The Role of Primary Cilia in the Crosstalk between the Ubiquitin–Proteasome System and Autophagy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
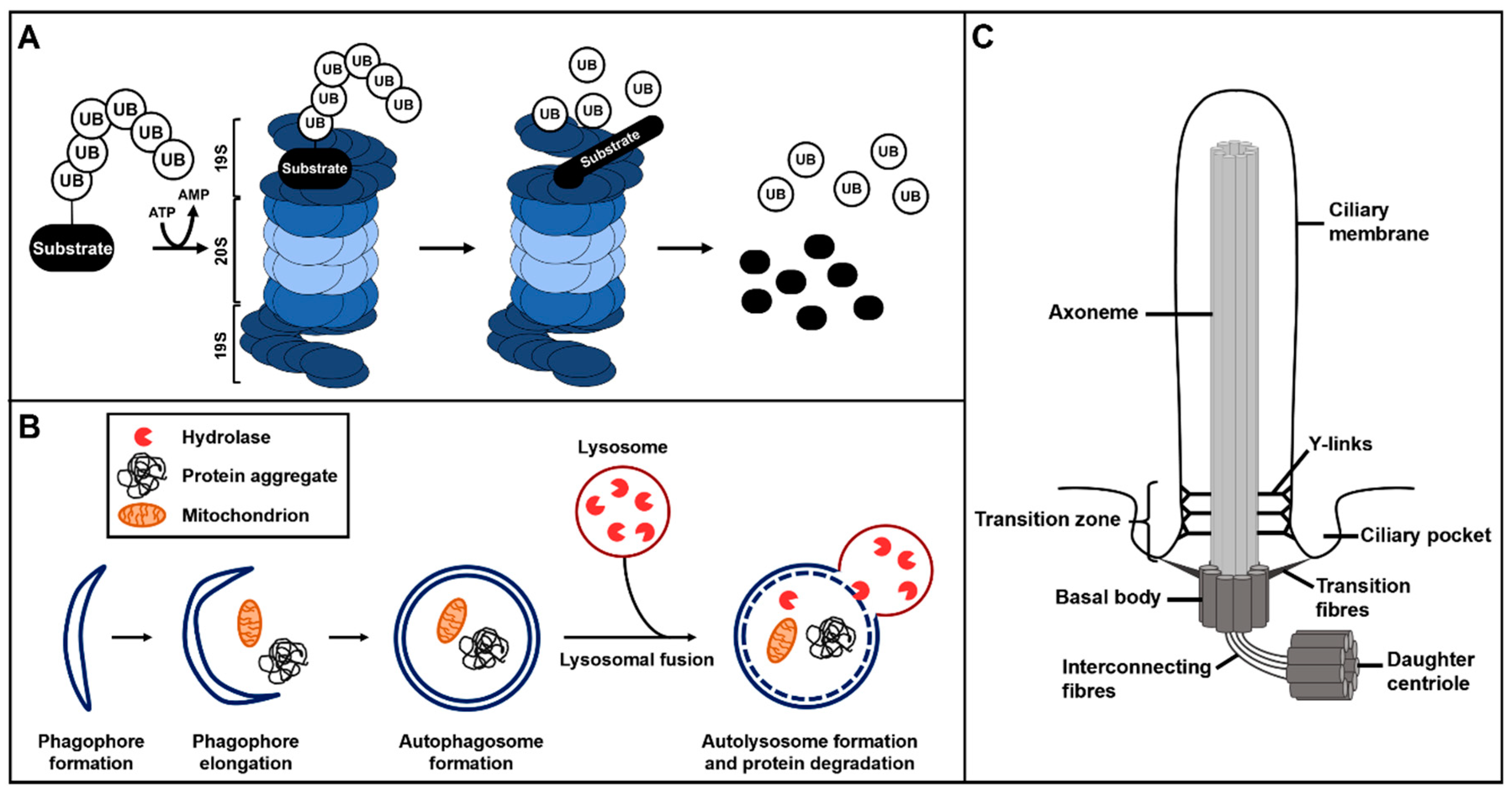
2. The Ubiquitin–Proteasome System and Autophagy
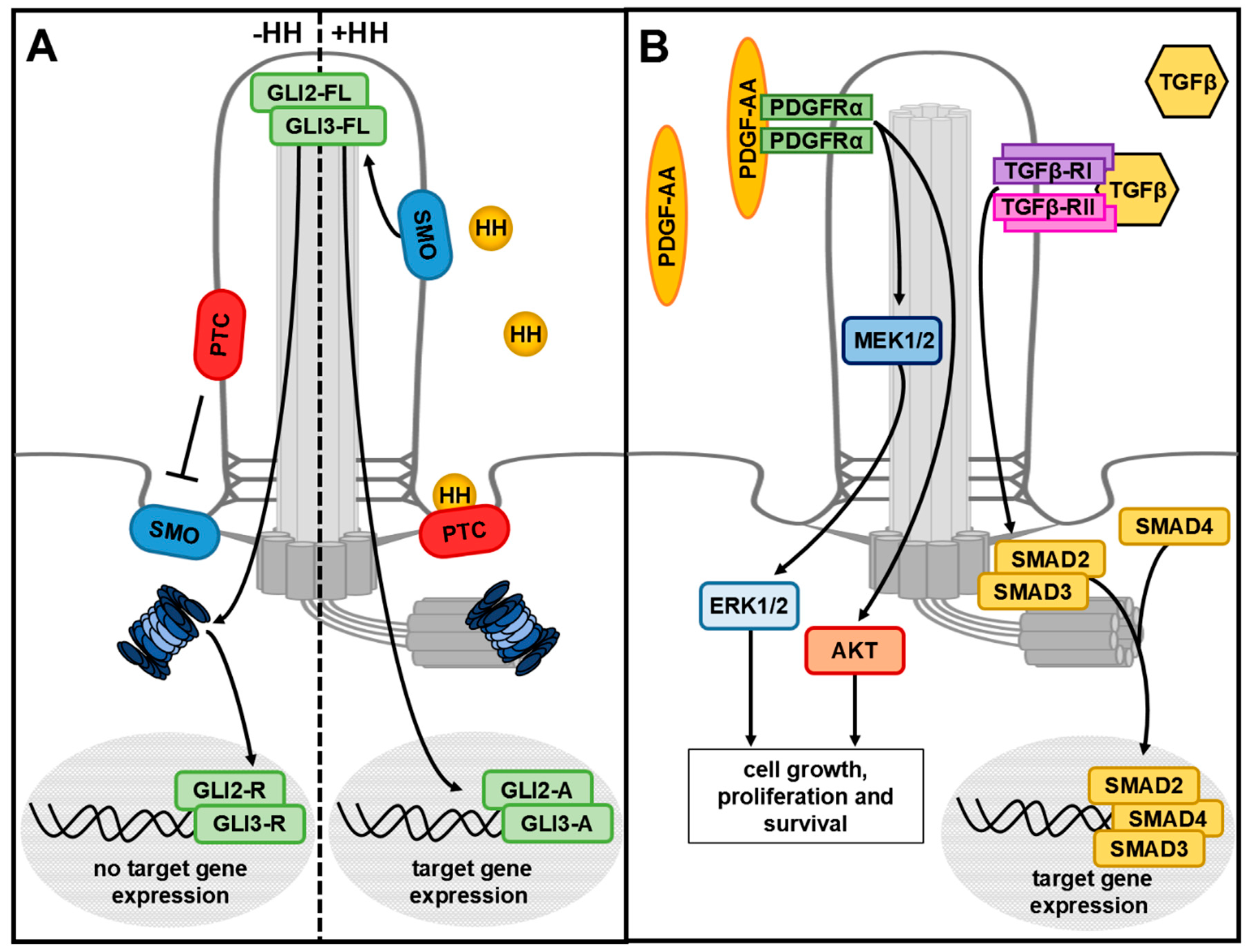
3. The Primary Cilium
4. Do the UPS and Autophagy Play a Role in the Development of Ciliopathies?
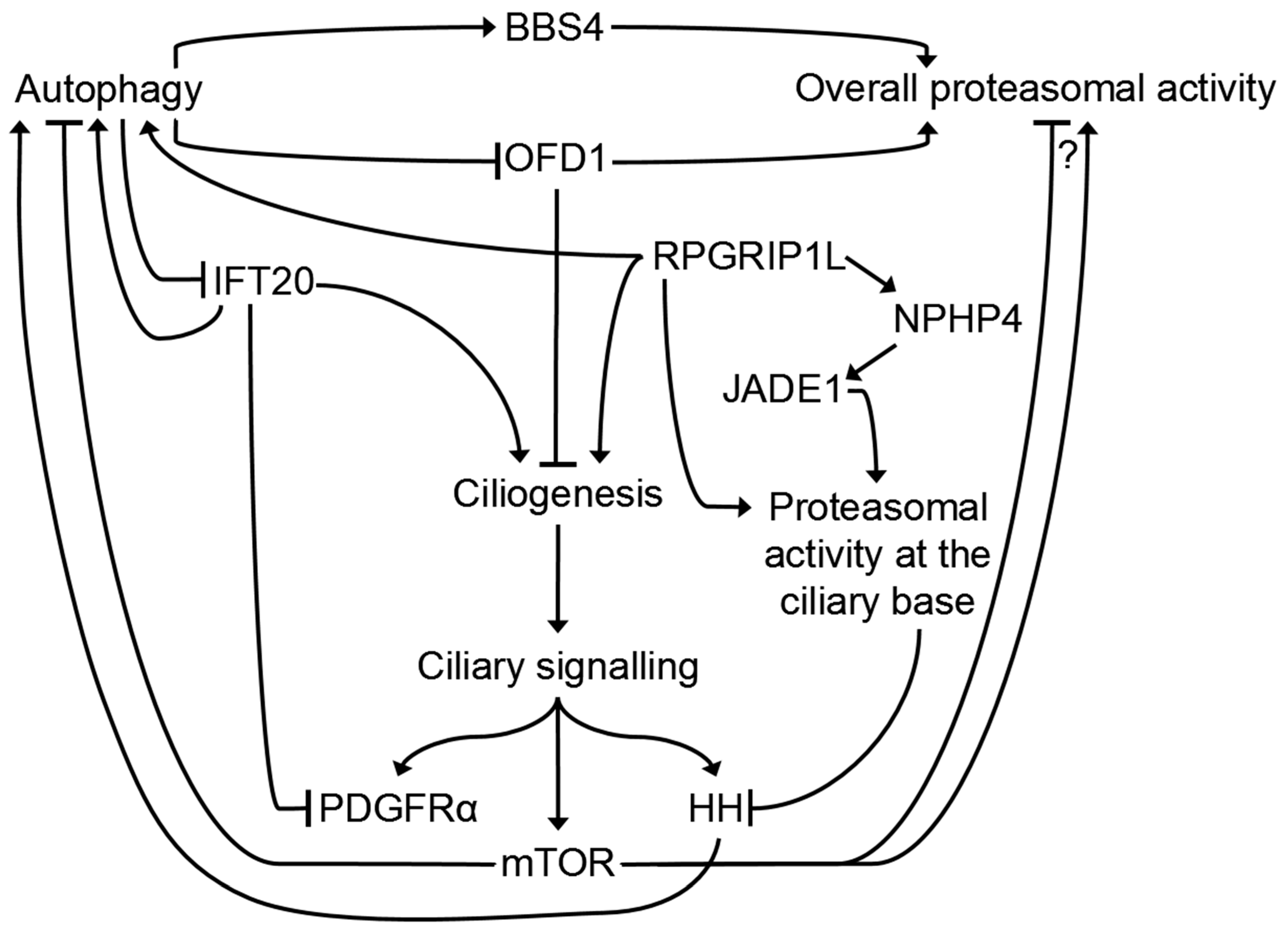
5. Which Role Does the Primary Cilium Play in the Crosstalk between the UPS and Autophagy?
6. Conclusions
Funding
Conflicts of Interest
References
- Irvine, G.; El-Agnaf, O.; Shankar, G.; Walsh, D. Protein aggregation in the brain: The molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef]
- Cox, D.; Raeburn, C.; Sui, X.; Hatters, D. Protein aggregation in cell biology: An aggregomics perspective of health and disease. In Seminars in Cell & Developmental Biology; pii: S1084-9521; Academic Press: New York, NY, USA, 2018. [Google Scholar]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Urbé, S.; Clague, M. Selective protein degradation in cell signalling. Semin. Cell Dev. Biol. 2012, 23, 509–514. [Google Scholar] [CrossRef]
- Koepp, D. Cell cycle regulation by protein degradation. Methods Mol. Biol. 2014, 1170, 61–73. [Google Scholar] [PubMed]
- Yao, T.; Ndoja, A. Regulation of gene expression by the ubiquitin-proteasome system. Semin. Cell Dev. Biol. 2012, 23, 523–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilienbaum, A. Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 2013, 4, 1–26. [Google Scholar]
- Gerhardt, C.; Leu, T.; Lier, J.; Rüther, U. The cilia-regulated proteasome and its role in the development of ciliopathies and cancer. Cilia 2016, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef]
- Le Bot, N. Autophagy: A new regulator of development. Nat. Cell Biol. 2007, 9, 741. [Google Scholar] [CrossRef]
- Ryter, S.; Cloonan, S.; Choi, A. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, C.; Wiegering, A.; Leu, T.; Rüther, U. Control of Hedgehog signalling by the cilia-regulated proteasome. J. Dev. Biol. 2016, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Lippai, M.; Szatmári, Z. Autophagy-from molecular mechanisms to clinical relevance. Cell Biol. Toxicol. 2017, 33, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Choi, A.; Ryter, S. Emerging role of selective autophagy in human diseases. Front. Pharmacol. 2014, 5, 244. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Aburto, M.; Hurlé, J.; Varela-Nieto, I.; Magariños, M. Autophagy during vertebrate development. Cells 2012, 1, 428–448. [Google Scholar] [CrossRef]
- Di Bartolomeo, S.; Nazio, F.; Cecconi, F. The role of autophagy during development in higher eukaryotes. Traffic 2010, 11, 1280–1289. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Breusing, N.; Arndt, J.; Voss, P.; Bresgen, N.; Wiswedel, I.; Gardemann, A.; Siems, W.; Grune, T. Inverse correlation of protein oxidation and proteasome activity in liver and lung. Mech. Ageing Dev. 2009, 130, 748–753. [Google Scholar] [CrossRef]
- Wang, X.; Robbins, J. Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell Cardiol. 2014, 71, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [PubMed]
- Costello, M.; Brennan, L.; Basu, S.; Chauss, D.; Mohamed, A.; Gilliland, K.; Johnsen, S.; Menko, A.; Kantorow, M. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp. Eye Res. 2013, 116, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Rajakaruna, S.; Reyes, B.; Van Bockstaele, E.; Menko, A. Suppression of MAPK/JNK-MTORC1 signaling leads to premature loss of organelles and nuclei by autophagy during terminal differentiation of lens fiber cells. Autophagy 2014, 10, 1193–1211. [Google Scholar] [CrossRef] [PubMed]
- Mellén, M.; de la Rosa, E.; Boya, P. The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ. 2008, 15, 1279–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.; Zou, Z.; Sun, Q.; Luby-Phelps, K.; Cheng, P.; Hogan, R.; Gilpin, C.; Levine, B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007, 128, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.; Peng, X.; Nagy, T.; Alcaraz, A.; Gu, H.; Guan, J. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J. Cell Biol. 2006, 175, 121–133. [Google Scholar] [CrossRef]
- Lee, E.; Koo, Y.; Ng, A.; Wei, Y.; Luby-Phelps, K.; Juraszek, A.; Xavier, R.; Cleaver, O.; Levine, B.; Amatruda, J. Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy 2014, 10, 572–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Wang, X.; Yu, J.; Wong, S.; Cheng, A.; Chan, F.; Ng, S.; Cho, C.; Sung, J.; Wu, W. A novel crosstalk between two major protein degradation systems. Autophagy 2013, 9, 1500–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannous, P.; Zhu, H.; Nemchenko, A.; Berry, J.; Johnstone, J.; Shelton, J.; Miller, F.J.; Rothermel, B.; Hill, J. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation 2008, 117, 3070–3078. [Google Scholar] [CrossRef]
- Demishtein, A.; Fraiberg, M.; Berko, D.; Tirosh, B.; Elazar, Z.; Navon, A. SQSTM1/p62-mediated autophagy compensates for loss of proteasome polyubiquitin recruiting capacity. Autophagy 2017, 13, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Huang, Z.; Wang, W.; Zhang, B.; Xu, Y.; Mao, Z.; Chen, L.; Hu, H.; Geng, Q. Proteasome inhibition promotes autophagy and protects from endoplasmic reticulum stress in rat alveolar macrophages exposed to hypoxia-reoxygenation injury. J. Cell. Physiol. 2018, 233, 6748–6758. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Zhang, J.; Wang, X.; Meng, F.; Li, W.; Luan, Y.; Ling, F.; Luo, Y. Inhibition of autophagy induced by proteasome inhibition increases cell death in human SHG-44 glioma cells. Acta Pharmacol. Sin. 2009, 30, 1046–1052. [Google Scholar] [CrossRef]
- Jiang, S.; Park, D.; Gao, Y.; Ravi, S.; Darley-Usmar, V.; Abraham, E.; Zmijewski, J. Participation of proteasome-ubiquitin protein degradation in autophagy and the activation of AMP-activated protein kinase. Cell. Signal. 2015, 27, 1186–1197. [Google Scholar] [CrossRef] [Green Version]
- Kyrychenko, V.; Nagibin, V.; Tumanovska, L.; Pashevin, D.; Gurianova, V.; Moibenko, A.; Dosenko, V.; Klionsky, D. Knockdown of PSMB7 induces autophagy in cardiomyocyte cultures: Possible role in endoplasmic reticulum stress. Pathobiology 2014, 81, 8–14. [Google Scholar] [CrossRef]
- Zhao, J.; Brault, J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.; Goldberg, A. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhai, B.; Gygi, S.; Goldberg, A. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [Green Version]
- Selimovic, D.; Porzig, B.; El-Khattouti, A.; Badura, H.; Ahmad, M.; Ghanjati, F.; Santourlidis, S.; Haikel, Y.; Hassan, M. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell. Signal. 2013, 25, 308–318. [Google Scholar] [CrossRef]
- Sha, Z.; Schnell, H.; Ruoff, K.; Goldberg, A. Rapid induction of p62 and GABARAPL1 upon proteasome inhibition promotes survival before autophagy activation. J. Cell Biol. 2018, 217, 1757–1776. [Google Scholar] [CrossRef]
- Sun, A.; Li, C.; Chen, R.; Huang, Y.; Chen, Q.; Cui, X.; Liu, H.; Thrasher, J.; Li, B. GSK-3β controls autophagy by modulating LKB1-AMPK pathway in prostate cancer cells. Prostate 2016, 76, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Cai, J.; Sun, L.; Li, Y.; Qu, J.; Snider, B.; Wu, S. Proteasome inhibitors activate autophagy involving inhibition of PI3K-Akt-mTOR pathway as an anti-oxidation defense in human RPE cells. PLoS ONE 2014, 9, e103364. [Google Scholar] [CrossRef]
- Wu, W.; Wu, Y.; Yu, L.; Li, Z.; Sung, J.; Cho, C. Induction of autophagy by proteasome inhibitor is associated with proliferative arrest in colon cancer cells. Biochem. Biophys. Res. Commun. 2008, 374, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, S.; Viollet, B.; Zou, M. Regulation of the proteasome by AMPK in endothelial cells: The role of O-GlcNAc transferase (OGT). PLoS ONE 2012, 7, e36717. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Dunner, K.J.; McConkey, D. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Fallon, J.; Beachy, P. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Schrader, E.; Harstad, K.; Holmgren, R.; Matouschek, A. A three-part signal governs differential processing of Gli1 and Gli3 proteins by the proteasome. J. Biol. Chem. 2011, 286, 39051–39058. [Google Scholar] [CrossRef] [PubMed]
- Weissman, A. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2001, 2, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.; Hristova, V.; Weissman, A. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Tu, D.; Brunger, A.; Ye, Y. A ubiquitin ligase transfers preformed polyubiquitin chains from a conjugating enzyme to a substrate. Nature 2007, 446, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Ravid, T.; Hochstrasser, M. Autoregulation of an E2 enzyme by ubiquitin-chain assembly on its catalytic residue. Nat. Cell Biol. 2007, 9, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Hilt, W. The proteasome: A proteolytic nanomachine of cell regulation and waste disposal. Biochim. Biophys. Acta 2004, 1695, 19–31. [Google Scholar] [CrossRef]
- Jung, T.; Grune, T. Structure of the proteasome. Prog. Mol. Biol. Transl. Sci. 2012, 109, 1–39. [Google Scholar]
- Liu, C.; Jacobson, A. Functions of the 19S complex in proteasomal degradation. Trends Biochem. Sci. 2013, 38, 103–110. [Google Scholar] [CrossRef]
- Coux, O.; Tanaka, K.; Goldberg, A. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996, 65, 801–847. [Google Scholar] [CrossRef]
- Gerdes, J.; Liu, Y.; Zaghloul, N.; Leitch, C.; Lawson, S.; Kato, M.; Beachy, P.; Beales, P.; DeMartino, G.; Fisher, S.; et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007, 39, 1350–1360. [Google Scholar] [CrossRef]
- Brooks, P.; Fuertes, G.; Murray, R.; Bose, S.; Knecht, E.; Rechsteiner, M.; Hendil, K.; Tanaka, K.; Dyson, J.; Rivett, J. Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochem. J. 2000, 346, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Wigley, W.; Fabunmi, R.; Lee, M.; Marino, C.; Muallem, S.; DeMartino, G.; Thomas, P. Dynamic association of proteasomal machinery with the centrosome. J. Cell Biol. 1999, 145, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, C.; Lier, J.; Burmühl, S.; Struchtrup, A.; Deutschmann, K.; Vetter, M.; Leu, T.; Reeg, S.; Grune, T.; Rüther, U. The transition zone protein Rpgrip1l regulates proteasomal activity at the primary cilium. J. Cell Biol. 2015, 210, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Jurek, A.; Amagasaki, K.; Gembarska, A.; Heldin, C.; Lennartsson, J. Negative and positive regulation of MAPK phosphatase 3 controls platelet-derived growth factor-induced Erk activation. J. Biol. Chem. 2009, 284, 4626–4634. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I. The ubiquitin-proteasome system and signal transduction pathways regulating Epithelial Mesenchymal transition of cancer. J. Biomed. Sci. 2012, 19, 67. [Google Scholar] [CrossRef]
- Liu, Y.; Tsai, I.; Morleo, M.; Oh, E.; Leitch, C.; Massa, F.; Lee, B.; Parker, D.; Finley, D.; Zaghloul, N.; et al. Ciliopathy proteins regulate paracrine signaling by modulating proteasomal degradation of mediators. J. Clin. Investig. 2014, 124, 2059–2070. [Google Scholar] [CrossRef] [Green Version]
- Farré, J.; Manjithaya, R.; Mathewson, R.; Subramani, S. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev. Cell 2008, 14, 365–376. [Google Scholar] [CrossRef]
- Kraft, C.; Deplazes, A.; Sohrmann, M.; Peter, M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 2008, 10, 602–610. [Google Scholar] [CrossRef]
- Orvedahl, A.; Sumpter, R.J.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef]
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.; Shani, V.; Engelender, S. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 18666–18671. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Klionsky, D. Mitochondria removal by autophagy. Autophagy 2011, 7, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Thurston, T.; Wandel, M.; von Muhlinen, N.; Foeglein, A.; Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012, 482, 414–418. [Google Scholar] [CrossRef]
- Arstila, A.; Trump, B. Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am. J. Pathol. 1968, 53, 687–733. [Google Scholar]
- Geng, J.; Klionsky, D. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ’Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef]
- Barth, S.; Glick, D.; Macleod, K. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Russell, R.; Tian, Y.; Yuan, H.; Park, H.; Chang, Y.; Kim, J.; Kim, H.; Neufeld, T.; Dillin, A.; Guan, K. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Pickart, C.; Eddins, M. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef]
- Sun, F.; Kanthasamy, A.; Anantharam, V.; Kanthasamy, A. Environmental neurotoxic chemicals-induced ubiquitin proteasome system dysfunction in the pathogenesis and progression of Parkinson’s disease. Pharmacol. Ther. 2007, 114, 327–344. [Google Scholar] [CrossRef]
- Thrower, J.; Hoffman, L.; Rechsteiner, M.; Pickart, C. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Wong, E.; Kirkpatrick, D.; Pletnikova, O.; Ko, H.; Tay, S.; Ho, M.; Troncoso, J.; Gygi, S.; Lee, M.; et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef]
- Hao, L.; Scholey, J. Intraflagellar transport at a glance. J. Cell Sci. 2009, 122, 889–892. [Google Scholar] [CrossRef] [Green Version]
- Gilula, N.; Satir, P. The ciliary necklace. A ciliary membrane specialization. J. Cell Biol. 1972, 53, 494–509. [Google Scholar] [CrossRef]
- Reiter, J.; Blacque, O.; Leroux, M. The base of the cilium: Roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012, 13, 608–618. [Google Scholar] [CrossRef]
- Czarnecki, P.G.; Shah, J.V. The ciliary transition zone: From morphology and molecules to medicine. Trends Cell Biol. 2012, 22, 201–210. [Google Scholar] [CrossRef]
- Garcia-Gonzalo, F.; Reiter, J. Scoring a backstage pass: Mechanisms of ciliogenesis and ciliary access. J. Cell Biol. 2012, 197, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Omran, H. NPHP proteins: Gatekeepers of the ciliary compartment. J. Cell Biol. 2010, 190, 715–717. [Google Scholar] [CrossRef]
- Benzing, T.; Schermer, B. Transition zone proteins and cilia dynamics. Nat. Genet. 2011, 43, 723–724. [Google Scholar] [CrossRef]
- Garcia-Gonzalo, F.; Reiter, J. Open Sesame: How Transition Fibers and the Transition Zone Control Ciliary Composition. Cold Spring Harb. Perspect. Biol. 2017, 9, a028134. [Google Scholar] [CrossRef]
- Jensen, V.; Leroux, M. Gates for soluble and membrane proteins, and two trafficking systems (IFT and LIFT), establish a dynamic ciliary signaling compartment. Curr. Opin. Cell Biol. 2017, 47, 83–91. [Google Scholar] [CrossRef]
- Betleja, E.; Cole, D. Ciliary trafficking: CEP290 guards a gated community. Curr. Biol. 2010, 20, R928–R931. [Google Scholar] [CrossRef]
- Craige, B.; Tsao, C.; Diener, D.; Hou, Y.; Lechtreck, K.; Rosenbaum, J.; Witman, G. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol. 2010, 190, 927–940. [Google Scholar] [CrossRef] [Green Version]
- Berbari, N.; O’Connor, A.; Haycraft, C.; Yoder, B. The primary cilium as a complex signaling center. Curr. Biol. 2009, 19, R526–R535. [Google Scholar] [CrossRef]
- Eggenschwiler, J.; Anderson, K. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007, 23, 345–373. [Google Scholar] [CrossRef]
- Corbit, K.; Aanstad, P.; Singla, V.; Norman, A.; Stainier, D.; Reiter, J. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef]
- Haycraft, C.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.; Yoder, B. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef]
- Schneider, L.; Clement, C.; Teilmann, S.; Pazour, G.; Hoffmann, E.; Satir, P.; Christensen, S. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005, 15, 1861–1866. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef]
- Clement, C.; Ajbro, K.; Koefoed, K.; Vestergaard, M.; Veland, I.; Henriques de Jesus, M.; Pedersen, L.; Benmerah, A.; Andersen, C.; Larsen, L.; et al. TGF-β signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 2013, 3, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Pedersen, L.; Christensen, S. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499–503. [Google Scholar] [Green Version]
- Chen, M.; Wilson, C.; Li, Y.; Law, K.; Lu, C.; Gacayan, R.; Zhang, X.; Hui, C.; Chuang, P. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef] [PubMed]
- Humke, E.; Dorn, K.; Milenkovic, L.; Scott, M.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [Green Version]
- Christensen, S.; Pedersen, S.; Satir, P.; Veland, I.; Schneider, L. The primary cilium coordinates signaling pathways in cell cycle control and migration during development and tissue repair. Curr. Top. Dev. Biol. 2008, 85, 261–301. [Google Scholar]
- Yun, S.; Lee, M.; Ryu, J.; Song, C.; Han, H. Role of HIF-1alpha and VEGF in human mesenchymal stem cell proliferation by 17beta-estradiol: Involvement of PKC, PI3K/Akt, and MAPKs. Am. J. Physiol. Cell Physiol. 2009, 296, 317–326. [Google Scholar] [CrossRef]
- Schild, C.; Wirth, M.; Reichert, M.; Schmid, R.; Saur, D.; Schneider, G. PI3K signaling maintains c-myc expression to regulate transcription of E2F1 in pancreatic cancer cells. Mol. Carcinog. 2009, 48, 1149–1158. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Reiter, J.; Leroux, M. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.; Beales, P. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef]
- McIntyre, J.; Williams, C.; Martens, J. Smelling the roses and seeing the light: Gene therapy for ciliopathies. Trends Biotechnol. 2013, 31, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, S.; Jung, Y.; Jung, E.; Kwon, H.; Kim, J. Eupatilin rescues ciliary transition zone defects to ameliorate ciliopathy-related phenotypes. J. Clin. Investig. 2018, 128, 3642–3648. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, J.; Davis, E.; Joiner, A.; Williams, C.; Tsai, I.; Jenkins, P.; McEwen, D.; Zhang, L.; Escobado, J.; Thomas, S.; et al. Gene therapy rescues cilia defects and restores olfactory function in a mammalian ciliopathy model. Nat. Med. 2012, 18, 1423–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.; Uytingco, C.; Green, W.; McIntyre, J.; Ukhanov, K.; Zimmerman, A.; Shively, D.; Zhang, L.; Nishimura, D.; Sheffield, V.; et al. Gene Therapeutic Reversal of Peripheral Olfactory Impairment in Bardet-Biedl Syndrome. Mol. Ther. 2017, 25, 904–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, G.; Barry, E.; Yu, D.; Lukason, M.; Cheng, S.; Scaria, A. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol. Ther. 2017, 25, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Ran, J.; Liu, M.; Li, D.; Li, Y.; Shi, X.; Meng, D.; Pan, J.; Ou, G.; Aneja, R.; et al. CYLD mediates ciliogenesis in multiple organs by deubiquitinating Cep70 and inactivating HDAC6. Cell Res. 2014, 24, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Mockel, A.; Obringer, C.; Hakvoort, T.; Seeliger, M.; Lamers, W.; Stoetzel, C.; Dollfus, H.; Marion, V. Pharmacological modulation of the retinal unfolded protein response in Bardet-Biedl syndrome reduces apoptosis and preserves light detection ability. J. Biol. Chem. 2012, 287, 37483–37494. [Google Scholar] [CrossRef]
- Ramsbottom, S.; Molinari, E.; Srivastava, S.; Silberman, F.; Henry, C.; Alkanderi, S.; Devlin, L.; White, K.; Steel, D.; Saunier, S.; et al. Targeted exon skipping of a CEP290 mutation rescues Joubert syndrome phenotypes in vitro and in a murine model. Proc. Natl. Acad. Sci. USA 2018, 115, 12489–12494. [Google Scholar]
- Green, W.; Uytingco, C.; Ukhanov, K.; Kolb, Z.; Moretta, J.; McIntyre, J.; Martens, J. Peripheral Gene Therapeutic Rescue of an Olfactory Ciliopathy Restores Sensory Input, Axonal Pathfinding, and Odor-Guided Behavior. J. Neurosci. 2018, 38, 7462–7475. [Google Scholar] [CrossRef]
- Shivanna, M.; Anand, M.; Chakrabarti, S.; Khanna, H. Ocular Ciliopathies: Genetic and mechanistic insights into developing therapies. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Stayner, C.; Brooke, D.; Bates, M.; Eccles, M. Targeted Therapies for Autosomal Dominant Polycystic Kidney Disease. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Uytingco, C.; Green, W.; Martens, J. Olfactory loss and dysfunction in ciliopathies: Molecular mechanisms and potential therapies. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Zhu, P.; Sieben, C.; Xu, X.; Harris, P.; Lin, X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 2017, 26, 158–172. [Google Scholar] [CrossRef]
- Tobin, J.; Beales, P. Restoration of renal function in zebrafish models of ciliopathies. Pediatr. Nephrol. 2008, 23, 2095–2099. [Google Scholar] [CrossRef] [Green Version]
- Heitman, J.; Movva, N.; Hall, M. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef]
- Kunz, J.; Henriquez, R.; Schneider, U.; Deuter-Reinhard, M.; Movva, N.; Hall, M. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 1993, 73, 585–596. [Google Scholar] [CrossRef]
- Brown, E.; Albers, M.; Shin, T.; Ichikawa, K.; Keith, C.; Lane, W.; Schreiber, S. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef]
- Chang, Y.; Juhász, G.; Goraksha-Hicks, P.; Arsham, A.; Mallin, D.; Muller, L.; Neufeld, T. Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem. Soc. Trans. 2009, 37, 232–236. [Google Scholar] [CrossRef]
- Ganley, I.; Lam, D.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef]
- Jung, C.; Jun, C.; Ro, S.; Kim, Y.; Otto, N.; Cao, J.; Kundu, M.; Kim, D. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.; Rubinsztein, D. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.; Saiki, S.; Siddiqi, F.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef]
- Stayner, C.; Shields, J.; Slobbe, L.; Shillingford, J.; Weimbs, T.; Eccles, M. Rapamycin-mediated suppression of renal cyst expansion in del34 Pkd1-/- mutant mouse embryos: An investigation of the feasibility of renal cyst prevention in the foetus. Nephrology (Carlton) 2012, 17, 739–747. [Google Scholar] [CrossRef]
- Shillingford, J.; Murcia, N.; Larson, C.; Low, S.; Hedgepeth, R.; Brown, N.; Flask, C.; Novick, A.; Goldfarb, D.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [Green Version]
- Gattone, V.N.; Sinders, R.; Hornberger, T.; Robling, A. Late progression of renal pathology and cyst enlargement is reduced by rapamycin in a mouse model of nephronophthisis. Kidney Int. 2009, 76, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Ravichandran, K.; Zafar, I.; Ozkok, A.; Edelstein, C. An mTOR kinase inhibitor slows disease progression in a rat model of polycystic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 45–53. [Google Scholar] [CrossRef]
- Tao, Y.; Kim, J.; Schrier, R.; Edelstein, C. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 46–51. [Google Scholar] [CrossRef]
- Wahl, P.; Serra, A.; Le Hir, M.; Molle, K.; Hall, M.; Wüthrich, R. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transplant. 2006, 21, 598–604. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, L.; Xiong, X.; Mao, Z.; Wang, L.; Mei, C. Mycophenolate mofetil versus Rapamycin in Han: SPRD rats with Polycystic Kidney Disease. Biol. Res. 2009, 42, 437–444. [Google Scholar] [CrossRef]
- Liu, Y.; Shao, Y.; He, Q. Sirolimus for treatment of autosomal-dominant polycystic kidney disease: A meta-analysis of randomized controlled trials. Transplant. Proc. 2014, 46, 66–74. [Google Scholar] [CrossRef]
- Stallone, G.; Infante, B.; Grandaliano, G.; Bristogiannis, C.; Macarini, L.; Mezzopane, D.; Bruno, F.; Montemurno, E.; Schirinzi, A.; Sabbatini, M.; et al. Rapamycin for treatment of type I autosomal dominant polycystic kidney disease (RAPYD-study): A randomized, controlled study. Nephrol. Dial. Transplant. 2012, 27, 3560–3567. [Google Scholar] [CrossRef]
- Anandh, U.; Chandrasekar, G.; Agarwal, V. Mammalian target of rapamycin inhibitors in a patient with polycystic kidney disease-1-tuberous sclerosis-2 contiguous gene syndrome. Saudi J. Kidney Dis. Transpl. 2018, 29, 1475–1479. [Google Scholar] [CrossRef]
- Li, A.; Fan, S.; Xu, Y.; Meng, J.; Shen, X.; Mao, J.; Zhang, L.; Zhang, X.; Moeckel, G.; Wu, D.; et al. Rapamycin treatment dose-dependently improves the cystic kidney in a new ADPKD mouse model via the mTORC1 and cell-cycle-associated CDK1/cyclin axis. J. Cell. Mol. Med. 2017, 21, 1619–1635. [Google Scholar] [CrossRef]
- Novalic, Z.; van der Wal, A.; Leonhard, W.; Koehl, G.; Breuning, M.; Geissler, E.; de Heer, E.; Peters, D. Dose-dependent effects of sirolimus on mTOR signaling and polycystic kidney disease. J. Am. Soc. Nephrol. 2012, 23, 842–853. [Google Scholar] [CrossRef]
- Korolchuk, V.; Menzies, F.; Rubinsztein, D. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef]
- Korolchuk, V.; Menzies, F.; Rubinsztein, D. A novel link between autophagy and the ubiquitin-proteasome system. Autophagy 2009, 5, 862–863. [Google Scholar] [CrossRef] [Green Version]
- Nam, T.; Han, J.; Devkota, S.; Lee, H. Emerging Paradigm of Crosstalk between Autophagy and the Ubiquitin-Proteasome System. Mol. Cells 2017, 40, 897–905. [Google Scholar]
- Ji, C.; Kwon, Y. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar] [Green Version]
- Kocaturk, N.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Tang, Z.; Lin, M.; Stowe, T.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Livingston, M.; Su, Y.; Dong, Z. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy 2015, 11, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pampliega, O.; Orhon, I.; Patel, B.; Sridhar, S.; Díaz-Carretero, A.; Beau, I.; Codogno, P.; Satir, B.; Satir, P.; Cuervo, A. Functional interaction between autophagy and ciliogenesis. Nature 2013, 502, 194–200. [Google Scholar] [CrossRef]
- Struchtrup, A.; Wiegering, A.; Stork, B.; Rüther, U.; Gerhardt, C. The ciliary protein RPGRIP1L governs autophagy independently of its proteasome-regulating function at the ciliary base in mouse embryonic fibroblasts. Autophagy 2018, 14, 567–583. [Google Scholar] [CrossRef] [Green Version]
- Mahuzier, A.; Gaudé, H.; Grampa, V.; Anselme, I.; Silbermann, F.; Leroux-Berger, M.; Delacour, D.; Ezan, J.; Montcouquiol, M.; Saunier, S.; et al. Dishevelled stabilization by the ciliopathy protein Rpgrip1l is essential for planar cell polarity. J. Cell Biol. 2012, 198, 927–940. [Google Scholar] [CrossRef] [Green Version]
- Wiegering, A.; Dildrop, R.; Kalfhues, L.; Spychala, A.; Kuschel, S.; Lier, J.; Zobel, T.; Dahmen, S.; Leu, T.; Struchtrup, A.; et al. Cell type-specific regulation of ciliary transition zone assembly in vertebrates. EMBO J. 2018, 37, e97791. [Google Scholar] [CrossRef]
- Borgal, L.; Habbig, S.; Hatzold, J.; Liebau, M.; Dafinger, C.; Sacarea, I.; Hammerschmidt, M.; Benzing, T.; Schermer, B. The Ciliary Protein Nephrocystin-4 Translocates the Canonical Wnt-Regulator Jade-1 to the Nucleus to Negatively Regulate Beta-Catenin Signaling. J. Biol. Chem. 2012, 287, 25370–25380. [Google Scholar] [CrossRef]
- Schmid, F.; Schou, K.; Vilhelm, M.; Holm, M.; Breslin, L.; Farinelli, P.; Larsen, L.; Andersen, J.; Pedersen, L.; Christensen, S. IFT20 modulates ciliary PDGFRα signaling by regulating the stability of Cbl E3 ubiquitin ligases. J. Cell Biol. 2018, 217, 151–161. [Google Scholar] [CrossRef]
- Saxton, R.; Sabatini, D. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Nicholatos, J.; Dreier, J.; Ricoult, S.; Widenmaier, S.; Hotamisligil, G.; Kwiatkowski, D.; Manning, B. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014, 513, 440–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Manning, B. Zhang & Manning reply. Nature 2016, 529, E2–E3. [Google Scholar] [PubMed] [Green Version]
- Zhao, J.; Garcia, G.; Goldberg, A. Control of proteasomal proteolysis by mTOR. Nature 2016, 529, E1–E2. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiegering, A.; Rüther, U.; Gerhardt, C. The Role of Primary Cilia in the Crosstalk between the Ubiquitin–Proteasome System and Autophagy. Cells 2019, 8, 241. https://doi.org/10.3390/cells8030241
Wiegering A, Rüther U, Gerhardt C. The Role of Primary Cilia in the Crosstalk between the Ubiquitin–Proteasome System and Autophagy. Cells. 2019; 8(3):241. https://doi.org/10.3390/cells8030241
Chicago/Turabian StyleWiegering, Antonia, Ulrich Rüther, and Christoph Gerhardt. 2019. "The Role of Primary Cilia in the Crosstalk between the Ubiquitin–Proteasome System and Autophagy" Cells 8, no. 3: 241. https://doi.org/10.3390/cells8030241
APA StyleWiegering, A., Rüther, U., & Gerhardt, C. (2019). The Role of Primary Cilia in the Crosstalk between the Ubiquitin–Proteasome System and Autophagy. Cells, 8(3), 241. https://doi.org/10.3390/cells8030241