The Use of Pluripotent Stem Cell-Derived Organoids to Study Extracellular Matrix Development during Neural Degeneration

Abstract
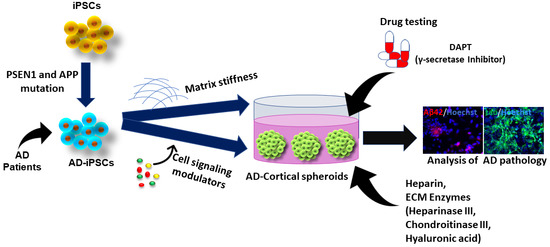
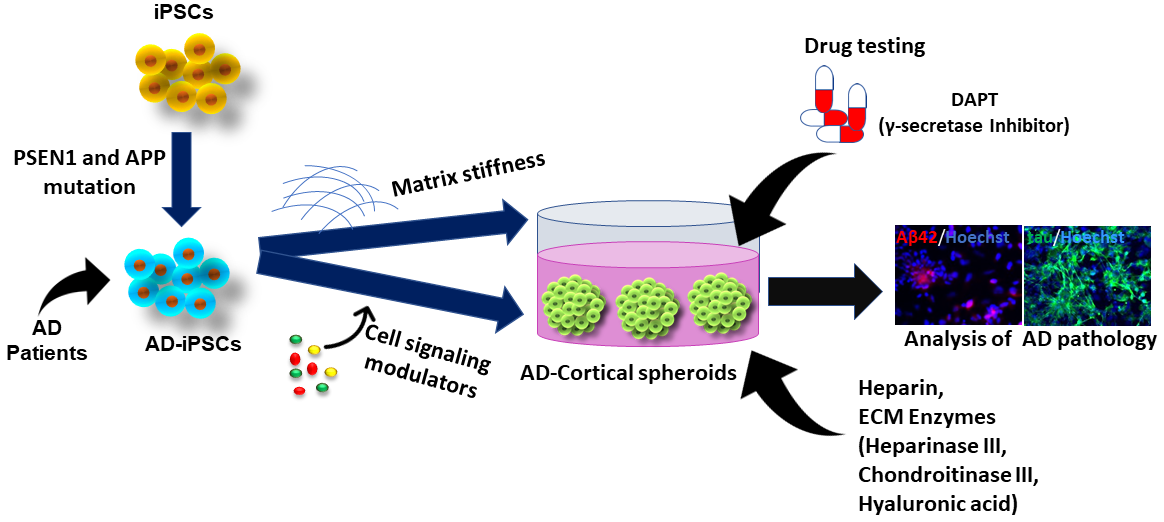
:
1. Introduction of Alzheimer’s Disease Pathology
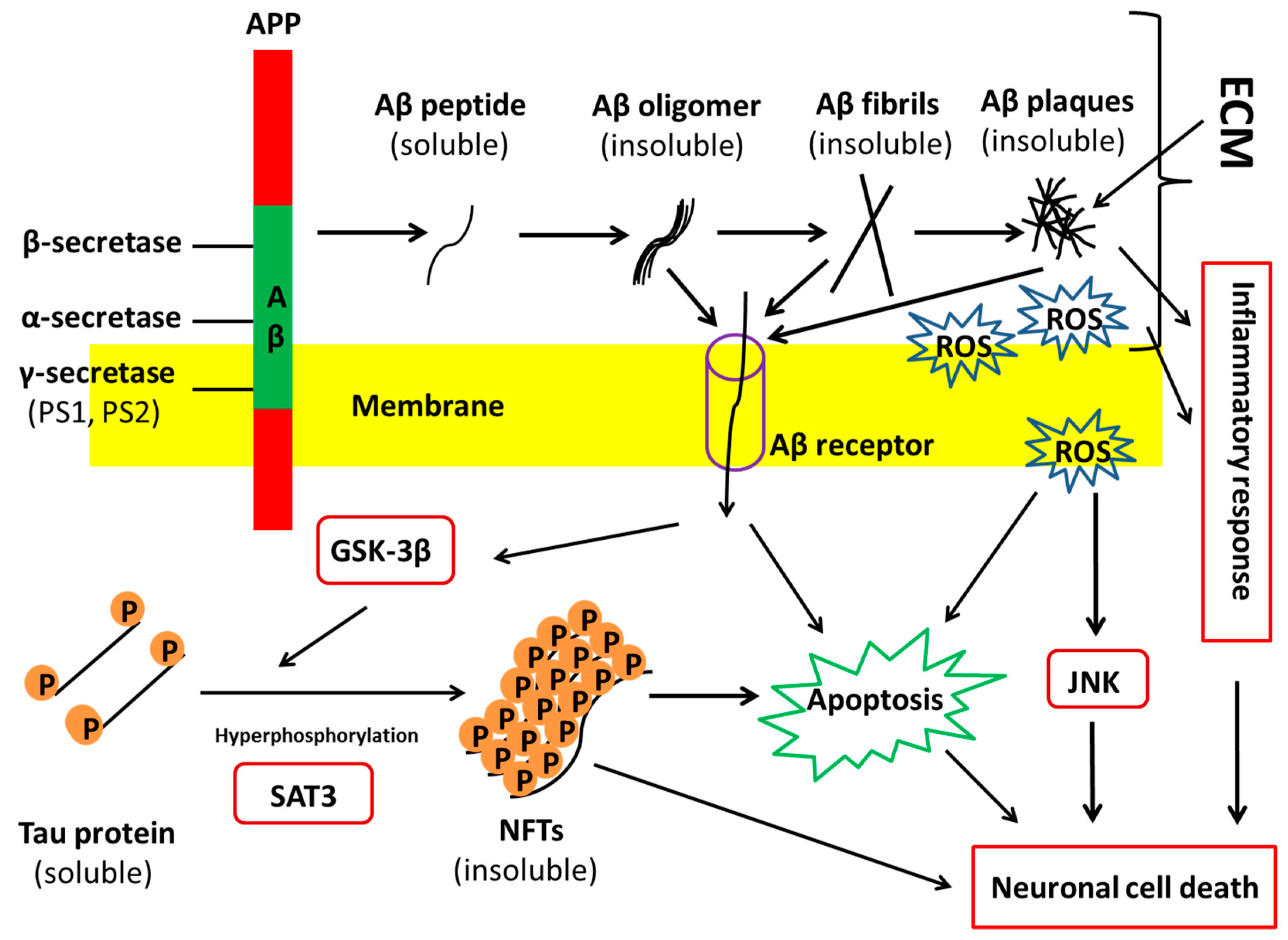
1.1. Alzheimer’s Disease Pathology and Progression
1.2. Current Challenges and the Demand for a Good AD Model
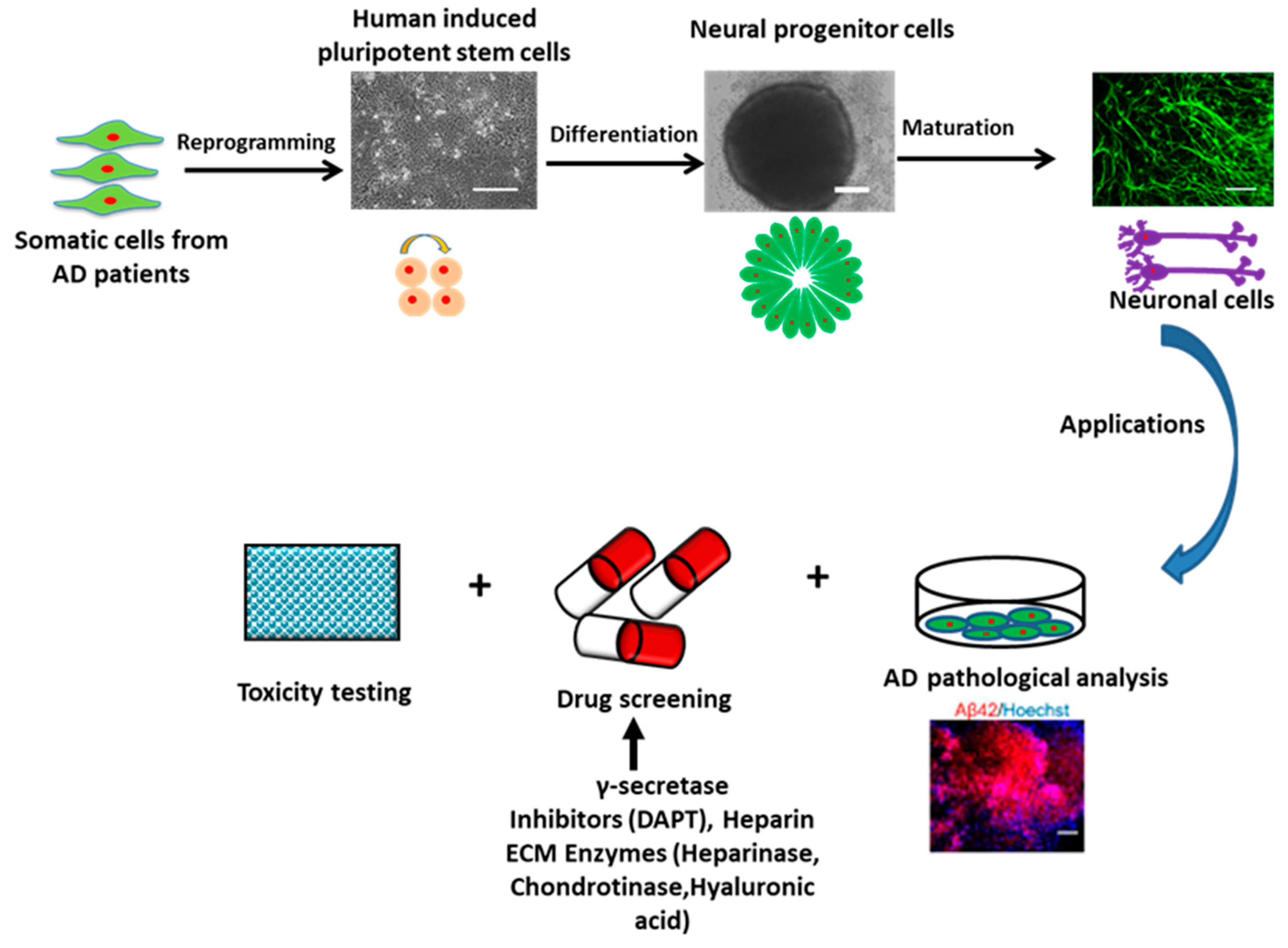
2. Human Pluripotent Stem Cells for Modeling AD
2.1. AD Models Using hPSCs
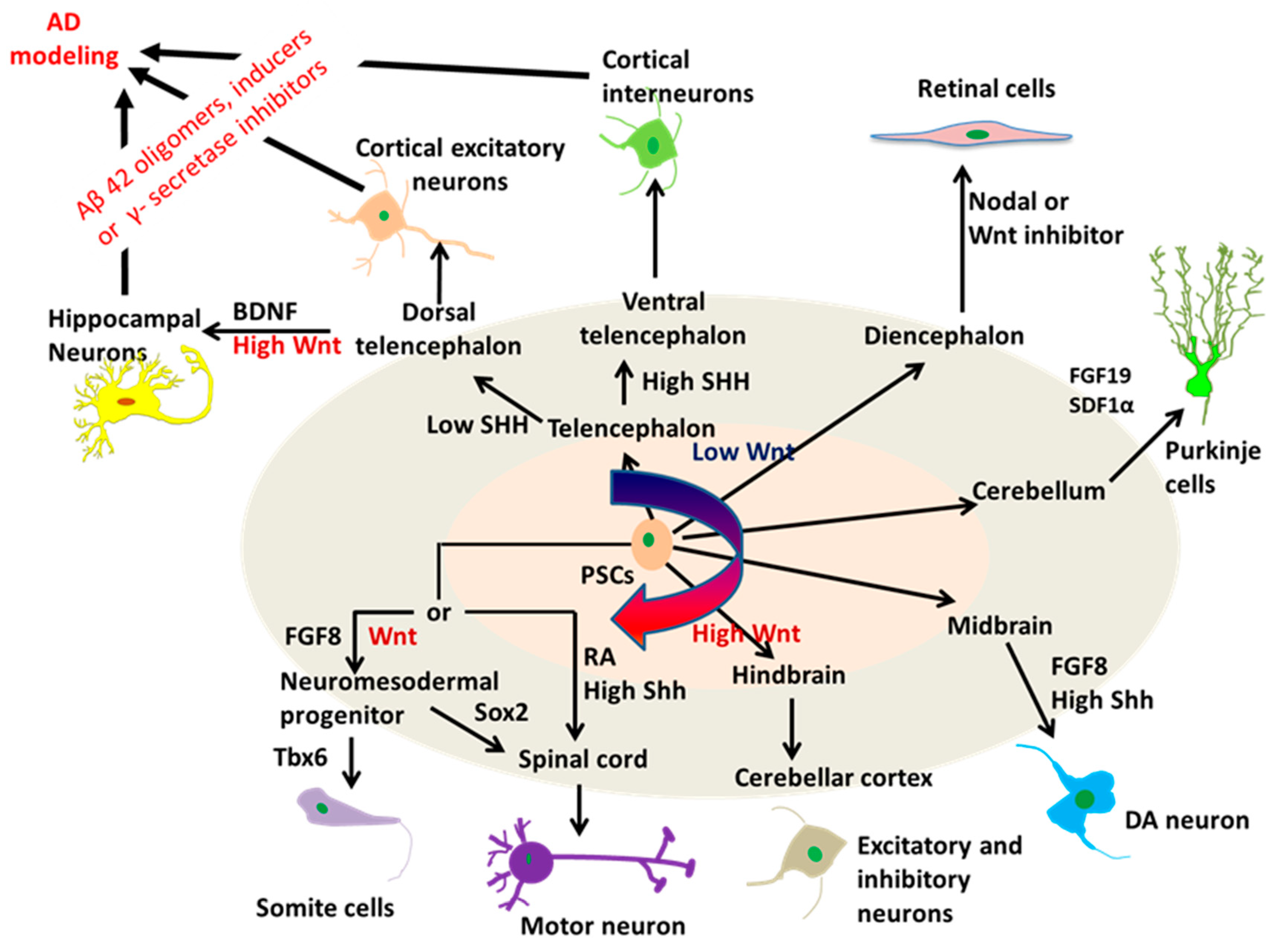
2.2. Neural Tissue Patterning of hPSCs
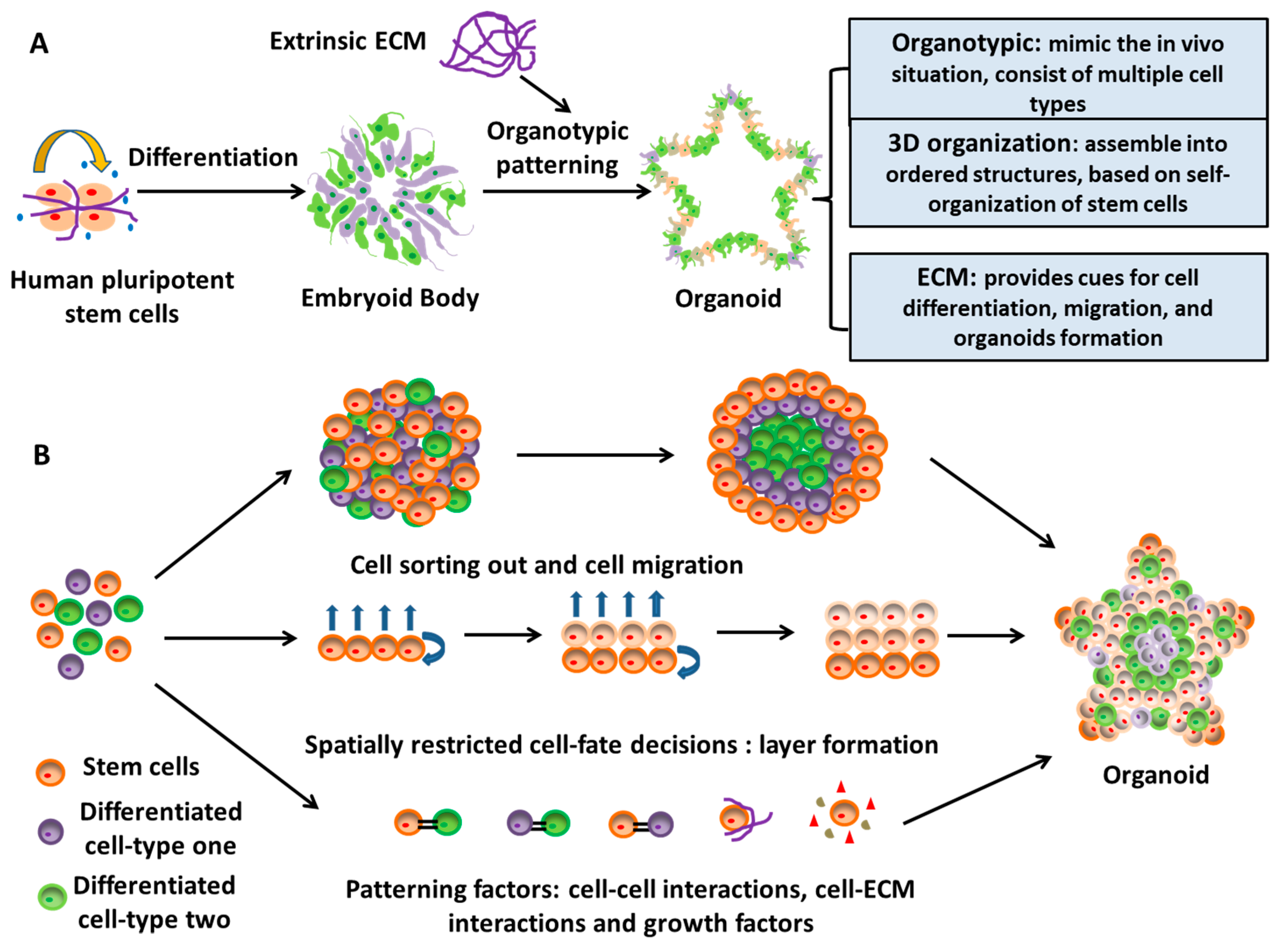
2.3. A Novel Neural Patterning Method: Organoid Technology
2.4. Effects of ECMs on Neural Patterning of hPSCs
3. Proteoglycans in the ECMs of AD Brain
3.1. Chondroitin Sulfate Proteoglycans (CSPGs) in Brain Development
3.2. Heparin/HSPG in Brain Development
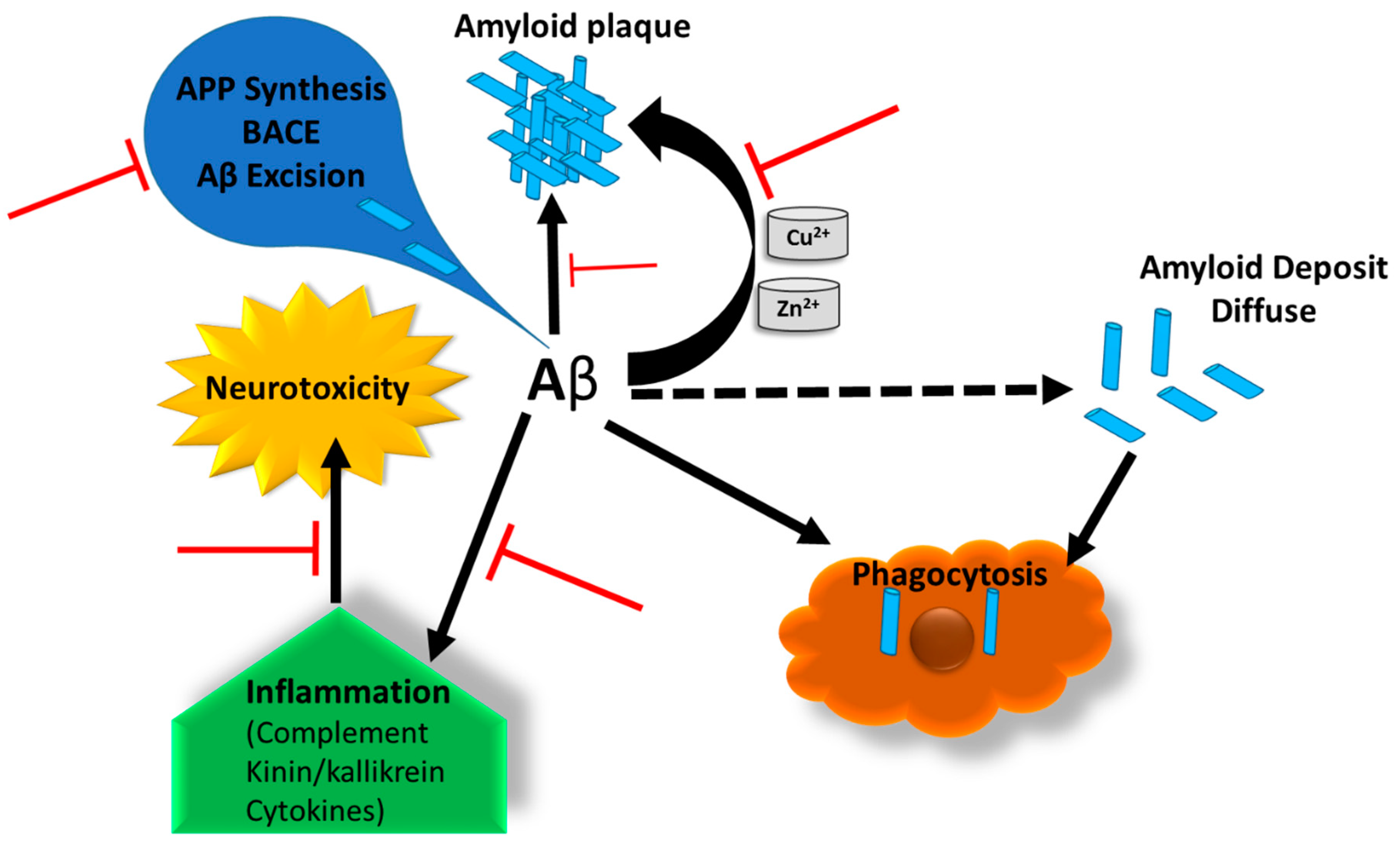
3.3. Impacts of CSPG on AD Pathology
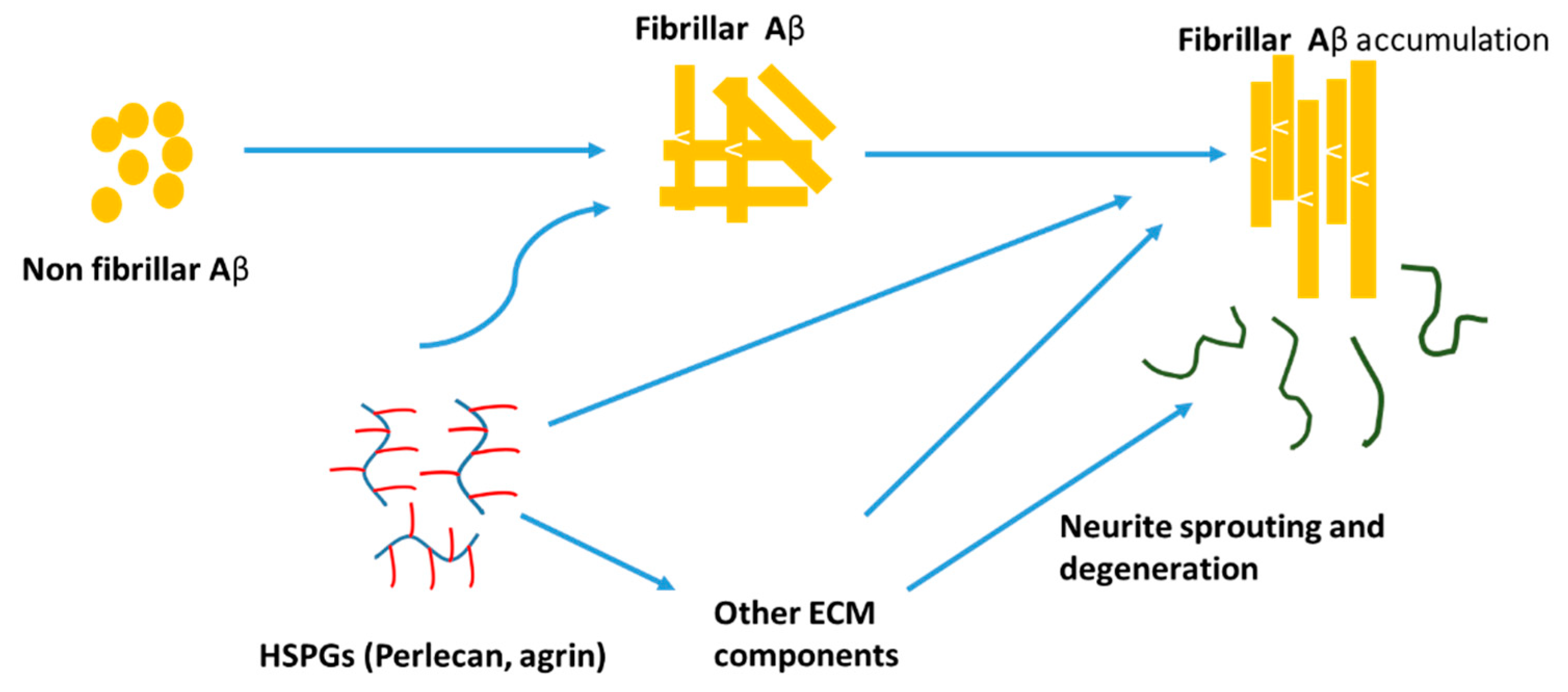
3.4. Impacts of Heparin/HSPGs on AD Pathology
3.5. Heparin-Based Therapy for Neural Degeneration
4. Studying ECM Effects in hiPSC-Derived Forebrain Organoids
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An English translation of Alzheimer’s 1907 paper, “Über eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dementia: A Public Health Priority; World Health Organization: Genève, Switzerland, 2012. [Google Scholar]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M. Neuroprotective therapeutics for Alzheimer’s disease: Progress and prospects. Trends Pharmacol. Sci. 2011, 32, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Wilhelmus, M.M.; Heljasvaara, R.; Pihlajaniemi, T.; Wesseling, P.; de Waal, R.M.; Verbeek, M.M. Collagen XVIII: A Novel Heparan Sulfate Proteoglycan Associated with Vascular Amyloid Depositions and Senile Plaques in Alzheimer’s Disease Brains. Brain Pathol. 2002, 12, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Meighan, S.E.; Meighan, P.C.; Choudhury, P.; Davis, C.J.; Olson, M.L.; Zornes, P.A.; Wright, J.W.; Harding, J.W. Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J. Neurochem. 2006, 96, 1227–1241. [Google Scholar] [CrossRef] [Green Version]
- Mlekusch, R.; Humpel, C. Matrix metalloproteinases-2 and-3 are reduced in cerebrospinal fluid with low beta-amyloid1–42 levels. Neurosci. Lett. 2009, 466, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Jiang, Z.-F. Accumulated amyloid-β peptide and hyperphosphorylated tau protein: Relationship and links in Alzheimer’s disease. J. Alzheimer Dis. 2009, 16, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Köpke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.D.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [PubMed]
- Palmer, A.M. Pharmacotherapy for Alzheimer’s disease: Progress and prospects. Trends Pharmacol. Sci. 2002, 23, 426–433. [Google Scholar] [CrossRef]
- Prokop, S.; Miller, K.R.; Heppner, F.L. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 461–477. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rüb, U.; Schultz, C.; Tredici, K.D. Vulnerability of cortical neurons to Alzheimer’s and Parkinson’s diseases. J. Alzheimer Dis. 2006, 9, 35–44. [Google Scholar] [CrossRef]
- Xu, X.-H.; Zhong, Z. Disease modeling and drug screening for neurological diseases using human induced pluripotent stem cells. Acta Pharmacol. Sin. 2013, 34, 755. [Google Scholar] [CrossRef]
- Duff, K.; Eckman, C.; Zehr, C.; Yu, X.; Prada, C.-M.; Perez-Tur, J.; Hutton, M.; Buee, L.; Harigaya, Y.; Yager, D. Increased amyloid-β42 (43) in brains of mice expressing mutant presenilin 1. Nature 1996, 383, 710. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.-H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Cedazo-Minguez, A. Successful therapies for Alzheimer’s disease: Why so many in animal models and none in humans? Front. Pharmacol. 2014, 5, 146. [Google Scholar] [CrossRef]
- Nam, H.; Lee, K.-H.; Nam, D.-H.; Joo, K.M. Adult human neural stem cell therapeutics: Current developmental status and prospect. World J. Stem Cells 2015, 7, 126. [Google Scholar] [CrossRef] [PubMed]
- Jakel, R.J.; Schneider, B.L.; Svendsen, C.N. Using human neural stem cells to model neurological disease. Nat. Rev. Genet. 2004, 5, 136. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narazaki, G.; Uosaki, H.; Teranishi, M.; Okita, K.; Kim, B.; Matsuoka, S.; Yamanaka, S.; Yamashita, J.K. Directed and systematic differentiation of cardiovascular cells from mouse induced pluripotent stem cells. Circulation 2008, 118, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Pavoni, S.; Jarray, R.; Nassor, F.; Guyot, A.C.; Cottin, S.; Rontard, J.; Mikol, J.; Mabondzo, A.; Deslys, J.P.; Yates, F. Small-molecule induction of Abeta-42 peptide production in human cerebral organoids to model Alzheimer’s disease associated phenotypes. PLoS ONE 2018, 13, e0209150. [Google Scholar] [CrossRef] [PubMed]
- Vazin, T.; Ball, K.A.; Lu, H.; Park, H.; Ataeijannati, Y.; Head-Gordon, T.; Poo, M.M.; Schaffer, D.V. Efficient derivation of cortical glutamatergic neurons from human pluripotent stem cells: A model system to study neurotoxicity in Alzheimer’s disease. Neurobiol. Dis. 2014, 62, 62–72. [Google Scholar] [CrossRef]
- Yan, Y.; Bejoy, J.; Xia, J.; Guan, J.; Zhou, Y.; Li, Y. Neural patterning of human induced pluripotent stem cells in 3-D cultures for studying biomolecule-directed differential cellular responses. Acta Biomater. 2016, 42, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.; Tamboli, I.Y.; Mertens, J.; Wunderlich, P.; Ladewig, J.; Stuber, K.; Esselmann, H.; Wiltfang, J.; Brustle, O.; Walter, J. Presenilin-1 L166P mutant human pluripotent stem cell-derived neurons exhibit partial loss of gamma-secretase activity in endogenous amyloid-beta generation. Am. J. Pathol. 2012, 180, 2404–2416. [Google Scholar] [CrossRef]
- Woodruff, G.; Young, J.E.; Martinez, F.J.; Buen, F.; Gore, A.; Kinaga, J.; Li, Z.; Yuan, S.H.; Zhang, K.; Goldstein, L.S. The presenilin-1 DeltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013, 5, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 2017, 168, 427–441.e421. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dreau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Wicklund, L.; Leao, R.N.; Stromberg, A.M.; Mousavi, M.; Hovatta, O.; Nordberg, A.; Marutle, A. Beta-amyloid 1-42 oligomers impair function of human embryonic stem cell-derived forebrain cholinergic neurons. PLoS ONE 2010, 5, e15600. [Google Scholar] [CrossRef]
- Yahata, N.; Asai, M.; Kitaoka, S.; Takahashi, K.; Asaka, I.; Hioki, H.; Kaneko, T.; Maruyama, K.; Saido, T.C.; Nakahata, T.; et al. Anti-Abeta drug screening platform using human iPS cell-derived neurons for the treatment of Alzheimer’s disease. PLoS ONE 2011, 6, e25788. [Google Scholar] [CrossRef]
- Mertens, J.; Stuber, K.; Poppe, D.; Doerr, J.; Ladewig, J.; Brustle, O.; Koch, P. Embryonic stem cell-based modeling of tau pathology in human neurons. Am. J. Pathol. 2013, 182, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef]
- Shi, Y.C.; Kirwan, P.; Smith, J.; MacLean, G.; Orkin, S.H.; Livesey, F.J. A Human Stem Cell Model of Early Alzheimer’s Disease Pathology in Down Syndrome. Sci. Transl. Med. 2012, 4, 124ra29. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Stuber, K.; Wunderlich, P.; Ladewig, J.; Kesavan, J.C.; Vandenberghe, R.; Vandenbulcke, M.; van Damme, P.; Walter, J.; Brustle, O.; et al. APP processing in human pluripotent stem cell-derived neurons is resistant to NSAID-based gamma-secretase modulation. Stem Cell Rep. 2013, 1, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Sproul, A.A.; Jacob, S.; Pre, D.; Kim, S.H.; Nestor, M.W.; Navarro-Sobrino, M.; Santa-Maria, I.; Zimmer, M.; Aubry, S.; Steele, J.W.; et al. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS ONE 2014, 9, e84547. [Google Scholar] [CrossRef]
- Mahairaki, V.; Ryu, J.; Peters, A.; Chang, Q.; Li, T.; Park, T.S.; Burridge, P.W.; Talbot, C.C., Jr.; Asnaghi, L.; Martin, L.J.; et al. Induced pluripotent stem cells from familial Alzheimer’s disease patients differentiate into mature neurons with amyloidogenic properties. Stem Cells Dev. 2014, 23, 2996–3010. [Google Scholar] [CrossRef]
- Duan, L.S.; Bhattacharyya, B.J.; Belmadani, A.; Pan, L.L.; Miller, R.J.; Kessler, J.A. Stem cell derived basal forebrain cholinergic neurons from Alzheimer’s disease patients are more susceptible to cell death. Mol. Neurodegen. 2014, 9, 3. [Google Scholar] [CrossRef]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Human. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Waltz, S.; Woodruff, G.; Ouyang, J.; Israel, M.A.; Herrera, C.; Sarsoza, F.; Tanzi, R.E.; Koo, E.H.; Ringman, J.M.; et al. Effect of potent gamma-secretase modulator in human neurons derived from multiple presenilin 1-induced pluripotent stem cell mutant carriers. JAMA Neurol. 2014, 71, 1481–1489. [Google Scholar] [CrossRef]
- Lee, J.K.; Jin, H.K.; Park, M.H.; Kim, B.R.; Lee, P.H.; Nakauchi, H.; Carter, J.E.; He, X.; Schuchman, E.H.; Bae, J.S. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J. Exp. Med. 2014, 211, 1551–1570. [Google Scholar] [CrossRef] [Green Version]
- Young, J.E.; Boulanger-Weill, J.; Williams, D.A.; Woodruff, G.; Buen, F.; Revilla, A.C.; Herrera, C.; Israel, M.A.; Yuan, S.H.; Edland, S.D.; et al. Elucidating molecular phenotypes caused by the SORL1 Alzheimer’s disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 2015, 16, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Evans, L.D.; Andersson, T.; Portelius, E.; Smith, J.; Dias, T.B.; Saurat, N.; McGlade, A.; Kirwan, P.; Blennow, K.; et al. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015, 11, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Hossini, A.M.; Megges, M.; Prigione, A.; Lichtner, B.; Toliat, M.R.; Wruck, W.; Schroter, F.; Nuernberg, P.; Kroll, H.; Makrantonaki, E.; et al. Induced pluripotent stem cell-derived neuronal cells from a sporadic Alzheimer’s disease donor as a model for investigating AD-associated gene regulatory networks. BMC Genom. 2015, 16, 84. [Google Scholar]
- Armijo, E.; Gonzalez, C.; Shahnawaz, M.; Flores, A.; Davis, B.; Soto, C. Increased susceptibility to Abeta toxicity in neuronal cultures derived from familial Alzheimer’s disease (PSEN1-A246E) induced pluripotent stem cells. Neurosci. Lett. 2017, 639, 74–81. [Google Scholar] [CrossRef]
- Balez, R.; Steiner, N.; Engel, M.; Munoz, S.S.; Lum, J.S.; Wu, Y.; Wang, D.; Vallotton, P.; Sachdev, P.; O’Connor, M.; et al. Neuroprotective effects of apigenin against inflammation, neuronal excitability and apoptosis in an induced pluripotent stem cell model of Alzheimer’s disease. Sci. Rep. 2016, 6, 31450. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.-C.; Muratore, C.R.; Gierahn, T.M.; Sullivan, S.E.; Srikanth, P.; De Jager, P.L.; Love, J.C.; Young-Pearse, T.L. Single-Cell Detection of Secreted Aβ and sAPPα from Human IPSC-Derived Neurons and Astrocytes. J. Neurosci. 2016, 36, 1730–1746. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Velazquez Sanchez, C.; Chen, M.; Morin, P.J.; Wells, J.M.; Hanlon, E.B.; Xia, W. Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0163072. [Google Scholar] [CrossRef]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [PubMed]
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis. 2017, 8, e2696. [Google Scholar] [CrossRef] [PubMed]
- Muratore, C.R.; Zhou, C.; Liao, M.; Fernandez, M.A.; Taylor, W.M.; Lagomarsino, V.N.; Pearse, R.V., 2nd; Rice, H.C.; Negri, J.M.; He, A.; et al. Cell-type Dependent Alzheimer’s Disease Phenotypes: Probing the Biology of Selective Neuronal Vulnerability. Stem Cell Rep. 2017, 9, 1868–1884. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Imamura, K.; Funayama, M.; Tsukita, K.; Miyake, M.; Ohta, A.; Woltjen, K.; Nakagawa, M.; Asada, T.; Arai, T.; et al. iPSC-Based Compound Screening and In Vitro Trials Identify a Synergistic Anti-amyloid beta Combination for Alzheimer’s Disease. Cell Rep. 2017, 21, 2304–2312. [Google Scholar] [CrossRef]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Teglasi, A.; Bock, I.; Giudice, M.L.; Tancos, Z.; Molnar, K.; Laszlo, L.; et al. Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, S.; Gubert Olive, M.; Shakirzyanova, A.; Leskela, S.; Sarajarvi, T.; Viitanen, M.; et al. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef]
- Hu, N.W.; Corbett, G.T.; Moore, S.; Klyubin, I.; O’Malley, T.T.; Walsh, D.M.; Livesey, F.J.; Rowan, M.J. Extracellular Forms of Abeta and Tau from iPSC Models of Alzheimer’s Disease Disrupt Synaptic Plasticity. Cell Rep. 2018, 23, 1932–1938. [Google Scholar] [CrossRef]
- Yan, Y.; Song, L.; Bejoy, J.; Zhao, J.; Kanekiyo, T.; Bu, G.; Zhou, Y.; Li, Y. Modelling neurodegenerative microenvironment using cortical organoids derived from human stem cells. Tissue Eng. Part A 2018, 24, 1125–1137. [Google Scholar] [CrossRef]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef]
- Chen, M.; Lee, H.K.; Moo, L.; Hanlon, E.; Stein, T.; Xia, W. Common proteomic profiles of induced pluripotent stem cell-derived three-dimensional neurons and brain tissue from Alzheimer patients. J. Proteom. 2018, 182, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kundig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-beta and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018, 27, 121–130. [Google Scholar] [CrossRef]
- Young, J.E.; Fong, L.K.; Frankowski, H.; Petsko, G.A.; Small, S.A.; Goldstein, L.S.B. Stabilizing the Retromer Complex in a Human Stem Cell Model of Alzheimer’s Disease Reduces TAU Phosphorylation Independently of Amyloid Precursor Protein. Stem Cell Rep. 2018, 10, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, D.A.; Korn, O.; Virshup, I.; Wells, C.A.; Wolvetang, E.J. The Impact of APP on Alzheimer-like Pathogenesis and Gene Expression in Down Syndrome iPSC-Derived Neurons. Stem Cell Rep. 2018, 11, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Cui, Y.; Ducy, P.; Karsenty, G.; Franceschi, R.T. Ascorbic acid-dependent activation of the osteocalcin promoter in MC3T3-E1 preosteoblasts: Requirement for collagen matrix synthesis and the presence of an intact OSE2 sequence. Mol. Endocrinol. 1997, 11, 1103–1113. [Google Scholar] [CrossRef]
- Hu, W.; Qiu, B.; Guan, W.; Wang, Q.; Wang, M.; Li, W.; Gao, L.; Shen, L.; Huang, Y.; Xie, G.; et al. Direct Conversion of Normal and Alzheimer’s Disease Human Fibroblasts into Neuronal Cells by Small Molecules. Cell Stem Cell 2015, 17, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Espuny-Camacho, I.; Arranz, A.M.; Fiers, M.; Snellinx, A.; Ando, K.; Munck, S.; Bonnefont, J.; Lambot, L.; Corthout, N.; Omodho, L.; et al. Hallmarks of Alzheimer’s Disease in Stem-Cell-Derived Human Neurons Transplanted into Mouse Brain. Neuron 2017, 93, 1066–1081.e1068. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ward, M.E.; Chen, R.; Liu, K.; Tracy, T.E.; Chen, X.; Xie, M.; Sohn, P.D.; Ludwig, C.; Meyer-Franke, A.; et al. Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Rep. 2017, 9, 1221–1233. [Google Scholar] [CrossRef]
- Harasta, A.E.; Ittner, L.M. Alzheimer’s Disease: Insights from Genetic Mouse Models and Current Advances in Human IPSC-Derived Neurons. Adv. Neurobiol. 2017, 15, 3–29. [Google Scholar]
- Jorfi, M.; D’Avanzo, C.; Tanzi, R.E.; Kim, D.Y.; Irimia, D. Human Neurospheroid Arrays for In Vitro Studies of Alzheimer’s Disease. Sci. Rep. 2018, 8, 2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, L.S.; Reyna, S.; Woodruff, G. Probing the secrets of Alzheimer’s disease using human-induced pluripotent stem cell technology. Neurotherapeutics 2015, 12, 121–125. [Google Scholar] [CrossRef]
- Vazin, T.; Ashton, R.S.; Conway, A.; Rode, N.A.; Lee, S.M.; Bravo, V.; Healy, K.E.; Kane, R.S.; Schaffer, D.V. The effect of multivalent Sonic hedgehog on differentiation of human embryonic stem cells into dopaminergic and GABAergic neurons. Biomaterials 2014, 35, 941–948. [Google Scholar] [CrossRef]
- Du, Z.-W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.-L.; Zhong, X.; Fan, F.; Zhang, S.-C. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 2015, 6, 6626. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N. Cerebral organoids model human brain development and microcephaly. Mov. Disord. 2014, 29, 185. [Google Scholar] [CrossRef] [PubMed]
- Paşca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.-Y.; O’rourke, N.A.; Nguyen, K.D. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Guo, M.; Martins-Taylor, K.; Wang, X.; Zhang, Z.; Park, J.W.; Zhan, S.; Kronenberg, M.S.; Lichtler, A.; Liu, H.-X. Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS ONE 2010, 5, e11853. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, K.; Sone, T.; Ibata, K.; Fujimori, K.; Yuzaki, M.; Akamatsu, W. Controlling the Regional Identity of hPSC-Derived Neurons to Uncover Neuronal Subtype Specificity of Neurological Disease Phenotypes. Stem Cell Rep. 2015, 5, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Kirwan, P.; Livesey, F.J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 2012, 7, 1836. [Google Scholar] [CrossRef]
- Suzuki, I.K.; Vanderhaeghen, P. Is this a brain which I see before me? Modeling human neural development with pluripotent stem cells. Development 2015, 142, 3138–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.A.; Waldau, B. Generation of human vascularized brain organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Goncalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, R.A.; Leopoldi, A.; Aichinger, M.; Wick, N.; Hantusch, B.; Novatchkova, M.; Taubenschmid, J.; Hammerle, M.; Esk, C.; Bagley, J.A.; et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature 2019, 565, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.R.; Leung, A.W. Next generation organoids for biomedical research and applications. Biotechnol. Adv. 2018, 36, 132–149. [Google Scholar] [CrossRef]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, C.; Ma, T. In vitro organogenesis from pluripotent stem cells. Organogenesis 2014, 10, 159–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Mead, B.E.; Safaee, H.; Langer, R.; Karp, J.M.; Levy, O. Engineering stem cell organoids. Cell Stem Cell 2016, 18, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 10. [Google Scholar] [CrossRef]
- Passier, R.; Orlova, V.; Mummery, C. Complex tissue and disease modeling using hiPSCs. Cell Stem Cell 2016, 18, 309–321. [Google Scholar] [CrossRef]
- Zhang, S.-C.; Wernig, M.; Duncan, I.D.; Brüstle, O.; Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1129. [Google Scholar] [CrossRef]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasail, Y. Self-Organized Formation of Polarized Cortical Tissues from ESCs and Its Active Manipulation by Extrinsic Signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.; Simonini, M.V.; Palejev, D.; Tomasini, L.; Coppola, G.; Szekely, A.M.; Horvath, T.L.; Vaccarino, F.M. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12770–12775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-dependent dysregulation of GABA/Glutamate neuron differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef]
- Amin, N.D.; Pasca, S.P. Building Models of Brain Disorders with Three-Dimensional Organoids. Neuron 2018, 100, 389–405. [Google Scholar] [CrossRef]
- Yan, Y.; Song, L.; Madinya, J.; Ma, T.; Li, Y. Derivation of cortical spheroids from human induced pluripotent stem cells in a suspension bioreactor. Tissue Eng. Part A 2018, 24, 418–431. [Google Scholar] [CrossRef]
- Edri, R.; Gal, I.; Noor, N.; Harel, T.; Fleischer, S.; Adadi, N.; Green, O.; Shabat, D.; Heller, L.; Shapira, A.; et al. Personalized Hydrogels for Engineering Diverse Fully Autologous Tissue Implants. Adv. Mater. 2019, 31, e1803895. [Google Scholar] [CrossRef] [PubMed]
- Sart, S.; Yan, Y.; Li, Y.; Lochner, E.; Zeng, C.; Ma, T.; Li, Y. Crosslinking of extracellular matrix scaffolds derived from pluripotent stem cell aggregates modulates neural differentiation. Acta Biomater. 2016, 30, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Wang, Y.; Kokovay, E.; Lin, G.; Chuang, S.-M.; Goderie, S.K.; Roysam, B.; Temple, S. Adult SVZ stem cells lie in a vascular niche: A quantitative analysis of niche cell-cell interactions. Cell Stem Cell 2008, 3, 289–300. [Google Scholar] [CrossRef]
- Yan, Y.; Martin, L.M.; Bosco, D.B.; Bundy, J.L.; Nowakowski, R.S.; Sang, Q.-X.A.; Li, Y. Differential effects of acellular embryonic matrices on pluripotent stem cell expansion and neural differentiation. Biomaterials 2015, 73, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejoy, J.; Wang, Z.; Bijonowski, B.; Yang, M.; Ma, T.; Sang, Q.X.; Li, Y. Differential effects of heparin and hyaluronic acid on neural patterning of human induced pluripotent stem cells. ACS Biomater. Sci. Eng. 2018, 4, 4354–4366. [Google Scholar] [CrossRef]
- Yan, Y.; Li, Y.; Song, L.; Zeng, C.; Li, Y. Pluripotent stem cell expansion and neural differentiation in 3-D scaffolds of tunable Poisson’s ratio. Acta Biomater. 2017, 49, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Mooney, D.J.; Zandstra, P.W. Growth factors, matrices, and forces combine and control stem cells. Science 2009, 324, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Sen, S.; Sweeney, L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef]
- Saha, K.; Keung, A.J.; Irwin, E.F.; Li, Y.; Little, L.; Schaffer, D.V.; Healy, K.E. Substrate modulus directs neural stem cell behavior. Biophys. J. 2008, 95, 4426–4438. [Google Scholar] [CrossRef]
- Leipzig, N.D.; Shoichet, M.S. The effect of substrate stiffness on adult neural stem cell behavior. Biomaterials 2009, 30, 6867–6878. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, C.R.; Kamm, R.D. 3D matrix microenvironment for targeted differentiation of embryonic stem cells into neural and glial lineages. Biomaterials 2013, 34, 5995–6007. [Google Scholar] [CrossRef] [PubMed]
- Lantoine, J.; Grevesse, T.; Villers, A.; Delhaye, G.; Mestdagh, C.; Versaevel, M.; Mohammed, D.; Bruyere, C.; Alaimo, L.; Lacour, S.P.; et al. Matrix stiffness modulates formation and activity of neuronal networks of controlled architectures. Biomaterials 2016, 89, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madl, C.M.; LeSavage, B.L.; Dewi, R.E.; Dinh, C.B.; Stowers, R.S.; Khariton, M.; Lampe, K.J.; Nguyen, D.; Chaudhuri, O.; Enejder, A.; et al. Maintenance of neural progenitor cell stemness in 3D hydrogels requires matrix remodelling. Nat. Mater. 2017, 16, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, X.; Janairo, R.R.R.; Kwong, G.; Tsou, A.D.; Chu, J.S.; Wang, A.; Yu, J.; Wang, D.; Li, S. Matrix stiffness modulates the differentiation of neural crest stem cells in vivo. J. Cell. Physiol. 2019, 234, 7569–7578. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.Q.; Gao, X.; Wang, A.; Yang, Y.; Liu, S.; Yu, Z.; Song, G.B.; Zhao, H.C. Substrate stiffness affects neural network activity in an extracellular matrix proteins dependent manner. Colloids Surf. B Biointerfaces 2018, 170, 729–735. [Google Scholar] [CrossRef]
- Srinivasan, A.; Chang, S.Y.; Zhang, S.; Toh, W.S.; Toh, Y.C. Substrate stiffness modulates the multipotency of human neural crest derived ectomesenchymal stem cells via CD44 mediated PDGFR signaling. Biomaterials 2018, 167, 153–167. [Google Scholar] [CrossRef]
- Keung, A.J.; Asuri, P.; Kumar, S.; Schaffer, D.V. Soft microenvironments promote the early neurogenic differentiation but not self-renewal of human pluripotent stem cells. Integr. Biol. 2012, 4, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Discher, D.E.; Janmey, P.; Wang, Y.-l. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef]
- Pagliara, S.; Franze, K.; McClain, C.R.; Wylde, G.; Fisher, C.L.; Franklin, R.J.M.; Kabla, A.J.; Keyser, U.F.; Chalut, K.J. Auxetic nuclei in embryonic stem cells exiting pluripotency. Nat. Mater. 2014, 13, 638–644. [Google Scholar] [CrossRef]
- Evans, N.D.; Minelli, C.; Gentleman, E.; LaPointe, V.; Patankar, S.N.; Kallivretaki, M.; Chen, X.; Roberts, C.J.; Stevens, M.M. Substrate stiffness affects early differentiation events in embryonic stem cells. Eur. Cells Mater. 2009, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Arha, M.; Choudhary, S.; Ashton, R.S.; Bhatia, S.R.; Schaffer, D.V.; Kane, R.S. The influence of hydrogel modulus on the proliferation and differentiation of encapsulated neural stem cells. Biomaterials 2009, 30, 4695–4699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, F.; Na, S.; Li, D.; Poh, Y.-C.; Tanaka, T.S.; Wang, F.; Wang, N. Material properties of the cell dictate stress-induced spreading and differentiation in embryonic stem cells. Nat. Mater. 2010, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Musah, S.; Morin, S.A.; Wrighton, P.J.; Zwick, D.B.; Jin, S.; Kiessling, L.L. Glycosaminoglycan-binding hydrogels enable mechanical control of human pluripotent stem cell self-renewal. ACS Nano 2012, 6, 10168–10177. [Google Scholar] [CrossRef] [PubMed]
- Zoldan, J.; Karagiannis, E.D.; Lee, C.Y.; Anderson, D.G.; Langer, R.; Levenberg, S. The influence of scaffold elasticity on germ layer specification of human embryonic stem cells. Biomaterials 2011, 32, 9612–9621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshi, A.; Nakashima, Y.; Nakano, H.; Eaimkhong, S.; Evseenko, D.; Reed, J.; Stieg, A.Z.; Gimzewski, J.K.; Nakano, A. Rigid Microenvironments Promote Cardiac Differentiation Of Mouse And Human Embryonic Stem Cells. Circ. Res. 2013, 113, 1. [Google Scholar] [CrossRef]
- Eroshenko, N.; Ramachandran, R.; Yadavalli, V.K.; Rao, R.R. Effect of substrate stiffness on early human embryonic stem cell differentiation. J. Biol. Eng. 2013, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Lim, V.Y.; Shen, J.Y.; Tan, W.; Rajendran, D.; Luo, S.C.; Gao, S.J.; Wan, A.C.A.; Ying, J.Y. Extracellular Matrix-Mediated Differentiation of Human Embryonic Stem Cells: Differentiation to Insulin-Secreting Beta Cells. Tissue Eng. Part A 2014, 20, 424–433. [Google Scholar] [CrossRef]
- Hazeltine, L.B.; Badur, M.G.; Lian, X.; Das, A.; Han, W.; Palecek, S.P. Temporal impact of substrate mechanics on differentiation of human embryonic stem cells to cardiomyocytes. Acta Biomater. 2014, 10, 604–612. [Google Scholar] [CrossRef]
- Maldonado, M.; Wong, L.Y.; Echeverria, C.; Ico, G.; Low, K.; Fujimoto, T.; Johnson, J.K.; Nam, J. The effects of electrospun substrate-mediated cell colony morphology on the self-renewal of human induced pluripotent stem cells. Biomaterials 2015, 50, 10–19. [Google Scholar] [CrossRef]
- Ribeiro, A.J.; Ang, Y.S.; Fu, J.D.; Rivas, R.N.; Mohamed, T.M.; Higgs, G.C.; Srivastava, D.; Pruitt, B.L. Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc. Natl. Acad. Sci. USA 2015, 112, 12705–12710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, N.; Tasnim, F.; Yue, C.; Qu, Y.; Phan, D.; Choudhury, Y.; Tan, M.-H.; Yu, H. Substrate Stiffness Modulates the Maturation of Human Pluripotent Stem-Cell-Derived Hepatocytes. ACS Biomater. Sci. Eng. 2016, 2, 1649–1657. [Google Scholar] [CrossRef]
- Caiazzo, M.; Okawa, Y.; Ranga, A.; Piersigilli, A.; Tabata, Y.; Lutolf, M.P. Defined three-dimensional microenvironments boost induction of pluripotency. Nat. Mater. 2016, 15, 344–352. [Google Scholar] [CrossRef]
- Przybyla, L.; Lakins, J.N.; Weaver, V.M. Tissue Mechanics Orchestrate Wnt-Dependent Human Embryonic Stem Cell Differentiation. Cell Stem Cell 2016, 19, 462–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, T.; Barner, S.; Candiello, J.; Kumta, P.N.; Banerjee, I. Capsule stiffness regulates the efficiency of pancreatic differentiation of human embryonic stem cells. Acta Biomater. 2016, 35, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordonez-Moran, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef]
- Wang, B.; Tu, X.; Wei, J.; Wang, L.; Chen, Y. Substrate elasticity dependent colony formation and cardiac differentiation of human induced pluripotent stem cells. Biofabrication 2018, 11, 015005. [Google Scholar] [CrossRef]
- Dorsey, T.B.; Kim, D.; Grath, A.; James, D.; Dai, G. Multivalent biomaterial platform to control the distinct arterial venous differentiation of pluripotent stem cells. Biomaterials 2018, 185, 1–12. [Google Scholar] [CrossRef]
- Fu, J.; Chuah, Y.J.; Liu, J.; Tan, S.Y.; Wang, D.-A. Respective Effects of Gelatin-Coated Polydimethylsiloxane (PDMS) Substrates on Self-renewal and Cardiac Differentiation of Induced Pluripotent Stem Cells (iPSCs). ACS Biomater. Sci. Eng. 2018, 4, 4321–4330. [Google Scholar] [CrossRef]
- Hirata, M.; Yamaoka, T. Effect of stem cell niche elasticity/ECM protein on the self-beating cardiomyocyte differentiation of induced pluripotent stem (iPS) cells at different stages. Acta Biomater. 2018, 65, 44–52. [Google Scholar] [CrossRef]
- Goetzke, R.; Franzen, J.; Ostrowska, A.; Vogt, M.; Blaeser, A.; Klein, G.; Rath, B.; Fischer, H.; Zenke, M.; Wagner, W. Does soft really matter? Differentiation of induced pluripotent stem cells into mesenchymal stromal cells is not influenced by soft hydrogels. Biomaterials 2018, 156, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.G.; Gil, C.H.; Seo, J.; Park, S.J.; Subbiah, R.; Jung, T.H.; Kim, J.S.; Jeong, Y.H.; Chung, H.M.; Lee, J.H.; et al. Mechanotransduction of human pluripotent stem cells cultivated on tunable cell-derived extracellular matrix. Biomaterials 2018, 150, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Bandtlow, C.E.; Zimmermann, D.R. Proteoglycans in the developing brain: New conceptual insights for old proteins. Physiol. Rev. 2000, 80, 1267–1290. [Google Scholar] [CrossRef] [PubMed]
- Galtrey, C.M.; Fawcett, J.W. The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Res. Rev. 2007, 54, 1–18. [Google Scholar] [CrossRef]
- Schwartz, N.B.; Domowicz, M. Proteoglycans in brain development. Glycoconj. J. 2004, 21, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [PubMed]
- Oohira, A.; Shuo, T.; Tokita, Y.; Nakanishi, K.; Aono, S. Neuroglycan C, a brain-specific part-time proteoglycan, with a particular multidomain structure. Glycoconj. J. 2004, 21, 53–57. [Google Scholar] [CrossRef]
- Senkov, O.; Andjus, P.; Radenovic, L.; Soriano, E.; Dityatev, A. Neural ECM molecules in synaptic plasticity, learning, and memory. Prog. Brain Res. 2014, 214, 53–80. [Google Scholar]
- Howell, M.D.; Bailey, L.A.; Cozart, M.A.; Gannon, B.M.; Gottschall, P.E. Hippocampal administration of chondroitinase ABC increases plaque-adjacent synaptic marker and diminishes amyloid burden in aged APPswe/PS1dE9 mice. Acta Neuropathol. Commun. 2015, 3, 54. [Google Scholar] [CrossRef]
- Perry, G.; Siedlak, S.L.; Richey, P.; Kawai, M.; Cras, P.; Kalaria, R.N.; Galloway, P.G.; Scardina, J.M.; Cordell, B.; Greenberg, B.D.; et al. Association of heparan sulfate proteoglycan with the neurofibrillary tangles of Alzheimer’s disease. J. Neurosci. 1991, 11, 3679–3683. [Google Scholar] [CrossRef]
- Castillo, G.M.; Ngo, C.; Cummings, J.; Wight, T.N.; Snow, A.D. Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer’s disease, accelerates A beta fibril formation, and maintains A beta fibril stability. J. Neurochem. 1997, 69, 2452–2465. [Google Scholar] [CrossRef] [PubMed]
- Cotman, S.L.; Halfter, W.; Cole, G.J. Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer’s disease brain. Mol. Cell Neurosci. 2000, 15, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Sandwall, E.; O’Callaghan, P.; Zhang, X.; Lindahl, U.; Lannfelt, L.; Li, J.P. Heparan sulfate mediates amyloid-beta internalization and cytotoxicity. Glycobiology 2010, 20, 533–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.P.; Galvis, M.L.; Gong, F.; Zhang, X.; Zcharia, E.; Metzger, S.; Vlodavsky, I.; Kisilevsky, R.; Lindahl, U. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc. Natl. Acad. Sci. USA 2005, 102, 6473–6477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leveugle, B.; Scanameo, A.; Ding, W.; Fillit, H. Binding of heparan sulfate glycosaminoglycan to beta-amyloid peptide: inhibition by potentially therapeutic polysulfated compounds. Neuroreport 1994, 5, 1389–1392. [Google Scholar] [PubMed]
- Giulian, D.; Haverkamp, L.J.; Yu, J.; Karshin, W.; Tom, D.; Li, J.; Kazanskaia, A.; Kirkpatrick, J.; Roher, A.E. The HHQK domain of beta-amyloid provides a structural basis for the immunopathology of Alzheimer’s disease. J. Biol. Chem. 1998, 273, 29719–29726. [Google Scholar] [CrossRef]
- Lindahl, B.; Westling, C.; Gimenez-Gallego, G.; Lindahl, U.; Salmivirta, M. Common binding sites for beta-amyloid fibrils and fibroblast growth factor-2 in heparan sulfate from human cerebral cortex. J. Biol. Chem. 1999, 274, 30631–30635. [Google Scholar] [CrossRef]
- Yang, L.; Liu, C.C.; Zheng, H.; Kanekiyo, T.; Atagi, Y.; Jia, L.; Wang, D.; N’Songo, A.; Can, D.; Xu, H.; et al. LRP1 modulates the microglial immune response via regulation of JNK and NF-kappaB signaling pathways. J. Neuroinflamm. 2016, 13, 304. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, E1965. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Frederickson, R.C.; Brunden, K.R. Neuroglial-mediated immunoinflammatory responses in Alzheimer’s disease: Complement activation and therapeutic approaches. Neurobiol. Aging 1996, 17, 781–787. [Google Scholar] [CrossRef]
- Bergamaschini, L.; Rossi, E.; Vergani, C.; De Simoni, M.G. Alzheimer’s disease: Another target for heparin therapy. Sci. World J. 2009, 9, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschini, L.; Rossi, E.; Storini, C.; Pizzimenti, S.; Distaso, M.; Perego, C.; De Luigi, A.; Vergani, C.; De Simoni, M.G. Peripheral treatment with enoxaparin, a low molecular weight heparin, reduces plaques and beta-amyloid accumulation in a mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 4181–4186. [Google Scholar] [CrossRef] [PubMed]
- Scholefield, Z.; Yates, E.A.; Wayne, G.; Amour, A.; McDowell, W.; Turnbull, J.E. Heparan sulfate regulates amyloid precursor protein processing by BACE1, the Alzheimer’s beta-secretase. J. Cell Biol. 2003, 163, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Bejoy, J.; Song, L.; Wang, Z.; Sang, Q.X.; Zhou, Y.; Li, Y. Neuroprotective Activities of Heparin, Heparinase III, and Hyaluronic Acid on the Aβ42-treated Forebrain Spheroids Derived from Human Stem Cells. ACS Biomater. Sci. Eng. 2018, 4, 2922–2933. [Google Scholar] [CrossRef]
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Neural Types | AD Phenotypes | Ref. |
|---|---|---|---|
| Chemicals Induced AD-Phenotypes Using hPSCs | |||
| Human ESC-derived neurons treated with Aβ42 oligomers | 3D neurospheres and 2D, basal forebrain cholinergic neurons expressing ChAT and β-tubulin III | Aβ oligomers suppressed the number of functional neurons | Wicklund et al., 2010 [31] |
| HiPSC-derived neurons treated with β-secretase (BSI) and γ-secretase inhibitor (GSI) and NSAID | 2D, forebrain neurons expressing FOXG1 and TBR1 (62%), CTIP2 (12%), Cux1 (83%) SATB (46%) at day 52 | Differentiated neuronal cells expressed Aβ40 and Aβ42. BSI, GSI, and NSAID partially or fully blocked Aβ production in the hiPSCs-derived neuronal cells | Yahata et al., 2011 [32] |
| Human ESC and hiPSC-derived neurons treated with Aβ42 oligomers | 2D, cortical glutamatergic neurons | Aβ oligomers yielded cell culture age-dependent binding of Aβ and cell death in the glutamatergic populations | Vazin et al., 2014 [25] |
| HiPSC-derived neurons treated with Aβ1-42 oligomers | 3D neurospheres, cortical glutamatergic neurons, and motor neurons | Aβ oligomers caused less cell viability, more caspase expression and higher ROS levels on cortical excitatory neurons population. GSK-3β inhibitor may attenuate Aβ-induced cytotoxicity | Yan et al., 2016 [26] |
| HiPSC-derived neurons induced by Aβ42 inducer (Aftin5) | 3D cortical organoids, neurons expressing NeuN, NCAM, MAP2, and CTIP2 | Increased secretion of Aβ42 and the Aβ42/40 ratio | Pavoni et al., 2018 [24] |
| Overexpression of AD-related gene in hPSCs using genetic modification | |||
| PSEN1 L166P mutant hPSC-derived neurons treated with γ-secretase inhibitor (DAPT) and NSAID | 2D, hPSC-derived neural stem cells (NSCs) expressing Nestin and β-tubulin III | DAPT reduced secretion of both Aβ42 and Aβ40. NSAID reduced secretion of Aβ42. PSEN1 1L166P mutation resulted in elevated Aβ42/40 ratio. | Koch et al., 2012 [27] |
| PSEN1 (∆E9) mutant hiPSCs | 2D, hPSC-derived neural progenitor cells (NPCs) expressing Nestin and tau | The PS1 ∆E9 mutation increases the Aβ42/Aβ40 ratio in human neurons by decreasing Aβ40 | Woodruff et al., 2013 [28] |
| Human ESC-derived neurons model tau pathology | 2D, neurons expressing Nestin, DACH1, SOX2, β-tubulin III, and tau | P-tau impaired the transport of mitochondria and led to axonal degeneration and cell death | Mertens et al., 2013 [33] |
| HPSC-derived neurons co-cultured with ApoE secreted glia | 2D, human neurons generated by forced expression of neurogenin-2 (Ngn2), expressing MAP2 and NeuN | ApoE secreted by glia stimulates neuronal Aβ40 and Aβ42 production with an ApoE4 > ApoE3 > ApoE2 potency rank order | Huang et al., 2017 [29] |
| Human NPCs and hiPSC-derived cells overexpressed APP (K670N/M671L and V717I) mutations | 3D microfluidic platform, tri-culture of neurons, astrocytes, and microglia | Increased Aβ aggregation and p-tau formation, induced microglia recruitment and axonal cleavage. Increased chemokines and cytokines. | Park et al., 2018 [30] |
| AD patient-derived iPSCs | |||
| FAD-hiPSCs with PSEN1/2 mutations | 2D, neurons expressing β-tubulin III (about 80%) and MAP2 | Change in APP processing; increased Aβ42 secretion; responding to γ-secretase inhibitors and modulators. | Yagi et al., 2011 [34] |
| FAD-hiPSCs from a patient with Down’s syndrome (Trisomy 21 defect) | 2D, cortical glutamatergic neurons expressing TBR1, CTIP2, SATB and β-tubulin III | Increased Aβ peptide production, Intracellular and extracellular Aβ42 aggregates. Decreased Aβ40/Aβ42 with γ-secretase inhibitors. Tau hyper-phosphorylation in cell bodies and dendrites. Neuronal cell death. | Shi et al., 2012 [35] |
| FAD-hiPSCs with APP gene duplications and SAD-hiPSCs | 2D, FACS-purified neurons expressing β-tubulin III (>90%) and MAP2 | Neurons from AD patients had higher levels of Aβ40, p-tau, and active glycogen synthase kinase-3β (aGSK-3β). β-secretase inhibitors, not γ-secretase inhibitors, reduced p-tau and aGSK-3b. | Israel et al., 2012 [36] |
| FAD-hiPSCs with APP mutations and SAD-hiPSCs | 2D, cortical neurons expressing β-tubulin III, MAP2, TBR1 and SATB2, and astrocytes expressing GFAP | Intracellular Aβ oligomer formation; reduced extracellular Aβ peptides. | Kondo et al., 2013 [37] |
| FAD-hiPSCs with APP or PSEN1 mutations | 2D, neural stem cells (NSCs) expressing Nestin SOX2, ZO1, β-tubulin III, and MAP2 | Increased the Aβ42/Aβ40 ratio compared to healthy control. With high concentrations of γ-secretase inhibitors (NSAID-based GSMs drugs), Aβ42/Aβ40 ratio was decreased. | Mertens et al., 2013 [38] |
| FAD-hiPSCs with PSEN1 mutations | 2D, NPCs expressing β-tubulin III | Increased the Aβ42/Aβ40 ratio. | Sproul et al., 2014 [39] |
| FAD-hiPSCs with PSEN1 (A246E) mutations | 3D EB-based, neurons expressing Nestin, PAX6, FOXG1, TBR1, STAB2, β-tubulin III, and MAP2 | Increased the Aβ42/Aβ40 ratio, increased expression of FOXG1, mGluR1, and SYT1. | Mahairaki et al., 2014 [40] |
| FAD-hiPSCs with PSEN1 and AG mutations and SAD-hiPSCs with APOE3/E4 mutations | Basal forebrain cholinergic neurons expressing MAP2, ChAT, and VaChT | Elevated Aβ42. With γ-secretase inhibitors, Aβ40 was increased and calcium transient was increased. | Duan et al., 2014 [41] |
| FAD-hiPSCs with APP mutations | 3D EB-based, forebrain neurons expressing MAP2, tau, β-tubulin III, Cux1, TBR1, vGlut1 | Increased Aβ42: Aβ40; Decreased APPsα: APPsβ, γ-secretase inhibitor blocked APPs, β cleavage. Increased total tau and p-tau (Ser262) d100. Aβ antibodies blocked, increased total tau. | Muratore et al., 2014 [42] |
| FAD-hiPSCs with PSEN1 (A246E, H163R or M146L) mutations | 2D, neurons expressing Nestin, PAX6 and SOX1 | Increased the Aβ42/Aβ40 ratio compared to healthy control. Reduced Aβ42 and Aβ38 by γ-secretase inhibitor-GSM4. | Liu et al., 2014 [43] |
| FAD-hiPSCs with PSEN1 mutations | 3D EB based, neurons expressing β-tubulin III | Increased the Aβ42 secretion level. Elevated acid sphingomyelinase (ASM) levels. ASM levels restored by ASM siRNA treatment. | Lee et al., 2014 [44] |
| SAD-hiPSCs with SOR1 variants | 2D, FACS-purified neurons expressing Nestin and MAP2 | Altered induction of SORL1 expression; altered Aβ peptide production. | Young et al., 2015 [45] |
| FAD-hiPSCs with PSEN1 or APP mutations | 2D, cortical excitatory neurons expressing tau | Increased the Aβ42 secretion level. Increased intracellular tau and phosphorylated tau levels. | Moore et al., 2015 [46] |
| SAD-hiPSCs with APP mutations | 2D, neurons expressing Nestin, PAX6 and β-tubulin III | Increased phosphor-tau (p-tau) and active glycogen synthase kinase-3β (aGSK-3β).Reduced p-tau by γ-secretase inhibitor. | Hossini et al., 2015 [47] |
| FAD-hiPSCs with PSEN1 (A246E) mutations and SAD-hiPSC mutations | 2D, neurons expressing Nestin, SOX2, MAP2, and β-tubulin III | Increased Aβ42 for FAD-hiPSCs-derived neurons. | Armijo et al., 2016 [48] |
| FAD-hiPSCs with PSEN1 (P117R)/APOE3/3 mutations and SAD-hiPSCs with APOE3/E4 mutations | 3D neurospheres, neural cells expressing GFAP, and MAP2 | Reduced neurites length and neuronal viability. Elevated levels of nitrite and apoptosis. Hyper-excitable Ca+ signaling phenotype. Protected neurites and cell viability by treatment of apigenin. | Balez et al., 2016 [49] |
| FAD-hiPSCs with APP (V717I) mutations | 3D EB based, forebrain neurons expressing GABA, PVB, and MAP2 | Elevated levels of Aβ and sAPPα. | Liao et al., 2016 [50] |
| SAD-hiPSCs | 3D neuro-spheroid, cortical neurons expressing PAX6, MAP2, NeuN and β-tubulin III | 3D spheroids recapitulated both amyloid β and tau pathology. Reduced Aβ42 and Aβ40 production both in 2D and 3D neurons with BACE1 or γ-secretase inhibitors. | Lee et al., 2016 [51] |
| FAD-human iPSCs with APP or PS1 mutations | 3D brain organoids, neuronal cells expressing SOX2, and MAP2 | 3D organoids recapitulated amyloid β, tau pathology, and endosome abnormalities. Reduced amyloid and tau pathology with β-and γ-secretase inhibitors. | Raja et al., 2016 [52] |
| FAD-hiPSCs with PSEN1 (M146L) mutations and SAD-hiPSCs with APOE4 mutations | 2D differentiation; cortical neurons and astrocytes | Reduced morphological heterogeneity in astrocytes. | Jones et al., 2017 [53] |
| FAD-hiPSCs with APP (V717I) mutations | 3D EB-based differentiation, caudal neurons expressing HOXB4 and rostral neurons expressing TBR1 | Reduced the Aβ42/Aβ40 ratio but increased the Aβ38/Aβ42 ratio for caudal neurons. Higher levels of total and phosphor-tau for rostral neuronal fate. | Muratore et al., 2017 [54] |
| FAD-hiPSCs with PSEN1 (M146L, G384A, H163R or A246E), APP (V717I) mutations and SAD-hiPSCs with APOE4 mutations | 2D, human cortical neurons (iN cells) generated by force expression of neurogenin-2 (Ngn2), iN cells expressing SATB2, MAP2, vGlut1, and TBR2 | iPSC-based screening of pharmaceutical compounds for Aβ phenotypes; anti-Aβ cocktail decreased toxic Aβ levels in neurons derived from patients’ cells. A combination of existing drugs synergistically improved Aβ phenotypes of AD. | Kondo et al., 2017 [55] |
| FAD-hiPSCs with PSEN1 mutations and SAD-hiPSCs with unknown mutations | 2D, cholinergic neurons (VAChT), dopaminergic neurons (TH), GABAergic neurons (GAD2/GAD1), and glutamatergic neurons (vGlut1/2) | Increased levels of extracellular Aβ40 and Aβ42 for FAD and SAD samples. Increased the Aβ42/Aβ40 ratio for FAD sample. Increased levels of p-tau and GSK3β. | Ochalek et al., 2017 [56] |
| FAD-hiPSCs with PSEN1 (∆E9) mutations | 3D EB-based differentiation, astrocytes expressing GFAP and S100β | AD astrocytes increased Aβ42 production, altered cytokine release, dysregulated Ca2+ homeostasis, increased oxidative stress and reduced lactate secretion. | Oksanen et al., 2017 [57] |
| FAD-hiPSC with PSEN1 and APP duplication or hiPSCs from Down’s syndrome (Trisomy 21) | 2D, cortical neurons expressing TBR1, and MAP2 | Synaptic dysfunction (long-term potentiation) caused by PSEN1 and APP duplication secretomes was mediated by Aβ peptides, whereas trisomy 21 neuronal secretomes induced dysfunction through extracellular tau. | Hu et al., 2018 [58] |
| FAD-hiPSCs with PSEN1 (M146V) mutation | 3D cortical organoids, neurons expressing TBR1, SATB2, BRN2, and MAP2 | 3D organoids recapitulated Aβ, tau pathology, and neuronal cell death. Reduced amyloid β with DAPT, heparin and heparinase. | Yan et al., 2018 [59] |
| FAD-hiPSC with PSEN1 (A246E) or hiPSCs from Down’s syndrome (Trisomy 21) | 3D cortical organoids, neurons expressing NeuN, SATB2, TBR1, and MAP2 | Accumulation of Aβ and tau aggregates and induced cellular apoptosis AD organoids. | Gonzalez et al., 2018 [60] |
| SAD-hiPSCs from APOE4/E3 mutations | 3D organoids, neurons, astrocytes, and microglia-like cells | APOE4 organoids displayed increased Aβ aggregation and hyperphosphorylation of tau. | Lin et al., 2018 [61] |
| SAD-hiPSCs from unknown mutations | 3D neuro-spheroid, neurons | AD organoids neuronal dysfunction was similar to AD brain tissue by mass spectrometry-based proteomics analysis. | Chen et al., 2018 [62] |
| SAD-hiPSCs from APOE4/E3 mutations | 2D, neuronal cells expressing MAP2 | Showed aberrant mitochondrial function. Increased levels of ROS and DNA damage. Increased levels of oxidative phosphorylation chain complexes. | Birnbaum et al., 2018 [63] |
| FAD-hiPSCs and SAD-hiPSCs | 2D, FACS-purified neurons | Reduced tau phosphorylation by retromer stabilization. | Young et al., 2018 [64] |
| HiPSCs from a Down’s syndrome patient by controlling APP gene copy number | 2D, cortical neurons | Higher APP gene dosage increased Aβ production, altered the Aβ42/Aβ40 ratio and caused deposition of the pyroglutamate (E3)-containing amyloid aggregates. | Ovchinnikov et al., 2018 [65] |
| SAD-hiPSCs from APOE4/4 or APOE3/3 mutations | 2D, cortical neurons and GABAergic neurons | APOE4 increased Aβ production in human neurons, APOE4-expressing neurons had higher levels of tau phosphorylation. | Wang et al., 2018 [66] |
| FAD-hiPSCs with APP duplication mutants | 2D, FACS-purified neurons | Neuronal cholesteryl esters (CE) regulated the proteasome-dependent degradation of p-tau, CE-mediated Aβ secretion by a cholesterol-binding down in APP, A CYP46A1-CE-tau axis was identified as an early pathway. | van der Kant et al., 2019 [67] |
| Cell Source | Range of Modulus and Substrates | Effect on Morphology, Proliferation, and Differentiation | Reference |
|---|---|---|---|
| Neural progenitor cells | 0.1 kPa–10 kPa; PA gels based vmIPNs | Soft gel (100–500 Pa) favored neurons, harder gel (1–10 kPa) promoted glial cells. | Saha et al., 2009 [111] |
| Neural progenitor cells | 1–20 kPa; MAC substrates | <1 kPa favored neuronal differentiation; <3.5 kPa supported astrocyte, >7kPa favored oligodendrocyte. | Leipzig et al., 2009 [112] |
| Mouse ESCs | 41–2700 kPa; collagen coated PDMS surface | Increasing substrate stiffness from 41–2700 kPa promoted cell spreading, proliferation, mesendodermal and osteogenic differentiation. | Evans et al., 2009 [122] |
| Rat neural stem cells | 180–20,000 Pa; 3D alginate hydrogel scaffolds | The rate of proliferation of neural stem cells decreased with an increase in the modulus of the hydrogels. Lower stiffness enhanced neural differentiation. | Banerjee et al., 2009 [123] |
| Mouse ESCs | 0.6 kPa; PA gel substrates | Soft substrate supported self-renewal | Chowdhury et al., 2010 [124] |
| Human ESCs and iPSCs | 0.7–10 kPa; GAG-binding hydrogel | The stiff (10 kPa) hydrogel maintained cell proliferation and pluripotency. | Musah et al., 2012 [125] |
| Human ESCs | 0.05–7 MPa, 3D PLLA, PLGA, PCL or PEGDA scaffold coated with matrigel | 50 to 100 kPa supported ectoderm differentiation; 100 to 1000 kPa supported endoderm differentiation; 1.5 to 6 MPa supported mesoderm differentiation. | Zoldan et al., 2011 [126] |
| Human ESCs and iPSCs | 0.1–75 kPa; matrigel-coated PA gels | Soft matrix (0.1 kPa) promoted early neural differentiation. | Keung et al., 2012 [119] |
| Human ESCs | 1 kPa, 10 kPa, 3 GPa; PDMS substrates | Rigid matrix promoted cardiac differentiation. | Arshi et al., 2013 [127] |
| Mouse ESCs | 0–1.5 kPa, 3D collagen-I, Matrigel, or HA hydrogel | <0.3 kPa less neurite outgrowth and supported glial cell; 0.5 to 1 kPa more neurite outgrowth and supported neurons. | Kothapalli et al., 2013 [113] |
| Human ESCs | 0.078–1.167 MPa; PDMS substrates | Increased stiffness upregulated mesodermal differentiation. | Eroshenko et al., 2013 [128] |
| Human ESCs | 1.3 kPa, 2.1 kPa, 3.5 kPa; HA hydrogel | Stiffness of 1.2 kPa was the best to support pancreatic β-cell differentiation. | Narayanan et al., 2014 [129] |
| Human ESCs | 4–80 kPa; PA hydrogels | Stiffness of 50 kPa was the best for cardiomyocyte differentiation. Stiffness impacted the initial differentiation of hESCs to mesendoderm, while it did not impact differentiation of cardiac progenitor cells to cardiomyocytes. | Hazeltine et al., 2014 [130] |
| Human iPSCs | 19–193 kPa; 3D PCL, PET, PEKK or PCU electrospun fibers | The substrate stiffness was inversely related to the sphericity of hiPSC colonies. | Maldonado et al., 2015 [131] |
| HPSCs | 6 kPa, 10 kPa, 35 kPa; Matrigel micropatterns | High stiffness (35 kPa) induced myofibril defects of hPSC-derived cardiomyocytes and decreased mechanical output. | Ribeiro et al., 2015 [132] |
| hPSC-derived hepatocytes (hPSC-Heps) | 20, 45, 140 kPa; collagen-coated PA hydrogels substrates | On softer substrates, the hPSC-Heps formed compact colonies while on stiffer substrates they formed a diffuse monolayer. Albumin production correlated inversely with stiffness. | Mittal et al., 2016 [133] |
| Rat cortical neurons (RCN) | 5 kPa (soft), PA gels; 500 kPa (stiff), PDMS substrates; | Soft substrates enhanced cortical neurons migration. Stiff substrates increased synaptic activity. | Lantoine et al., 2016 [114] |
| Mouse ESCs and iPSCs | 300–1200 Pa; 3D PEG hydrogels | Stiffness and other biophysical effectors promoted somatic-cell reprogramming and iPSC generation; lower modulus (300–600 Pa) showed higher reprogramming efficiency. | Caiazzo et al., 2016 [134] |
| Human ESCs | 400 Pa, 60 kPa; PA hydrogels | On stiff substrates, β-catenin degradation inhibits mesodermal differentiation of human ESCs. | Przybyla et al., 2016 [135] |
| Human ESCs | 1–100 kPa; barium alginate capsules | Stiffness of 4–7 kPa supported cell proliferation and higher stiffness suppressed cell growth. Increased stiffness promoted endoderm differentiation, while suppressed pancreatic induction. About 3.9 kPa was the best for pancreatic differentiation. | Richardson et al., 2016 [136] |
| Mouse intestinal stem cells (ISC) | 300 Pa, 700 Pa, 1.3 kPa, 1.7 kPa; PEG hydrogels | Higher stiffness enhanced ISC expansion. Lower stiffness supported ISC differentiation and organoid formation. | Gjorevski et al., 2016 [137] |
| Mouse neural progenitor cells (NPC) | 0.5–50 kPa; 3D elastin-like protein hydrogels | In stiffness from 0.5 to 50 kPa, NPC stemness maintenance did not correlate with initial hydrogel stiffness. | Madl et al., 2017 [115] |
| Mouse ESCs and hiPSCs | 10–100 kPa; 3D PU scaffolds | Scaffolds with proper stiffness, Poisson’s ratio and pore structure enhanced neural differentiation of PSCs. | Yan et al., 2017 [108] |
| Human iPSCs | 3–168 kPa; PDMS substrates | Elasticity of substrates significantly affected cell colony formation. Intermediate substrate elasticity of about 9 kPa is preferable to reach an EB-like aggregation and optimal for cardiac differentiation. | Wang et al., 2018 [138] |
| Mouse ESCs | 3.4 kPa, 64 kPa, 144 kPa; PEGDA or PEG hydrogel substrates | Soft hydrogel (3.4 kPa) showed strong cell attachment and a growth pattern similar to 2D surface. Stiff hydrogel (144 kPa) supported a 3D aggregation. | Dorsey et al., 2018 [139] |
| Mouse iPSCs | 0–2.4 MPa; PDMS substrates | Stiffer substrate supported pluripotency of iPSCs. Softer substrate promoted cardiac differentiation. | Fu et al., 2018 [140] |
| Neural crest stem cells (NCSCs) from hiPSCs | 1kPa, 15 kPa, 1 GPa; PA gel substrates | >50 kPa promoted smooth muscle cells from NCSCs, <15 kPa promoted glial cells from NCSCs. | Zhu et al., 2018 [116] |
| Mouse hippocampal neurons | 2.13 kPa, 22.1 kPa; PDMS substrates | Stiff substrate enhanced voltage-gated Ca2+ channel currents in neurons. | Wen et al., 2018 [117] |
| Neural crest stem cells (NCSCs) derived from hESCs | 3.3 kPa, 1.7 MPa, 1 GPa; PDMS substrates | Soft substrate increased differentiation of ectodermal mesenchymal stem cells (MSCs) from NCSCs via CD44 mediated PDGFR signaling. | Srinivasan et al., 2018 [118] |
| iPSCs and neonatal rat cardiomyocytes | 9, 20, 180 kPa; PA gel substrates | Cardiac differentiation preferred rigid substrates, and beating behavior preferred soft substrate. | Hirata et al., 2018 [141] |
| Human iPSCs | About 24 Pa, fibrin-based gel substrates (human platelet lysate gel); >1 GPa, tissue culture plastics | Soft substrates did not impact on differentiation of iPSCs into MSCs. | Goetzke et al., 2018 [142] |
| Human ESCs | 118 ± 51 Pa, 800 ± 180 Pa, 5600 ± 1100 Pa, and 8900 ± 1500 Pa; decellularized fibroblast-derived matrices crosslinked by genipin | Soft matrix supported cell migration and induced EMT of hPSC. Stiff matrix supported cell pluripotency and suppressed EMT of hPSCs. | Kim et al., 2018 [143] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Y.; Bejoy, J.; Marzano, M.; Li, Y. The Use of Pluripotent Stem Cell-Derived Organoids to Study Extracellular Matrix Development during Neural Degeneration. Cells 2019, 8, 242. https://doi.org/10.3390/cells8030242
Yan Y, Bejoy J, Marzano M, Li Y. The Use of Pluripotent Stem Cell-Derived Organoids to Study Extracellular Matrix Development during Neural Degeneration. Cells. 2019; 8(3):242. https://doi.org/10.3390/cells8030242
Chicago/Turabian StyleYan, Yuanwei, Julie Bejoy, Mark Marzano, and Yan Li. 2019. "The Use of Pluripotent Stem Cell-Derived Organoids to Study Extracellular Matrix Development during Neural Degeneration" Cells 8, no. 3: 242. https://doi.org/10.3390/cells8030242
APA StyleYan, Y., Bejoy, J., Marzano, M., & Li, Y. (2019). The Use of Pluripotent Stem Cell-Derived Organoids to Study Extracellular Matrix Development during Neural Degeneration. Cells, 8(3), 242. https://doi.org/10.3390/cells8030242