Lamina Associated Domains and Gene Regulation in Development and Cancer


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Transcriptional Repression at the NL
3. LAD Organization and Chromatin Features
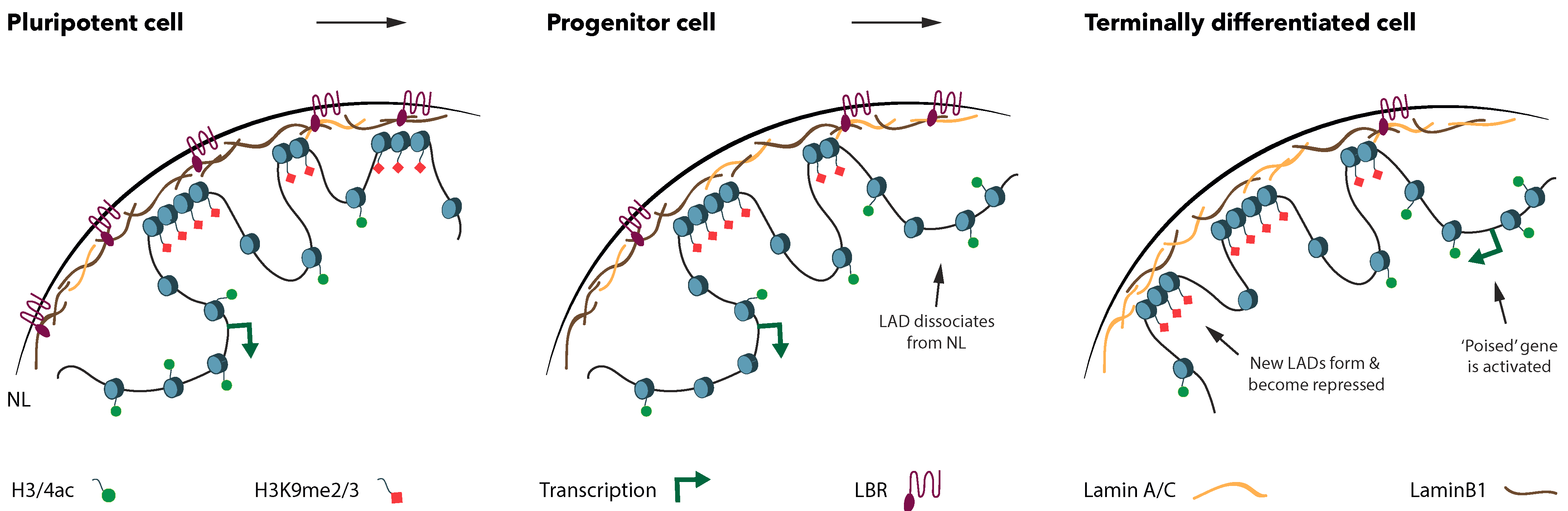
4. Spatial Dynamics of Lineage-Specific Genes in Differentiation
5. The Changes in NL Organization in Development
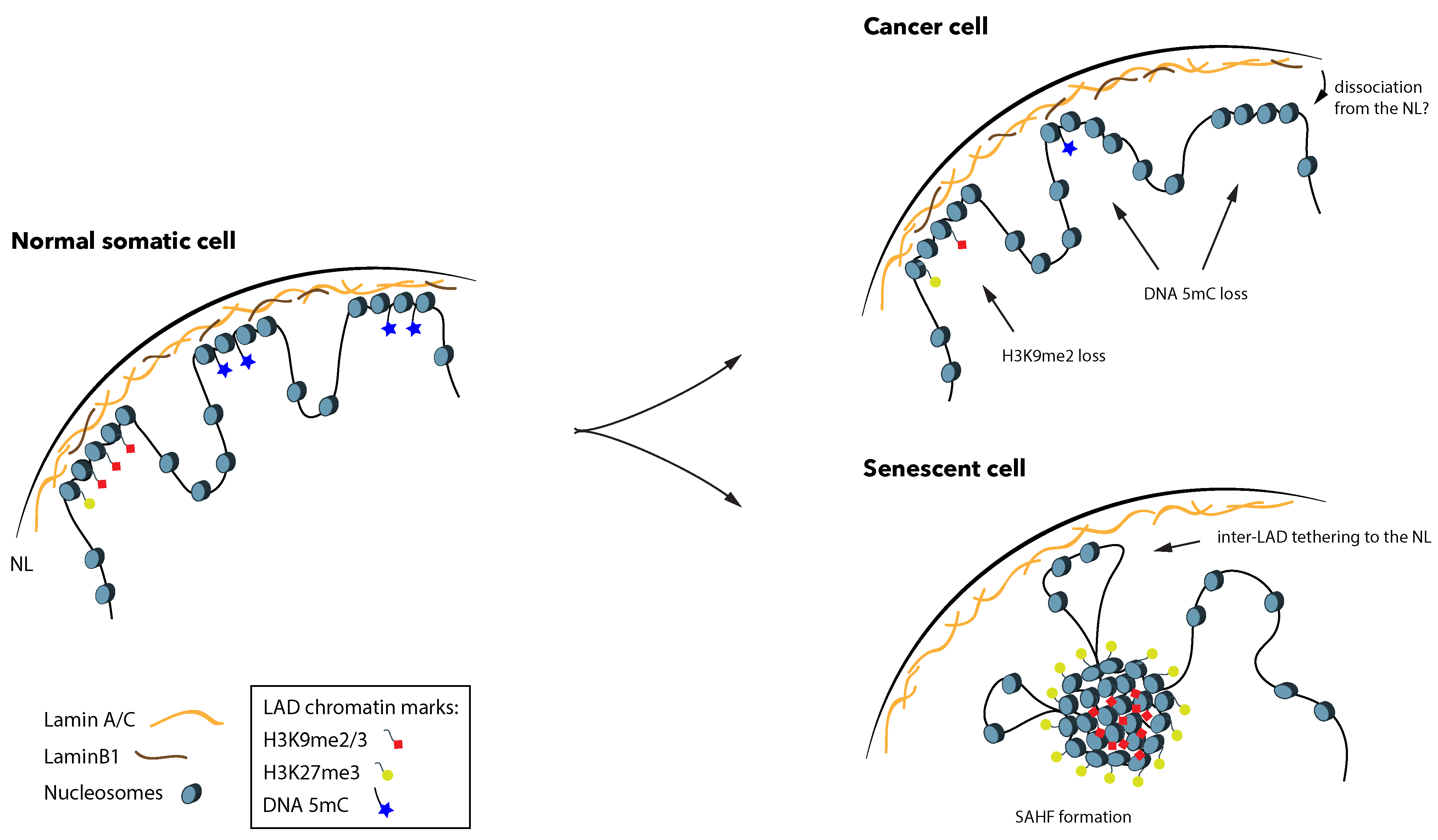
6. LADs Are Potential Major Players in Cancer
7. LADs Are Linked to Loss of DNA Methylation in Cancer
8. LADs Are Extensively Redistributed during Cellular Senescence
9. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Foisner, R. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Ann. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.; Stewart, C.L. The nuclear lamins: Flexibility in function. Nat. Rev. Mol. Cell Biol. 2013, 14, 13–24. [Google Scholar] [CrossRef]
- De Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear lamins: Thin filaments with major functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Rober, R.-A.; Weber, K.; Osborn, M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: A developmental study. Development 1989, 105, 365–378. [Google Scholar] [PubMed]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Pickersgill, H.; Kalverda, B.; de Wit, E.; Talhout, W.; Fornerod, M.; van Steensel, B. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat. Genet. 2006, 38, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; Belmont, A.S. Lamina-associated domains: Links with chromosome architecture, heterochromatin, and gene repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef]
- Vogel, M.J.; Peric-Hupkes, D.; Van Steensel, B. Detection of in vivo protein–DNA interactions using DamID in mammalian cells. Nat. Protoc. 2007, 2, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Kind, J.; Pagie, L.; de Vries, S.S.; Nahidiazar, L.; Dey, S.S.; Bienko, M.; Zhan, Y.; Lajoie, B.; de Graaf, C.A.; Amendola, M. Genome-wide maps of nuclear lamina interactions in single human cells. Cell 2015, 163, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.; Solovei, I.; Brugman, W.; Gräf, S.; Flicek, P.; Kerkhoven, R.M.; van Lohuizen, M. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 2010, 38, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, K.; Egelhofer, T.A.; Strome, S.; Lieb, J.D. Caenorhabditis elegans chromosome arms are anchored to the nuclear membrane via discontinuous association with LEM-2. Genome Biol. 2010, 11, R120. [Google Scholar] [CrossRef] [PubMed]
- Van Bemmel, J.G.; Pagie, L.; Braunschweig, U.; Brugman, W.; Meuleman, W.; Kerkhoven, R.M.; van Steensel, B. The Insulator Protein SU(HW) Fine-Tunes Nuclear Lamina Interactions of the Drosophila Genome. PLoS ONE 2010, 5, e15013. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yao, J. Identifying Novel Transcriptional and Epigenetic Features of Nuclear Lamina-associated Genes. Sci. Rep. 2017, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Kind, J.; Pagie, L.; Ortabozkoyun, H.; Boyle, S.; de Vries, S.S.; Janssen, H.; Amendola, M.; Nolen, L.D.; Bickmore, W.A.; van Steensel, B. Single-cell dynamics of genome-nuclear lamina interactions. Cell 2013, 153, 178–192. [Google Scholar] [CrossRef]
- Pindyurin, A.V.; Ilyin, A.A.; Ivankin, A.V.; Tselebrovsky, M.V.; Nenasheva, V.V.; Mikhaleva, E.A.; Pagie, L.; van Steensel, B.; Shevelyov, Y.Y. The large fraction of heterochromatin in Drosophila neurons is bound by both B-type lamin and HP1a. Epigenet. Chromatin 2018, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Németh, A.; Conesa, A.; Santoyo-Lopez, J.; Medina, I.; Montaner, D.; Péterfia, B.; Solovei, I.; Cremer, T.; Dopazo, J.; Längst, G. Initial genomics of the human nucleolus. PLoS Genet. 2010, 6, e1000889. [Google Scholar] [CrossRef]
- Németh, A.; Längst, G. Genome organization in and around the nucleolus. Trends Genet. 2011, 27, 149–156. [Google Scholar] [CrossRef]
- Meister, P.; Towbin, B.D.; Pike, B.L.; Ponti, A.; Gasser, S.M. The spatial dynamics of tissue-specific promoters during C. elegans development. Genes Dev. 2010, 24, 766–782. [Google Scholar] [CrossRef]
- Reddy, K.; Zullo, J.; Bertolino, E.; Singh, H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 2008, 452, 243–247. [Google Scholar] [CrossRef]
- Finlan, L.E.; Sproul, D.; Thomson, I.; Boyle, S.; Kerr, E.; Perry, P.; Ylstra, B.; Chubb, J.R.; Bickmore, W.A. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 2008, 4, e1000039. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, R.I.; Spector, D.L. A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J. Cell Biol. 2008, 180, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Bian, Q.; Khanna, N.; Alvikas, J.; Belmont, A.S. β-Globin cis-elements determine differential nuclear targeting through epigenetic modifications. J. Cell Biol. 2013, 203, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Zullo, J.M.; Demarco, I.A.; Piqué-Regi, R.; Gaffney, D.J.; Epstein, C.B.; Spooner, C.J.; Luperchio, T.R.; Bernstein, B.E.; Pritchard, J.K.; Reddy, K.L. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 2012, 149, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.; van der Zwalm, M.; Brueckner, L.; Comoglio, F.; van Schaik, T.; Pagie, L.; van Arensbergen, J.; van Steensel, B. Promoter-intrinsic and local chromatin features determine gene repression in lamina-associated domains. BioRxiv 2018, 464081. [Google Scholar]
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase separation drives heterochromatin domain formation. Nature 2017, 547, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 2017, 547, 236–240. [Google Scholar] [CrossRef]
- Therizols, P.; Illingworth, R.S.; Courilleau, C.; Boyle, S.; Wood, A.J.; Bickmore, W.A. Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science 2014, 346, 1238–1242. [Google Scholar] [CrossRef]
- Demmerle, J.; Koch, A.J.; Holaska, J.M. The Nuclear Envelope Protein Emerin Binds Directly to Histone Deacetylase 3 (HDAC3) and Activates HDAC3 Activity. J. Biol. Chem. 2012, 287, 22080–22088. [Google Scholar] [CrossRef]
- Demmerle, J.; Koch, A.J.; Holaska, J.M. Emerin and histone deacetylase 3 (HDAC3) cooperatively regulate expression and nuclear positions of MyoD, Myf5, and Pax7 genes during myogenesis. Chromosome Res. 2013, 21, 765–779. [Google Scholar] [CrossRef]
- Milon, B.C.; Cheng, H.; Tselebrovsky, M.V.; Lavrov, S.A.; Nenasheva, V.V.; Mikhaleva, E.A.; Shevelyov, Y.Y.; Nurminsky, D.I. Role of Histone Deacetylases in Gene Regulation at Nuclear Lamina. PLoS ONE 2012, 7, e49692. [Google Scholar] [CrossRef] [PubMed]
- Yokochi, T.; Poduch, K.; Ryba, T.; Lu, J.; Hiratani, I.; Tachibana, M.; Shinkai, Y.; Gilbert, D.M. G9a selectively represses a class of late-replicating genes at the nuclear periphery. Proc. Natl. Acad. Sci. USA 2009, 106, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sandoval, A.; Towbin, B.D.; Kalck, V.; Cabianca, D.S.; Gaidatzis, D.; Hauer, M.H.; Geng, L.; Wang, L.; Yang, T.; Wang, X.; et al. Perinuclear Anchoring of H3K9-Methylated Chromatin Stabilizes Induced Cell Fate in C. elegans Embryos. Cell 2015, 163, 1333–1347. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Hizume, K.; Kimura, H.; Takeyasu, K.; Haraguchi, T.; Hiraoka, Y. Lamin B Receptor Recognizes Specific Modifications of Histone H4 in Heterochromatin Formation. J. Biol. Chem. 2012, 287, 42654–42663. [Google Scholar] [CrossRef] [PubMed]
- Polioudaki, H.; Kourmouli, N.; Drosou, V.; Bakou, A.; Theodoropoulos, P.A.; Singh, P.B.; Giannakouros, T.; Georgatos, S.D. Histones H3/H4 form a tight complex with the inner nuclear membrane protein LBR and heterochromatin protein 1. EMBO Rep. 2001, 2, 920–925. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, G.; Welch, D.B.M.; Zwerger, M.; Olins, D.E. Lamin B receptor: Multi-tasking at the nuclear envelope. Nucleus 2010, 1, 53–70. [Google Scholar]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and Lamin A/C Sequentially Tether Peripheral Heterochromatin and Inversely Regulate Differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Poleshko, A.; Mansfield, K.M.; Burlingame, C.C.; Andrake, M.D.; Shah, N.R.; Katz, R.A. The human protein PRR14 tethers heterochromatin to the nuclear lamina during interphase and mitotic exit. Cell Rep. 2013, 5, 292–301. [Google Scholar] [CrossRef]
- Poleshko, A.; Shah, P.P.; Gupta, M.; Babu, A.; Morley, M.P.; Manderfield, L.J.; Ifkovits, J.L.; Calderon, D.; Aghajanian, H.; Sierra-Pagán, J.E.; et al. Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell 2017, 171, 573–587.e14. [Google Scholar] [CrossRef] [PubMed]
- See, K.; Lan, Y.; Rhoades, J.; Jain, R.; Smith, C.L.; Epstein, J.A. Lineage-specific reorganization of nuclear peripheral heterochromatin and H3K9me2 domains. Development 2019, 146, dev174078. [Google Scholar] [CrossRef]
- Robson, M.I.; Jose, I.; Czapiewski, R.; Lê Thành, P.; Booth, D.G.; Kelly, D.A.; Webb, S.; Kerr, A.R.; Schirmer, E.C. Tissue-specific gene repositioning by muscle nuclear membrane proteins enhances repression of critical developmental genes during myogenesis. Mol. Cell 2016, 62, 834–847. [Google Scholar] [CrossRef]
- Ugarte, F.; Sousae, R.; Cinquin, B.; Martin, E.W.; Krietsch, J.; Sanchez, G.; Inman, M.; Tsang, H.; Warr, M.; Passegué, E. Progressive chromatin condensation and H3K9 methylation regulate the differentiation of embryonic and hematopoietic stem cells. Stem Cell Rep. 2015, 5, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Gesson, K.; Rescheneder, P.; Skoruppa, M.P.; von Haeseler, A.; Dechat, T.; Foisner, R. A-type lamins bind both hetero-and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 2016, 26, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Taniura, H.; Glass, C.; Gerace, L. A chromatin binding site in the tail domain of nuclear lamins that interacts with core histones. J. Cell Biol. 1995, 131, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, X.; Zheng, Y. The nuclear lamina regulates germline stem cell niche organization via modulation of EGFR signaling. Cell Stem Cell 2013, 13, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Vergnes, L.; Péterfy, M.; Bergo, M.O.; Young, S.G.; Reue, K. Lamin B1 is required for mouse development and nuclear integrity. Proc. Natl. Acad. Sci. USA 2004, 101, 10428–10433. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef]
- Coffinier, C.; Jung, H.-J.; Nobumori, C.; Chang, S.; Tu, Y.; Barnes, R.H.; Yoshinaga, Y.; de Jong, P.J.; Vergnes, L.; Reue, K. Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol. Biol. Cell 2011, 22, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.-J.; Nobumori, C.; Goulbourne, C.N.; Tu, Y.; Lee, J.M.; Tatar, A.; Wu, D.; Yoshinaga, Y.; de Jong, P.J.; Coffinier, C.; et al. Farnesylation of lamin B1 is important for retention of nuclear chromatin during neuronal migration. Proc. Natl. Acad. Sci. USA 2013, 110, E1923–E1932. [Google Scholar] [CrossRef] [PubMed]
- Amendola, M.; van Steensel, B. Nuclear lamins are not required for lamina-associated domain organization in mouse embryonic stem cells. EMBO Rep. 2015, 16, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Sharov, A.A.; McDole, K.; Cheng, M.; Hao, H.; Fan, C.-M.; Gaiano, N.; Ko, M.S.; Zheng, Y. Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science 2011, 334, 1706–1710. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Zheng, X.; Zheng, Y. Proliferation and differentiation of mouse embryonic stem cells lacking all lamins. Cell Res. 2013, 23, 1420–1423. [Google Scholar] [CrossRef] [PubMed]
- Eckersley-Maslin, M.A.; Bergmann, J.H.; Lazar, Z.; Spector, D.L. Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus 2013, 4, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Hu, J.; Yue, S.; Kristiani, L.; Kim, M.; Sauria, M.; Taylor, J.; Kim, Y.; Zheng, Y. Lamins Organize the Global Three-Dimensional Genome from the Nuclear Periphery. Mol. Cell 2018, 71, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, B.A.; de Laat, W. Getting the genome in shape: The formation of loops, domains and compartments. Genome Biol. 2015, 16, 154. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Luperchio, T.R.; Sauria, M.E.G.; Hoskins, V.E.; Xianrong, W.; DeBoy, E.; Gaillard, M.-C.; Tsang, P.; Pekrun, K.; Ach, R.A.; Yamada, A.; et al. The repressive genome compartment is established early in the cell cycle before forming the lamina associated domains. BioRxiv 2018. [Google Scholar] [CrossRef]
- Clowney, E.J.; LeGros, M.A.; Mosley, C.P.; Clowney, F.G.; Markenskoff-Papadimitriou, E.C.; Myllys, M.; Barnea, G.; Larabell, C.A.; Lomvardas, S. Nuclear Aggregation of Olfactory Receptor Genes Governs Their Monogenic Expression. Cell 2012, 151, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-K.; Blanco, M.; Jackson, C.; Aznauryan, E.; Ollikainen, N.; Surka, C.; Chow, A.; Cerase, A.; McDonel, P.; Guttman, M. Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing. Science 2016, 354, 468–472. [Google Scholar] [CrossRef]
- McHugh, C.A.; Chen, C.-K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.L.; Feinberg, A.P. Higher order chromatin organization in cancer. Semin. Cancer Biol. 2013, 23, 109–115. [Google Scholar] [CrossRef]
- Zink, D.; Fischer, A.H.; Nickerson, J.A. Nuclear structure in cancer cells. Nat. Rev. Cancer 2004, 4, 677–687. [Google Scholar] [CrossRef]
- Wen, B.; Wu, H.; Shinkai, Y.; Irizarry, R.A.; Feinberg, A.P. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 2009, 41, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 2010, 6, 479–491. [Google Scholar] [CrossRef]
- Soufi, A.; Donahue, G.; Zaret, K.S. Facilitators and Impediments of the Pluripotency Reprogramming Factors’ Initial Engagement with the Genome. Cell 2012, 151, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011, 18, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, C.P.E.; van Dijk, C.M.; Tollenaar, R.A.E.M.; et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina–associated domains. Nat. Genet. 2011, 44, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Hon, G.C.; Hawkins, R.D.; Caballero, O.L.; Lo, C.; Lister, R.; Pelizzola, M.; Valsesia, A.; Ye, Z.; Kuan, S.; Edsall, L.E.; et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012, 22, 246–258. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.-H.; LeProust, E.M.; Park, I.-H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Dinh, H.Q.; Ramjan, Z.; Weisenberger, D.J.; Nicolet, C.M.; Shen, H.; Laird, P.W.; Berman, B.P. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018, 50, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Criscione, S.W.; Teo, Y.V.; Neretti, N. The Chromatin Landscape of Cellular Senescence. Trends Genet. 2016, 32, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Parry, A.J.; Narita, M. Old cells, new tricks: Chromatin structure in senescence. Mamm. Genome 2016, 27, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Courtois-Cox, S.; Jones, S.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.F.M.; Schlegelberger, B.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; VanderKraats, N.D.; Shah, P.P.; van Tuyn, J.; Singh Rai, T.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nuñez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Chandra, T.; Kirschner, K.; Thuret, J.-Y.; Pope, B.D.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.A.; Salama, R.; Carroll, T.; et al. Independence of Repressive Histone Marks and Chromatin Compaction during Senescent Heterochromatic Layer Formation. Mol. Cell 2012, 47, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Sadaie, M.; Salama, R.; Carroll, T.; Tomimatsu, K.; Chandra, T.; Young, A.R.J.; Narita, M.; Pérez-Mancera, P.A.; Bennett, D.C.; Chong, H.; et al. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2013, 27, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Ewels, P.A.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.W.; Kirschner, K.; Thuret, J.-Y.; Andrews, S.; Fraser, P.; Reik, W. Global Reorganization of the Nuclear Landscape in Senescent Cells. Cell Rep. 2015, 10, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Lenain, C.; de Graaf, C.A.; Pagie, L.; Visser, N.L.; de Haas, M.; de Vries, S.S.; Peric-Hupkes, D.; van Steensel, B.; Peeper, D.S. Massive reshaping of genome–nuclear lamina interactions during oncogene-induced senescence. Genome Res. 2017, 27, 1634–1644. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lochs, S.J.A.; Kefalopoulou, S.; Kind, J. Lamina Associated Domains and Gene Regulation in Development and Cancer. Cells 2019, 8, 271. https://doi.org/10.3390/cells8030271
Lochs SJA, Kefalopoulou S, Kind J. Lamina Associated Domains and Gene Regulation in Development and Cancer. Cells. 2019; 8(3):271. https://doi.org/10.3390/cells8030271
Chicago/Turabian StyleLochs, Silke J. A., Samy Kefalopoulou, and Jop Kind. 2019. "Lamina Associated Domains and Gene Regulation in Development and Cancer" Cells 8, no. 3: 271. https://doi.org/10.3390/cells8030271
APA StyleLochs, S. J. A., Kefalopoulou, S., & Kind, J. (2019). Lamina Associated Domains and Gene Regulation in Development and Cancer. Cells, 8(3), 271. https://doi.org/10.3390/cells8030271