA20422 | A07:71313165-71323809 + | 10645 | 1806 | 45.5 | 9 | 601 | 67.504 | 6.286 | −0.662 | Peroxisome |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Cheng, Y.; Chen, D.; Liu, D.; Hu, M.; Dong, J.; Zhang, X.; Song, L.; Shen, F. The Catalase Gene Family in Cotton: Genome-Wide Characterization and Bioinformatics Analysis. Cells 2019, 8, 86. https://doi.org/10.3390/cells8020086
Wang W, Cheng Y, Chen D, Liu D, Hu M, Dong J, Zhang X, Song L, Shen F. The Catalase Gene Family in Cotton: Genome-Wide Characterization and Bioinformatics Analysis. Cells. 2019; 8(2):86. https://doi.org/10.3390/cells8020086
Chicago/Turabian StyleWang, Wei, Yingying Cheng, Dongdong Chen, Dan Liu, Mengjiao Hu, Jie Dong, Xiaopei Zhang, Lirong Song, and Fafu Shen. 2019. "The Catalase Gene Family in Cotton: Genome-Wide Characterization and Bioinformatics Analysis" Cells 8, no. 2: 86. https://doi.org/10.3390/cells8020086
APA StyleWang, W., Cheng, Y., Chen, D., Liu, D., Hu, M., Dong, J., Zhang, X., Song, L., & Shen, F. (2019). The Catalase Gene Family in Cotton: Genome-Wide Characterization and Bioinformatics Analysis. Cells, 8(2), 86. https://doi.org/10.3390/cells8020086