The Roles of YAP/TAZ and the Hippo Pathway in Healthy and Diseased Skin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
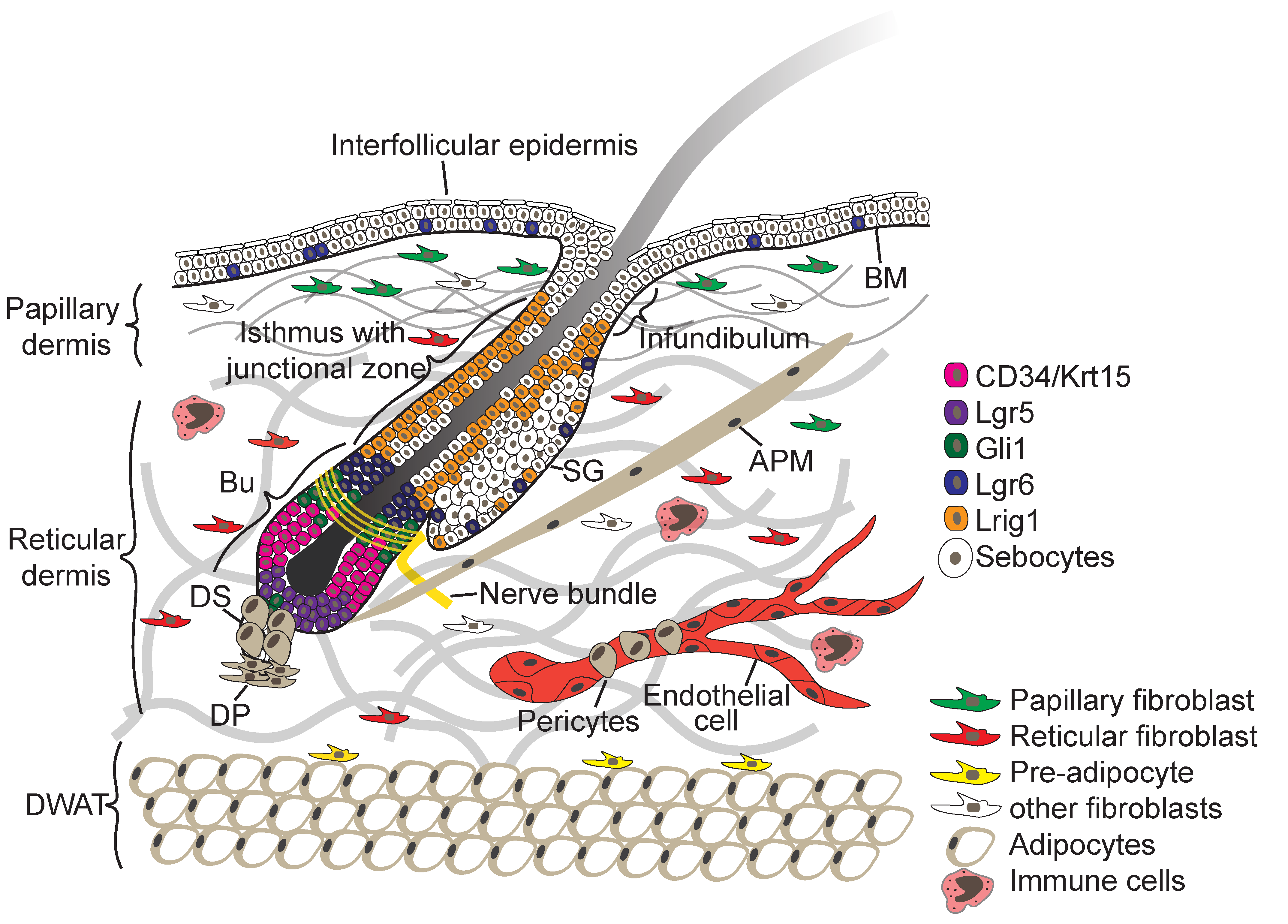
2. Skin Architecture
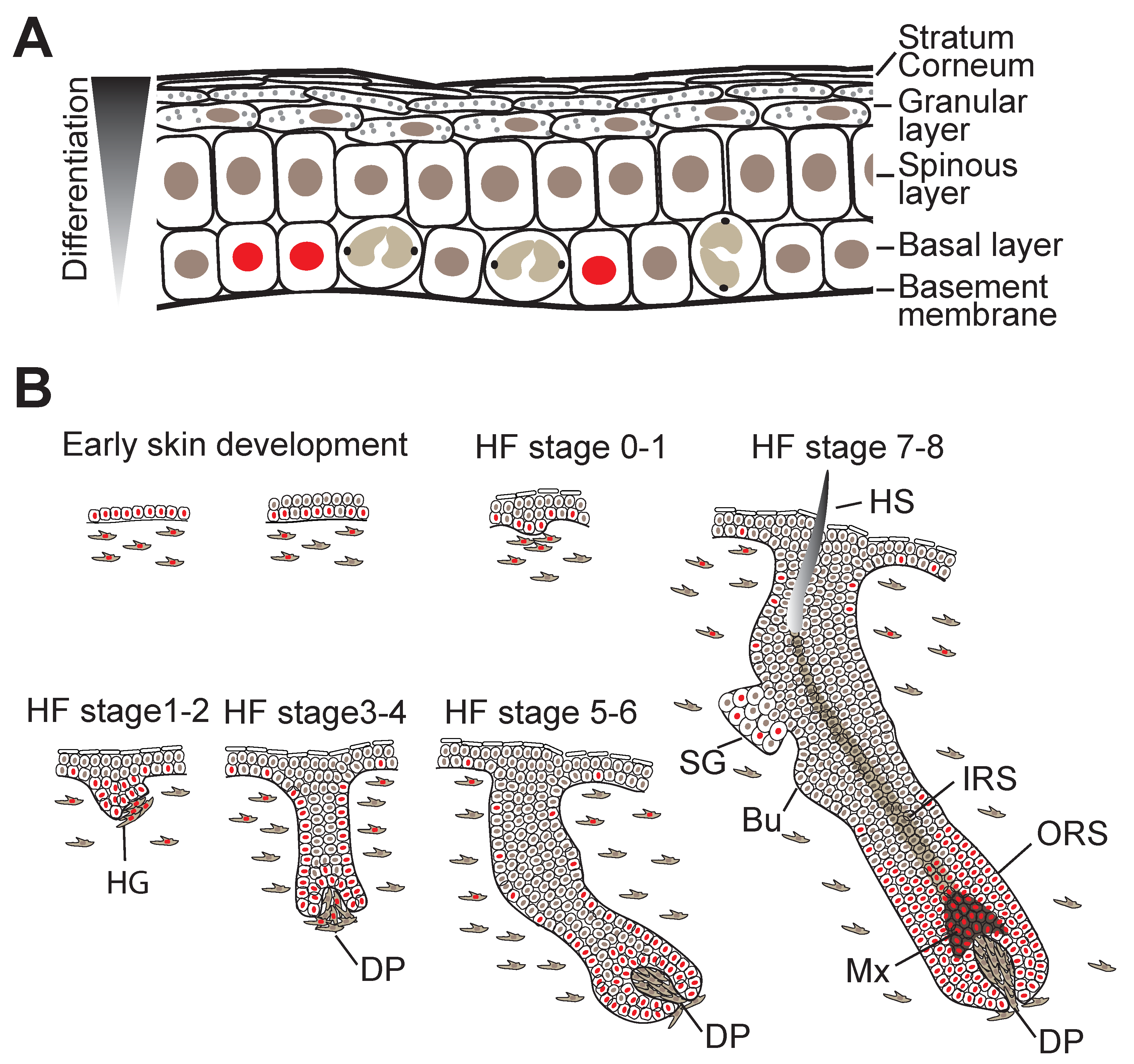
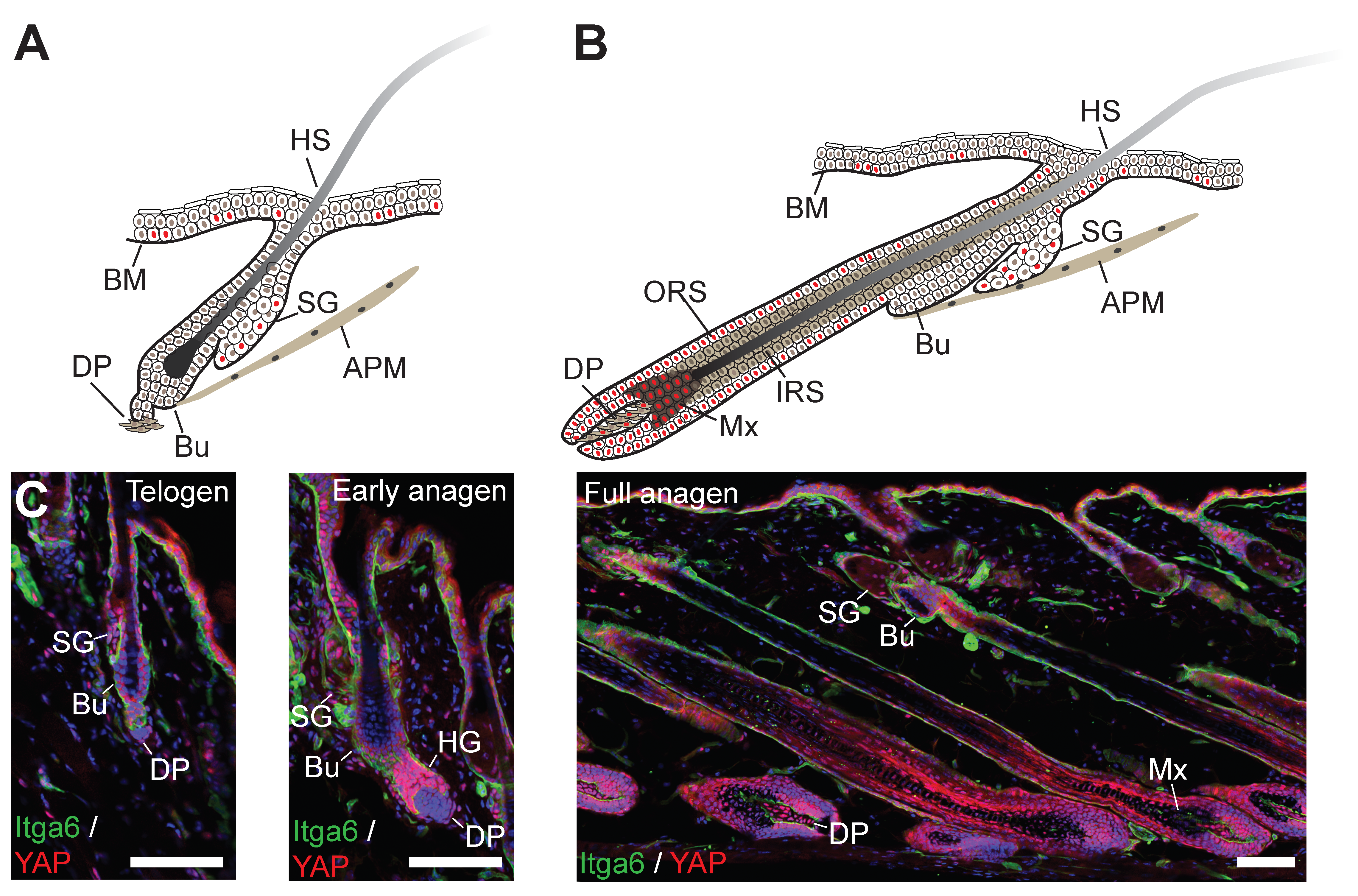
3. Skin Morphogenesis and Hair Follicle Development
4. Skin Homeostasis and Tissue Repair
5. Skin Cancer
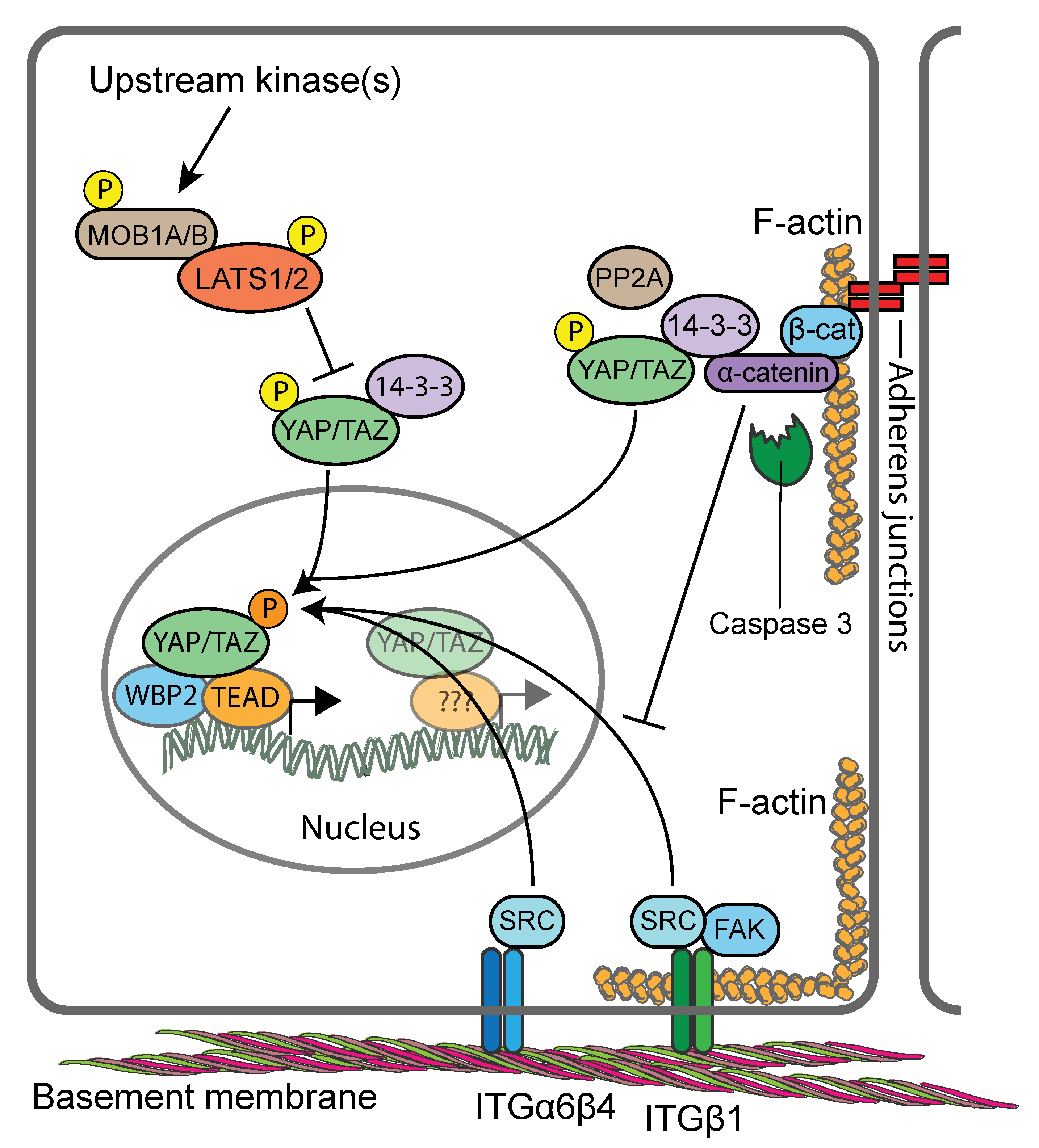
6. The Hippo Signalling Pathway
7. Expression of YAP and TAZ in Skin during Development, Homeostasis, Regeneration and Cancer
8. YAP/TAZ Drive Proliferation of Epidermal SCs and Fibroblasts during Development and Tissue Regeneration
9. YAP/TAZ Activity in Skin is Controlled by Hippo Signalling-Dependent and Independent Signalling Mechanisms
10. YAP and TAZ as Oncogenes in Skin Cancers
11. Targeting YAP and TAZ for Skin Cancer Treatment
12. Summary and Outlook
13. Materials and Methods
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Belokhvostova, D.; Berzanskyte, I.; Cujba, A.-M.; Jowett, G.; Marshall, L.; Prueller, J.; Watt, F.M. Homeostasis, regeneration and tumour formation in the mammalian epidermis. Int. J. Dev. Boil. 2018, 62, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Schepeler, T.; Page, M.E.; Jensen, K.B. Heterogeneity and plasticity of epidermal stem cells. Development 2014, 141, 2559–2567. [Google Scholar] [CrossRef] [Green Version]
- Rognoni, E.; Watt, F.M. Skin Cell Heterogeneity in Development, Wound Healing, and Cancer. Trends Cell Biol. 2018, 28, 709–722. [Google Scholar] [CrossRef]
- Dekoninck, S.; Blanpain, C. Stem cell dynamics, migration and plasticity during wound healing. Nat. Cell Boil. 2019, 21, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, K.A.U.; Fuchs, E. Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 2017, 43, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Fuchs, E. Stretching the limits: From homeostasis to stem cell plasticity in wound healing and cancer. Nat. Rev. Microbiol. 2018, 19, 311–325. [Google Scholar] [CrossRef]
- Rinkevich, Y.; Walmsley, G.G.; Hu, M.S.; Maan, Z.N.; Newman, A.M.; Drukker, M.; Januszyk, M.; Krampitz, G.W.; Gurtner, G.C.; Lorenz, H.P. Skin Fibrosis. Identification and Isolation of a Dermal Lineage with Intrinsic Fibrogenic Potential. Science 2015, 348, aaa2151. [Google Scholar] [CrossRef]
- Philippeos, C.; Telerman, S.; Oules, B.; Pisco, A.; Shaw, T.; Elgueta, R.; Lombardi, G.; Driskell, R.; Soldin, M.; Lynch, M.; et al. 1354 Spatial and single-cell transcriptional profiling identifies functionally distinct human dermal fibroblast subpopulations. J. Invest. Derm. 2018, 138, S230. [Google Scholar] [CrossRef] [Green Version]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferrón, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nat. Cell Boil. 2013, 504, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Tabib, T.; Morse, C.; Wang, T.; Chen, W.; Lafyatis, R. Sfrp2/Dpp4 and Fmo1/Lsp1 Define Major Fibroblast Populations in Human Skin. J. Invest. Derm. 2018, 138, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Rognoni, E.; Gomez, C.; Pisco, A.O.; Rawlins, E.L.; Simons, B.D.; Watt, F.M.; Driskell, R.R. Inhibition of Beta-Catenin Signalling in Dermal Fibroblasts Enhances Hair Follicle Regeneration During Wound Healing. Development 2016, 143, 2522–2535. [Google Scholar] [CrossRef]
- Green, A.C.; Olsen, C.M. Cutaneous Squamous Cell Carcinoma: An Epidemiological Review. Br. J. Derm. 2017, 177, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Burton, K.A.; Ashack, K.A.; Khachemoune, A. Cutaneous Squamous Cell Carcinoma: A Review of High-Risk and Metastatic Disease. Am. J. Clin. Derm. 2016, 17, 491–508. [Google Scholar] [CrossRef]
- Sánchez-Danés, A.; Blanpain, C. Deciphering the cells of origin of squamous cell carcinomas. Nat. Rev. Cancer 2018, 18, 549–561. [Google Scholar] [CrossRef]
- De Gruijl, F.R.; Tensen, C.P. Pathogenesis of Skin Carcinomas and a Stem Cell as Focal Origin. Front. Med. 2018, 5, 165. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Pasolli, H.A.; Fuchs, E. Dynamics Between Stem Cells, Niche and Progeny in the Hair Follicle. Cell 2011, 144, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzschmar, K.; Watt, F.M. Lineage Tracing. Cell 2012, 148, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Kretzschmar, K.; Watt, F.M. Markers of Epidermal Stem Cell Subpopulations in Adult Mammalian Skin. Cold Spring Harb Perspect Med. 2014, 4, a013631. [Google Scholar] [CrossRef] [PubMed]
- Khavari, P.A. Modelling cancer in human skin tissue. Nat. Rev. Cancer 2006, 6, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, P.; Guvendiren, M.; Chua, W.; Telerman, S.B.; Liakath-Ali, K.; Burdick, J.A.; Watt, F.M. Mimicking the topography of the epidermal–dermal interface with elastomer substrates. Integr. Boil. 2016, 8, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Walko, G.; Woodhouse, S.; Pisco, A.O.; Rognoni, E.; Liakath-Ali, K.; Lichtenberger, B.M.; Mishra, A.; Telerman, S.B.; Viswanathan, P.; Logtenberg, M.; et al. A genome-wide screen identifies YAP/WBP2 interplay conferring growth advantage on human epidermal stem cells. Nat. Commun. 2017, 8, 14744. [Google Scholar] [CrossRef] [Green Version]
- Jensen, U.B.; Lowell, S.; Watt, F.M. The spatial relationship between stem cells and their progeny in the basal layer of human epidermis: A new view based on whole-mount labelling and lineage analysis. Development 1999, 126, 2409–2418. [Google Scholar]
- Giangreco, A.; Goldie, S.J.; Failla, V.; Saintigny, G.; Watt, F.M. Human Skin Aging Is Associated with Reduced Expression of the Stem Cell Markers Beta1 Integrin and Mcsp. J. Invest. Derm. 2010, 130, 604–608. [Google Scholar] [CrossRef]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Invest. 2018, 128, 26–35. [Google Scholar] [CrossRef]
- Donati, G.; Proserpio, V.; Lichtenberger, B.M.; Natsuga, K.; Sinclair, R.; Fujiwara, H.; Watt, F.M. Epidermal Wnt/Beta-Catenin Signaling Regulates Adipocyte Differentiation Via Secretion of Adipogenic Factors. Proc. Natl. Acad Sci. USA 2014, 111, E1501–E1509. [Google Scholar] [CrossRef] [PubMed]
- Salzer, M.C.; Lafzi, A.; Berenguer-Llergo, A.; Youssif, C.; Castellanos, A.; Solanas, G.; Peixoto, F.O.; Attolini, C.S.-O.; Prats, N.; Aguilera, M.; et al. Identity Noise and Adipogenic Traits Characterize Dermal Fibroblast Aging. Cell 2018, 175, 1575–1590.e22. [Google Scholar] [CrossRef] [PubMed]
- Rognoni, E.; Pisco, A.O.; Hiratsuka, T.; Sipilä, K.H.; Belmonte, J.M.; Mobasseri, S.A.; Philippeos, C.; Dilão, R.; Watt, F.M. Fibroblast state switching orchestrates dermal maturation and wound healing. Mol. Syst. Boil. 2018, 14, e8174. [Google Scholar] [CrossRef]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Boil. 2009, 10, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.L.; Patel, D.M.; Green, K.J. Deconstructing the skin: Cytoarchitectural determinants of epidermal morphogenesis. Nat. Rev. Mol. Cell Boil. 2011, 12, 565–580. [Google Scholar] [CrossRef]
- Mishra, A.; Oules, B.; Pisco, A.O.; Ly, T.; Liakath-Ali, K.; Walko, G.; Viswanathan, P.; Tihy, M.; Nijjher, J.; Dunn, S.-J.; et al. A protein phosphatase network controls the temporal and spatial dynamics of differentiation commitment in human epidermis. eLife 2017, 6. [Google Scholar] [CrossRef]
- Jones, P.H.; Simons, B.D.; Watt, F.M. Sic Transit Gloria: Farewell to the Epidermal Transit Amplifying Cell? Cell Stem Cell 2007, 1, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrandon, Y.; Grasset, N.; Zaffalon, A.; Gorostidi, F.; Claudinot, S.; Droz-Georget, S.L.; Nanba, D.; Rochat, A. Capturing epidermal stemness for regenerative medicine. Semin. Cell Dev. Boil. 2012, 23, 937–944. [Google Scholar] [CrossRef]
- Jones, P.H.; Watt, F.M. Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell 1993, 73, 713–724. [Google Scholar] [CrossRef]
- Barrandon, Y.; Green, H. Three clonal types of keratinocyte with different capacities for multiplication. Proc. Acad. Sci. 1987, 84, 2302–2306. [Google Scholar] [CrossRef]
- Doupé, D.P.; Klein, A.M.; Simons, B.D.; Jones, P.H. The Ordered Architecture of Murine Ear Epidermis Is Maintained by Progenitor Cells with Random Fate. Dev. Cell 2010, 18, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sada, A.; Jacob, F.; Leung, E.; Wang, S.; White, B.S.; Shalloway, D.; Tumbar, T. Defining the cellular lineage hierarchy in the interfollicular epidermis of adult skin. Nat. Cell Boil. 2016, 18, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascré, G.; Dekoninck, S.; Drogat, B.; Youssef, K.K.; Brohée, S.; Sotiropoulou, P.A.; Simons, B.D.; Blanpain, C. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nat. Cell Boil. 2012, 489, 257–262. [Google Scholar] [CrossRef]
- Tan, D.W.M.; Jensen, K.B.; Trotter, M.W.B.; Connelly, J.T.; Broad, S.; Watt, F.M. Single-cell gene expression profiling reveals functional heterogeneity of undifferentiated human epidermal cells. Development 2013, 140, 1433–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.B.; Sedgewick, A.J.; Finnegan, A.I.; Harirchian, P.; Lee, J.; Kwon, S.; Fassett, M.S.; Golovato, J.; Gray, M.; Ghadially, R.; et al. Transcriptional Programming of Normal and Inflamed Human Epidermis at Single-Cell Resolution. Cell Rep. 2018, 25, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Joost, S.; Zeisel, A.; Jacob, T.; Sun, X.; La Manno, G.; Lonnerberg, P.; Linnarsson, S.; Kasper, M. Single-Cell Transcriptomics Reveals that Differentiation and Spatial Signatures Shape Epidermal and Hair Follicle Heterogeneity. Cell Syst. 2016, 3, 221–237.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishnubalaji, R.; Al-Nbaheen, M.; Kadalmani, B.; Aldahmash, A.; Ramesh, T. Skin-derived multipotent stromal cells – an archrival for mesenchymal stem cells. Cell Tissue Res. 2012, 350, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Dekoninck, S.; Rulands, S.; Lenglez, S.; Mascré, G.; Simons, B.D.; Blanpain, C. Defining stem cell dynamics and migration during wound healing in mouse skin epidermis. Nat. Commun. 2017, 8, 14684. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Gonzalez, D.G.; Guirao, B.; Boucher, J.D.; Cockburn, K.; Marsh, E.D.; Mesa, K.R.; Brown, S.; Rompolas, P.; Haberman, A.M.; et al. Tissue-Scale Coordination of Cellular Behaviour Promotes Epidermal Wound Repair in Live Mice. Nat. Cell Biol. 2017, 19, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Rognoni, E.; Hiratsuka, T.; Liakath-Ali, K.; Hoste, E.; Kar, G.; Kayikci, M.; Russell, R.; Kretzschmar, K.; Mulder, K.W.; et al. Wounding Induces Dedifferentiation of Epidermal Gata6(+) Cells and Acquisition of Stem Cell Properties. Nat. Cell Biol 2017, 19, 603–613. [Google Scholar] [CrossRef]
- Schmidt, B.A.; Horsley, V. Intradermal adipocytes mediate fibroblast recruitment during skin wound healing. Development 2013, 140, 1517–1527. [Google Scholar] [CrossRef] [Green Version]
- Gay, D.; Kwon, O.; Zhang, Z.; Spata, M.; Plikus, M.V.; Holler, P.D.; Ito, M.; Yang, Z.; Treffeisen, E.; Kim, C.D.; et al. Fgf9 from Dermal Gammadelta T Cells Induces Hair Follicle Neogenesis after Wounding. Nat. Med. 2013, 19, 916–923. [Google Scholar] [CrossRef]
- Lim, C.H.; Sun, Q.; Ratti, K.; Lee, S.-H.; Zheng, Y.; Takeo, M.; Lee, W.; Rabbani, P.; Plikus, M.V.; Cain, J.E.; et al. Hedgehog stimulates hair follicle neogenesis by creating inductive dermis during murine skin wound healing. Nat. Commun. 2018, 9, 4903. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Guerrero-Juarez, C.F.; Ito, M.; Li, Y.R.; Dedhia, P.H.; Zheng, Y.; Shao, M.; Gay, D.L.; Ramos, R.; Hsi, T.-C.; et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 2017, 355, 748–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didona, D.; Paolino, G.; Bottoni, U.; Cantisani, C. Non Melanoma Skin Cancer Pathogenesis Overview. Biomedicines 2018, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.R.; Santos, A.C.; Sanchez-Lopez, E.; Kovacevic, A.B.; Espina, M.; Calpena, A.C.; Veiga, F.J.; Garcia, M.L.; Souto, E.B. Neoplastic Multifocal Skin Lesions: Biology, Etiology, and Targeted Therapies for Nonmelanoma Skin Cancers. Skin Pharm. Physiol. 2018, 31, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Thieu, K.; Ruiz, M.E.; Owens, D.M. Cells of Origin and Tumor-Initiating Cells for Nonmelanoma Skin Cancers. Cancer Lett. 2013, 338, 82–88. [Google Scholar] [CrossRef]
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Melanoma: Genetic Abnormalities, Tumor Progression, Clonal Evolution and Tumor Initiating Cells. Med Sci. 2017, 5, 28. [Google Scholar] [Green Version]
- Nissinen, L.; Farshchian, M.; Riihilä, P.; Kähäri, V.-M. New perspectives on role of tumor microenvironment in progression of cutaneous squamous cell carcinoma. Cell Tissue Res. 2016, 365, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Guerra, L.; Odorisio, T.; Zambruno, G.; Castiglia, D. Stromal microenvironment in type VII collagen-deficient skin: The ground for squamous cell carcinoma development. Matrix Biol. 2017, 63, 1–10. [Google Scholar] [CrossRef]
- Arwert, E.N.; Hoste, E.; Watt, F.M. Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 2012, 12, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Z.; South, A.P. Tumour–stroma crosstalk in the development of squamous cell carcinoma. Int. J. Biochem. Cell Boil. 2014, 53, 450–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Zeisberg, M. Fibroblasts in Cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef]
- Hogervorst, M.; Rietveld, M.; De Gruijl, F.; El Ghalbzouri, A. A shift from papillary to reticular fibroblasts enables tumour–stroma interaction and invasion. Br. J. Cancer 2018, 118, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.-L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Moya, I.M.; Halder, G. Hippo–YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Boil. 2018, 1. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.-L. The Hippo Pathway: Biology and Pathophysiology. Annu. Biochem. 2018, 88. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [Green Version]
- Sharif, A.A.; Hergovich, A. The NDR/LATS protein kinases in immunology and cancer biology. Semin. Cancer Biol. 2018, 48, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Callus, B.A.; Finch-Edmondson, M.L.; Fletcher, S.; Wilton, S.D. YAPping about and not forgetting TAZ. Febs Lett. 2019, 593, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, S.W.; Lin, K.C.; Moore, J.L., 3rd; Tan, F.E.; Ma, S.; Ye, Z.; Qiu, Y.; Ren, B.; Guan, K.L. The Hippo Pathway Effector Proteins Yap and Taz Have Both Distinct and Overlapping Functions in the Cell. J. Biol. Chem. 2018, 293, 11230–11240. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; De Mello, V.; Mohamed, A.; Quiroga, H.P.O.; Al Bloshi, A.; Tremblay, A.M.; Von Kriegsheim, A.; Vargesson, N.; Matallanas, D.; Wackerhage, H.; et al. Common and Distinctive Functions of the Hippo Effectors Taz and Yap in Skeletal Muscle Stem Cell Function. Stem Cells 2017, 35, 1958–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.; Slattery, M.; Ma, L.; Crofts, A.; White, K.P.; Mann, R.S.; Irvine, K.D. Genome-wide association of Yorkie with chromatin and chromatin remodeling complexes. Cell Rep. 2013, 3, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.; Slattery, M.; Ma, L.; White, K.P.; Mann, R.S.; Irvine, K.D. Yorkie promotes transcription by recruiting a Histone methyltransferase complex. Cell Rep. 2014, 8, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Ikmi, A.; Gaertner, B.; Seidel, C.; Srivastava, M.; Zeitlinger, J.; Gibson, M.C. Molecular Evolution of the Yap/Yorkie Proto-Oncogene and Elucidation of Its Core Transcriptional Program. Mol. Boil. Evol. 2014, 31, 1375–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, G.G.; Carrara, M.; Yuan, W.-C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; De Laat, W.; et al. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Boil. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Bardet, A.F.; Roma, G.; Bergling, S.; Clay, I.; Ruchti, A.; Agarinis, C.; Schmelzle, T.; Bouwmeester, T.; Schübeler, D.; et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015, 11, e1005465. [Google Scholar] [CrossRef]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genome Res. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.-S. Transcriptional Co-repressor Function of the Hippo Pathway Transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.K.; Jang, J.W.; Bae, S.C. DNA Binding Partners of Yap/Taz. BMB Rep. 2018, 51, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nakagawa, M.; Olson, E.N.; Nakagawa, O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt–Oram syndrome. Proc. Acad. Sci. 2005, 102, 18034–18039. [Google Scholar] [CrossRef] [Green Version]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. Beta-Catenin-Driven Cancers Require a Yap1 Transcriptional Complex for Survival and Tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.W.; Kim, M.K.; Lee, Y.S.; Lee, J.W.; Kim, D.M.; Song, S.H.; Lee, J.Y.; Choi, B.Y.; Min, B.; Chi, X.Z.; et al. Rac-Lats1/2 Signaling Regulates Yap Activity by Switching between the Yap-Binding Partners Tead4 and Runx3. Oncogene 2017, 36, 999–1011. [Google Scholar] [CrossRef]
- Vitolo, M.I.; Anglin, I.E.; Mahoney, W.M.; Renoud, K.J.; Gartenhaus, R.B.; Bachman, K.E.; Passaniti, A. The RUNX2 transcription factor cooperates with the YES-associated protein, YAP65, to promote cell transformation. Cancer Biol. Ther. 2007, 6, 856–863. [Google Scholar] [CrossRef] [Green Version]
- Papaspyropoulos, A.; Bradley, L.; Thapa, A.; Leung, C.Y.; Toskas, K.; Koennig, D.; Pefani, D.-E.; Raso, C.; Grou, C.; Hamilton, G.; et al. RASSF1A uncouples Wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nat. Commun. 2018, 9, 424. [Google Scholar] [CrossRef] [Green Version]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The Transcriptional Coactivator Yes-Associated Protein Drives p73 Gene-Target Specificity in Response to DNA Damage. Mol. Cell 2005, 19, 429. [Google Scholar] [CrossRef]
- Strano, S.; Monti, O.; Baccarini, A.; Sudol, M.; Sacchi, A.; Blandino, G. Physical interaction with yes-associated protein enhances p73 transcriptional activity. J. Biol. Chem. 2001, 37, S279. [Google Scholar]
- Liu, X.; Li, H.; Rajurkar, M.; Li, Q.; Cotton, J.L.; Ou, J.; Zhu, L.J.; Goel, H.L.; Mercurio, A.M.; Park, J.-S.; et al. Tead and AP1 coordinate transcription and motility. Cell Rep. 2016, 14, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- Croci, O.; De Fazio, S.; Biagioni, F.; Donato, E.; Caganova, M.; Curti, L.; Doni, M.; Sberna, S.; Aldeghi, D.; Biancotto, C.; et al. Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genome Res. 2017, 31, 2017–2022. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plouffe, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.-S.; Lu, W.; Lu, S.; et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 2015, 6, 8357. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Wang, W.; Liu, B.; Deng, H.; Uster, E.; Pan, D. Identification of Happyhour/MAP4K as alternative Hpo/Mst-like kinases in the Hippo kinase cascade. Dev. Cell 2015, 34, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Li, S.; Mana-Capelli, S.; Flach, R.J.R.; Danai, L.V.; Amcheslavsky, A.; Nie, Y.; Kaneko, S.; Yao, X.; Chen, X.; et al. The conserved Misshapen-Warts-Yorkie pathway acts in enteroblasts to regulate intestinal stem cells in Drosophila. Dev. Cell 2014, 31, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genome Res. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A Coordinated Phosphorylation by Lats and Ck1 Regulates Yap Stability through Scf(Beta-Trcp). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef]
- Liu, C.Y.; Zha, Z.Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W.; et al. The Hippo Tumor Pathway Promotes Taz Degradation by Phosphorylating a Phosphodegron and Recruiting the Scf{Beta}-Trcp E3 Ligase. J. Biol. Chem. 2010, 285, 37159–37169. [Google Scholar] [CrossRef]
- Das, A.; Fischer, R.S.; Pan, D.; Waterman, C.M. YAP Nuclear Localization in the Absence of Cell-Cell Contact Is Mediated by a Filamentous Actin-dependent, Myosin II- and Phospho-YAP-independent Pathway during Extracellular Matrix Mechanosensing*. J. Boil. Chem. 2016, 291, 6096–6110. [Google Scholar] [CrossRef] [Green Version]
- Elbediwy, A.; Vanyai, H.; Diaz-De-La-Loza, M.-D.-C.; Frith, D.; Snijders, A.P.; Thompson, B.J. Enigma proteins regulate YAP mechanotransduction. J. Cell Sci. 2018, 131, jcs.221788. [Google Scholar] [CrossRef] [PubMed]
- Kofler, M.; Speight, P.; Little, D.; Di Ciano-Oliveira, C.; Szászi, K.; Kapus, A. Mediated nuclear import and export of TAZ and the underlying molecular requirements. Nat. Commun. 2018, 9, 4966. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.-L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410.e14. [Google Scholar] [CrossRef] [PubMed]
- Ege, N.; Dowbaj, A.M.; Jiang, M.; Howell, M.; Hooper, S.; Foster, C.; Jenkins, R.P.; Sahai, E. Quantitative Analysis Reveals that Actin and Src-Family Kinases Regulate Nuclear YAP1 and Its Export. Cell Syst. 2018, 6, 692–708.e13. [Google Scholar] [CrossRef]
- Manning, S.A.; Dent, L.G.; Kondo, S.; Zhao, Z.W.; Plachta, N.; Harvey, K.F. Dynamic Fluctuations in Subcellular Localization of the Hippo Pathway Effector Yorkie In Vivo. Curr. Boil. 2018, 28, 1651–1660.e4. [Google Scholar] [CrossRef] [PubMed]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.R.; et al. Yap1 Acts Downstream of Alpha-Catenin to Control Epidermal Proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef] [Green Version]
- Silvis, M.R.; Kreger, B.T.; Lien, W.H.; Klezovitch, O.; Rudakova, G.M.; Camargo, F.D.; Lantz, D.M.; Seykora, J.T.; Vasioukhin, V. Alpha-Catenin Is a Tumor Suppressor That Controls Cell Accumulation by Regulating the Localization and Activity of the Transcriptional Coactivator Yap1. Sci Signal. 2011, 4, ra33. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.-Y.; Yu, J.; Guan, K.-L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.-G.; Gumbiner, B.M. Adhesion to fibronectin regulates Hippo signaling via the FAK–Src–PI3K pathway. J. Cell Boil. 2015, 210, 503–515. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.-G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Acad. Sci. 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nat. Cell Boil. 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A Mechanical Checkpoint Controls Multicellular Growth through YAP/TAZ Regulation by Actin-Processing Factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Wada, K.-I.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [Green Version]
- Bao, M.; Xie, J.; Piruska, A.; Huck, W.T.S. 3D microniches reveal the importance of cell size and shape. Nat. Commun. 2017, 8, 1962. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-L.; Gajewski, K.M.; Hamaratoglu, F.; Bossuyt, W.; Sansores-Garcia, L.; Tao, C.; Halder, G. The apical-basal cell polarity determinant Crumbs regulates Hippo signaling in Drosophila. Proc. Acad. Sci. 2010, 107, 15810–15815. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Irvine, K.D. Regulation of Hippo Signaling by Jun Kinase Signaling During Compensatory Cell Proliferation and Regeneration, and in Neoplastic Tumors. Dev. Biol. 2011, 350, 139–151. [Google Scholar] [CrossRef]
- Zhou, B.; Flodby, P.; Luo, J.; Castillo, D.R.; Liu, Y.; Yu, F.X.; McConnell, A.; Varghese, B.; Li, G.; Chimge, N.O.; et al. Claudin-18-Mediated Yap Activity Regulates Lung Stem and Progenitor Cell Homeostasis and Tumorigenesis. J. Clin Invest. 2018, 128, 970–984. [Google Scholar] [CrossRef]
- Szymaniak, A.D.; Mahoney, J.E.; Cardoso, W.V.; Varelas, X. Crumbs3-mediated polarity directs airway epithelial cell fate through the Hippo pathway effector Yap. Dev. Cell 2015, 34, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narimatsu, M.; Samavarchi-Tehrani, P.; Varelas, X.; Wrana, J.L. Distinct Polarity Cues Direct Taz/Yap and Tgfbeta Receptor Localization to Differentially Control Tgfbeta-Induced Smad Signaling. Dev. Cell 2015, 32, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-C.; Graves, H.K.; Moya, I.M.; Tao, C.; Hamaratoglu, F.; Gladden, A.B.; Halder, G.; Hamaratoǧlu, F. Differential regulation of the Hippo pathway by adherens junctions and apical–basal cell polarity modules. Proc. Acad. Sci. 2015, 112, 1785–1790. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zheng, Y.; Dong, J.; Klusza, S.; Deng, W.-M.; Pan, D. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev. Cell 2010, 18, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamaratoglu, F.; Willecke, M.; Kango-Singh, M.; Nolo, R.; Hyun, E.; Tao, C.; Jafar-Nejad, H.; Halder, G. The Tumour-Suppressor Genes Nf2/Merlin and Expanded Act through Hippo Signalling to Regulate Cell Proliferation and Apoptosis. Nat. Cell Biol. 2006, 8, 27–36. [Google Scholar] [CrossRef]
- Elbediwy, A.; Thompson, B.J.; Vincent-Mistiaen, Z.I.; Vincent-Mistiaen, Z.I. YAP and TAZ in epithelial stem cells: A sensor for cell polarity, mechanical forces and tissue damage. BioEssays 2016, 38, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Koo, J.H.; Guan, K.-L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206. [Google Scholar] [CrossRef]
- Totaro, A.; Castellan, M.; Battilana, G.; Zanconato, F.; Azzolin, L.; Giulitti, S.; Cordenonsi, M.; Piccolo, S. YAP/TAZ link cell mechanics to Notch signalling to control epidermal stem cell fate. Nat. Commun. 2017, 8, 15206. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-García, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Boil. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Silvis, M.R.; Honaker, Y.; Lien, W.H.; Arron, S.T.; Vasioukhin, V. Alphae-Catenin Inhibits a Src-Yap1 Oncogenic Module That Couples Tyrosine Kinases and the Effector of Hippo Signaling Pathway. Genes Dev. 2016, 30, 798–811. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.; Yang, J.; DeRan, M.; Wu, C.; Su, A.I.; Bonamy, G.M.; Liu, J.; Peters, E.C.; Wu, X. Identification of Serum-Derived Sphingosine-1-Phosphate as a Small Molecule Regulator of YAP. Chem. Boil. 2012, 19, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.K.; Lu, S.Y.; Kang, S.-A.; Tan, H.J.; Li, Z.; Wee, Z.N.A.; Guan, J.S.; Chichili, V.P.R.; Sivaraman, J.; Putti, T.; et al. Wnt Signaling Promotes Breast Cancer by Blocking ITCH-Mediated Degradation of YAP/TAZ Transcriptional Coactivator WBP2. Cancer Res 2016, 76, 6278–6289. [Google Scholar] [CrossRef]
- Cai, J.; Maitra, A.; Anders, R.A.; Taketo, M.M.; Pan, D. Beta-Catenin Destruction Complex-Independent Regulation of Hippo-Yap Signaling by Apc in Intestinal Tumorigenesis. Genes Dev. 2015, 29, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. Yap/Taz Incorporation in the Beta-Catenin Destruction Complex Orchestrates the Wnt Response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, N.H.; Cho, E.S.; Yang, J.H.; Cha, Y.H.; Kang, H.E.; Yun, J.S.; Cho, S.B.; Lee, S.-H.; Paclíková, P.; et al. Dishevelled has a YAP nuclear export function in a tumor suppressor context-dependent manner. Nat. Commun. 2018, 9, 2301. [Google Scholar] [CrossRef]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [Green Version]
- Lamar, J.M.; Xiao, Y.; Norton, E.; Jiang, Z.G.; Gerhard, G.M.; Kooner, S.; Warren, J.S.A.; Hynes, R.O. Src Tyrosine Kinase Activates the Yap/Taz Axis and Thereby Drives Tumor Growth and Metastasis. J. Biol. Chem. 2019, 294, 2302–2317. [Google Scholar] [CrossRef]
- Yui, S.; Azzolin, L.; Maimets, M.; Pedersen, M.T.; Fordham, R.P.; Hansen, S.L.; Larsen, H.L.; Guiu, J.; Alves, M.R.; Rundsten, C.F.; et al. YAP/TAZ-Dependent Reprogramming of Colonic Epithelium Links ECM Remodeling to Tissue Regeneration. Cell Stem Cell 2018, 22, 35–49.e7. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Cao, X.; Dai, X.; Xu, L.; Guo, X.; Yan, H.; Zhu, C.; Zhou, Q.; Tang, M.; Xia, Z.; et al. Src Inhibits the Hippo Tumor Suppressor Pathway through Tyrosine Phosphorylation of Lats1. Cancer Res 2017, 77, 4868–4880. [Google Scholar] [CrossRef]
- Taniguchi, K.; Wu, L.-W.; Grivennikov, S.I.; De Jong, P.R.; Lian, I.; Yu, F.-X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP Module Links Inflammation to Epithelial Regeneration. Nat. Cell Boil. 2015, 519, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Kim, N.-G.; Gumbiner, B.M. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Acad. Sci. 2013, 110, 2569–2574. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J. Akt Phosphorylates the Yes-Associated Protein, YAP, to Induce Interaction with 14-3-3 and Attenuation of p73-Mediated Apoptosis. Mol. Cell 2003, 11, 11–23. [Google Scholar] [CrossRef]
- Zhang, H.; Pasolli, H.A.; Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Acad. Sci. 2011, 108, 2270–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beverdam, A.; Claxton, C.; Zhang, X.; James, G.; Harvey, K.F.; Key, B. Yap Controls Stem/Progenitor Cell Proliferation in the Mouse Postnatal Epidermis. J. Invest. Derm. 2013, 133, 1497–1505. [Google Scholar] [CrossRef] [Green Version]
- Akladios, B.; Mendoza-Reinoso, V.; Samuel, M.S.; Hardeman, E.C.; Khosrotehrani, K.; Key, B.; Beverdam, A. Epidermal Yap2-5sa-Deltac Drives Beta-Catenin Activation to Promote Keratinocyte Proliferation in Mouse Skin in Vivo. J. Invest. Derm. 2017, 137, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Yosefzon, Y.; Soteriou, D.; Feldman, A.; Kostić, L.; Koren, E.; Brown, S.; Ankawa, R.; Sedov, E.; Glaser, F.; Fuchs, Y. Caspase-3 Regulates YAP-Dependent Cell Proliferation and Organ Size. Mol. Cell 2018, 70, 573–587.e4. [Google Scholar] [CrossRef]
- Debaugnies, M.; Sánchez-Danés, A.; Rorive, S.; Raphaël, M.; Liagre, M.; Parent, M.; Brisebarre, A.; Salmon, I.; Blanpain, C. YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep. 2018, 19, e45809. [Google Scholar] [CrossRef]
- Iglesias-Bartolome, R.; Torres, D.; Marone, R.; Feng, X.; Martin, D.; Simaan, M.; Chen, M.; Weinstein, L.S.; Taylor, S.S.; Molinolo, A.A.; et al. Inactivation of a Galpha(S)-Pka Tumour Suppressor Pathway in Skin Stem Cells Initiates Basal-Cell Carcinogenesis. Nat. Cell Biol. 2015, 17, 793–803. [Google Scholar] [CrossRef]
- Roy, E.; Neufeld, Z.; Cerone, L.; Wong, H.Y.; Hodgson, S.; Livet, J.; Khosrotehrani, K. Bimodal behaviour of interfollicular epidermal progenitors regulated by hair follicle position and cycling. EMBO J. 2016, 35, 2658–2670. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Li, C.; Yang, J.; Wang, X.; Li, R.; Luo, S.; Li, Z.; Liu, J.; Liu, Z.; Zheng, Y. Yes-associated protein promotes the abnormal proliferation of psoriatic keratinocytes via an amphiregulin dependent pathway. Sci. Rep. 2018, 8, 14513. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through Yap and Taz Drives Fibroblast Activation and Fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef]
- Maglic, D.; Schlegelmilch, K.; Dost, A.F.; Panero, R.; Dill, M.T.; A Calogero, R.; Camargo, F.D. YAP-TEAD signaling promotes basal cell carcinoma development via a c-JUN/AP1 axis. EMBO J. 2018, 37, e98642. [Google Scholar] [CrossRef]
- Jia, J.; Li, C.; Luo, S.; Liu-Smith, F.; Yang, J.; Wang, X.; Wang, N.; Lai, B.; Lei, T.; Wang, Q.; et al. Yes-Associated Protein Contributes to the Development of Human Cutaneous Squamous Cell Carcinoma via Activation of RAS. J. Invest. Derm. 2016, 136, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Vincent-Mistiaen, Z.; Elbediwy, A.; Vanyai, H.; Cotton, J.; Stamp, G.; Nye, E.; Spencer-Dene, B.; Thomas, G.J.; Mao, J.; Thompson, B.; et al. YAP drives cutaneous squamous cell carcinoma formation and progression. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Xu, Y.; Qin, Z.; Robichaud, P.; Betcher, S.; Calderone, K.; He, T.; Johnson, T.M.; Voorhees, J.J.; Fisher, G.J. Elevated Yap and Its Downstream Targets Ccn1 and Ccn2 in Basal Cell Carcinoma: Impact on Keratinocyte Proliferation and Stromal Cell Activation. Am. J. Pathol. 2014, 184, 937–943. [Google Scholar] [CrossRef]
- Akladios, B.; Reinoso, V.M.; Cain, J.E.; Wang, T.; Lambie, D.L.; Watkins, D.N.; Beverdam, A. Positive regulatory interactions between YAP and Hedgehog signalling in skin homeostasis and BCC development in mouse skin in vivo. PLoS ONE 2017, 12, e0183178. [Google Scholar] [CrossRef]
- Nishio, M.; Hamada, K.; Kawahara, K.; Sasaki, M.; Noguchi, F.; Chiba, S.; Mizuno, K.; Suzuki, S.O.; Dong, Y.; Tokuda, M.; et al. Cancer susceptibility and embryonic lethality in Mob1a/1b double-mutant mice. J. Clin. Invest. 2012, 122, 4505–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappellesso, R.; Bellan, A.; Saraggi, D.; Salmaso, R.; Ventura, L.; Fassina, A. Yap Immunoreactivity Is Directly Related to Pilomatrixoma Size and Proliferation Rate. Arch. Derm. Res. 2015, 307, 379–383. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, J.Z.; Vergara, I.A.; Zhang, Y.; Szeto, P.; Yang, L.; Mintoff, C.; Colebatch, A.; McIntosh, L.; Mitchell, K.A.; et al. Somatic hypermutation of the YAP oncogene in a human cutaneous melanoma. Mol. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nallet-Staub, F.; Marsaud, V.; Li, L.; Gilbert, C.; Dodier, S.; Bataille, V.; Sudol, M.; Herlyn, M.; Mauviel, A. Pro-Invasive Activity of the Hippo Pathway Effectors Yap and Taz in Cutaneous Melanoma. J. Invest. Derm. 2014, 134, 123–132. [Google Scholar] [CrossRef]
- Wong, S.Y.; Reiter, J.F. Wounding mobilizes hair follicle stem cells to form tumors. Proc. Acad. Sci. 2011, 108, 4093–4098. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.C.; Eberl, M.; Vagnozzi, A.N.; Belkadi, A.; Veniaminova, N.A.; Verhaegen, M.E.; Bichakjian, C.K.; Ward, N.L.; Dlugosz, A.A.; Wong, S.Y. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 2015, 16, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Brakebusch, C.; Grose, R.; Quondamatteo, F.; Ramirez, A.; Jorcano, J.L.; Pirro, A.; Svensson, M.; Herken, R.; Sasaki, T.; Timpl, R.; et al. Skin and Hair Follicle Integrity Is Crucially Dependent on Beta 1 Integrin Expression on Keratinocytes. EMBO J. 2000, 19, 3990–4003. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-J.; Byun, M.R.; Furutani-Seiki, M.; Hong, J.-H.; Jung, H.-S. YAP and TAZ Regulate Skin Wound Healing. J. Inves. Derm. 2014, 134, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Chen, J.; Lim, Y.B.; Finch-Edmondson, M.L.; Seshachalam, V.P.; Qin, L.; Jiang, T.; Low, B.C.; Singh, H.; Lim, C.T.; et al. YAP Regulates Actin Dynamics through ARHGAP29 and Promotes Metastasis. Cell Rep. 2017, 19, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Nardone, G.; La Cruz, J.O.-D.; Vrbsky, J.; Martini, C.; Přibyl, J.; Skládal, P.; Pešl, M.; Caluori, G.; Pagliari, S.; Martino, F.; et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun. 2017, 8, 15321. [Google Scholar] [CrossRef] [Green Version]
- Um, J.; Yu, J.; Park, K.-S. Substance P accelerates wound healing in type 2 diabetic mice through endothelial progenitor cell mobilization and Yes-associated protein activation. Mol. Med. Rep. 2017, 15, 3035–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grannas, K.; Arngarden, L.; Lonn, P.; Mazurkiewicz, M.; Blokzijl, A.; Zieba, A.; Soderberg, O. Crosstalk between Hippo and Tgfbeta: Subcellular Localization of Yap/Taz/Smad Complexes. J. Mol Biol 2015, 427, 3407–3415. [Google Scholar] [CrossRef]
- Corley, S.M.; Mendoza-Reinoso, V.; Giles, N.; Singer, E.S.; Common, J.E.; Wilkins, M.R.; Beverdam, A. Plau and Tgfbr3 are YAP-regulated genes that promote keratinocyte proliferation. Cell Death 2018, 9, 1106. [Google Scholar] [CrossRef]
- Mendoza-Reinoso, V.; Beverdam, A. Epidermal Yap Activity Drives Canonical Wnt16/Beta-Catenin Signaling to Promote Keratinocyte Proliferation in Vitro and in the Murine Skin. Stem Cell Res. 2018, 29, 15–23. [Google Scholar] [CrossRef]
- Lim, X.; Tan, S.H.; Koh, W.L.C.; Chau, R.M.W.; Yan, K.S.; Kuo, C.J.; Van Amerongen, R.; Klein, A.M.; Nusse, R.; Koh, W.L.C.; et al. Interfollicular epidermal stem cells self-renew via autocrine Wnt signaling. Science 2013, 342, 1226–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.S.; Zhang, Y.; Xu, M.; Yang, Y.; Ito, M.; Peng, T.; Cui, Z.; Nagy, A.; Hadjantonakis, A.K.; Lang, R.A.; et al. Distinct Functions for Wnt/Beta-Catenin in Hair Follicle Stem Cell Proliferation and Survival and Interfollicular Epidermal Homeostasis. Cell Stem Cell 2013, 13, 720–733. [Google Scholar] [CrossRef]
- Lin, H.Y.; Yang, L.T. Differential Response of Epithelial Stem Cell Populations in Hair Follicles to Tgf-Beta Signaling. Dev. Biol. 2013, 373, 394–406. [Google Scholar] [CrossRef]
- Oshimori, N.; Fuchs, E. Paracrine Tgf-Beta Signaling Counterbalances Bmp-Mediated Repression in Hair Follicle Stem Cell Activation. Cell Stem Cell 2012, 10, 63–75. [Google Scholar] [CrossRef]
- Rognoni, E.; Widmaier, M.; Jakobson, M.; Ruppert, R.; Ussar, S.; Katsougkri, D.; Bottcher, R.T.; Lai-Cheong, J.E.; Rifkin, D.B.; McGrath, J.A.; et al. Kindlin-1 Controls Wnt and Tgf-Beta Availability to Regulate Cutaneous Stem Cell Proliferation. Nat. Med. 2014, 20, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Xia, W.; Fisher, G.J.; Voorhees, J.J.; Quan, T. Yap/Taz Regulates Tgf-Beta/Smad3 Signaling by Induction of Smad7 Via Ap-1 in Human Skin Dermal Fibroblasts. Cell Commun. Signal. 2018, 16, 18. [Google Scholar] [CrossRef]
- Mobasseri, S.A.; Zijl, S.; Salameti, V.; Walko, G.; Stannard, A.; Garcia-Manyes, S.; Watt, F.M. Patterning of human epidermal stem cells on undulating elastomer substrates reflects differences in cell stiffness. Acta Biomater. 2019, 87, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Samsonov, M.; Sokabe, M. Actomyosin contractility provokes contact inhibition in E-cadherin-ligated keratinocytes. Sci. Rep. 2017, 7, 46326. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, H.; Ferreira, M.; Donati, G.; Marciano, D.K.; Linton, J.M.; Sato, Y.; Hartner, A.; Sekiguchi, K.; Reichardt, L.F.; Watt, F.M. The basement membrane of hair follicle stem cells is a muscle cell niche. Cell 2011, 144, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Margadant, C.; Charafeddine, R.A.; Sonnenberg, A. Unique and redundant functions of integrins in the epidermis. Faseb J. 2010, 24, 4133–4152. [Google Scholar] [CrossRef]
- Totaro, A.; Castellan, M.; Di Biagio, D.; Piccolo, S. Crosstalk between YAP/TAZ and Notch Signaling. Trends Cell Biol. 2018, 28, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Muroyama, A.; Underwood, J.; Leylek, R.; Ray, S.; Soderling, S.H.; Lechler, T. Actin-related protein2/3 complex regulates tight junctions and terminal differentiation to promote epidermal barrier formation. Proc. Acad. Sci. 2013, 110, E3820–E3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiwanuka, E.; Andersson, L.; Caterson, E.J.; Junker, J.P.; Gerdin, B.; Eriksson, E. Ccn2 Promotes Keratinocyte Adhesion and Migration Via Integrin Alpha5beta1. Exp. Cell Res. 2013, 319, 2938–2946. [Google Scholar] [CrossRef]
- Walko, G.; Castañón, M.J.; Wiche, G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015, 360, 529–544. [Google Scholar] [CrossRef] [Green Version]
- Livshits, G.; Kobielak, A.; Fuchs, E. Governing epidermal homeostasis by coupling cell–cell adhesion to integrin and growth factor signaling, proliferation, and apoptosis. Proc. Acad. Sci. 2012, 109, 4886–4891. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Tibbitt, M.W.; Basta, L.; Anseth, K.S. Mechanical memory and dosing influence stem cell fate. Nat. Mater. 2014, 13, 645–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Y.; Kang, Q.; Zhao, Y.; Hu, X.; Li, N. Lats2 Modulates Adipocyte Proliferation and Differentiation via Hippo Signaling. PLoS ONE 2013, 8, e72042. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Yu, Q.; Gong, Y.; Chen, W.; Tong, Y.; Wang, Y.; Xu, H.; Shi, Y. Yes-Associated Protein (YAP) Promotes Tumorigenesis in Melanoma Cells Through Stimulation of Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1). Sci. Rep. 2017, 7, 15528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, P.; Liu, H.; Lamar, J.M.; Schindler, J.W.; Jiang, Z.-G.; O Hynes, R. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Acad. Sci. 2012, 109, E2441–E2450. [Google Scholar]
- Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Eckert, R.L. Inhibition of YAP function overcomes BRAF inhibitor resistance in melanoma cancer stem cells. Oncotarget 2017, 8, 110257–110272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.H.; Lee, J.K.; Jung, E.; Kim, J. Actin Remodeling Confers Braf Inhibitor Resistance to Melanoma Cells through Yap/Taz Activation. EMBO J. 2016, 35, 462–478. [Google Scholar] [CrossRef]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, C.G.; Kim, S.-K.; Shin, S.J.; Choe, E.A.; Park, S.-H.; Shin, E.-C.; Kim, J. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol. Res. 2018, 6, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sambandam, S.A.; Lu, H.J.; Thomson, A.; Kim, S.H.; Lu, H.; Xin, Y.; Lu, Q. 14-3-3sigma and P63 Play Opposing Roles in Epidermal Tumorigenesis. Carcinogenesis 2011, 32, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Sambandam, S.A.T.; Kasetti, R.B.; Xue, L.; Dean, D.C.; Lu, Q.; Li, Q. 14-3-3sigma Regulates Keratinocyte Proliferation and Differentiation by Modulating Yap1 Cellular Localization. J. Invest. Derm. 2015, 135, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Dufort, C.C.; Paszek, M.J.; Weaver, V.M. Balancing forces: Architectural control of mechanotransduction. Nat. Rev. Mol. Cell Boil. 2011, 12, 308–319. [Google Scholar] [CrossRef]
- Foster, C.T.; Gualdrini, F.; Treisman, R. Mutual dependence of the MRTF–SRF and YAP–TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genome Res. 2017, 31, 2361–2375. [Google Scholar] [CrossRef] [Green Version]
- Mo, J.-S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.-S.; Guan, K.-L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Boil. 2015, 17, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140.e10. [Google Scholar] [CrossRef]
- Santinon, G.; Brian, I.; Pocaterra, A.; Romani, P.; Franzolin, E.; Rampazzo, C.; Bicciato, S.; Dupont, S. dNTP metabolism links mechanical cues and YAP/TAZ to cell growth and oncogene-induced senescence. EMBO J. 2018, 37, e97780. [Google Scholar] [CrossRef]
- Puig, S.; Berrocal, A. Management of high-risk and advanced basal cell carcinoma. Clin. Transl. Oncol. 2015, 17, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Henriques, V.; Martins, T.; Link, W.; Ferreira, B.I. The Emerging Therapeutic Landscape of Advanced Melanoma. Curr. Pharm. Des. 2018, 24, 249–558. [Google Scholar] [CrossRef] [PubMed]
- Chong, K.; Daud, A.; Ortiz-Urda, S.; Arron, S.T. Cutting Edge in Medical Management of Cutaneous Oncology. Semin. Cutan. Med. Surg. 2012, 31, 140–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spallone, G.; Botti, E.; Costanzo, A. Targeted Therapy in Nonmelanoma Skin Cancers. Mol. Cell. Basis Metastasis: Road Ther. 2011, 3, 2255–2273. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Battilana, G.; Cordenonsi, M.; Piccolo, S. YAP/TAZ as therapeutic targets in cancer. Curr. Opin. Pharmacol. 2016, 29, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. Yap/Taz at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Halder, G. The Two Faces of Hippo: Targeting the Hippo Pathway for Regenerative Medicine and Cancer Treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef]
- Yang, X.; Daifallah, A.E.M.; Shankar, S.; Beer, J.; Marshall, C.; Dentchev, T.; Seykora, F.; D’Armas, S.; Hahn, J.; Lee, V.; et al. Topical kinase inhibitors induce regression of cutaneous squamous cell carcinoma. Exp. Dermatol. 2019. [Google Scholar] [CrossRef]
- Gibault, F.; Sturbaut, M.; Bailly, F.; Melnyk, P.; Cotelle, P. Targeting Transcriptional Enhanced Associate Domains (Teads). J. Med. Chem. 2018, 61, 5057–5072. [Google Scholar] [CrossRef] [PubMed]
- Santucci, M.; Vignudelli, T.; Ferrari, S.; Mor, M.; Scalvini, L.; Bolognesi, M.L.; Uliassi, E.; Costi, M.P. The Hippo Pathway and YAP/TAZ–TEAD Protein–Protein Interaction as Targets for Regenerative Medicine and Cancer Treatment. J. Med. Chem. 2015, 58, 4857–4873. [Google Scholar] [CrossRef]
- Holden, J.K.; Cunningham, C.N. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Bum-Erdene, K.; Zhou, D.; Gonzalez-Gutierrez, G.; Ghozayel, M.K.; Si, Y.; Xu, D.; Shannon, H.E.; Bailey, B.J.; Corson, T.W.; Pollok, K.E.; et al. Small-Molecule Covalent Modification of Conserved Cysteine Leads to Allosteric Inhibition of the TEAD⋅Yap Protein-Protein Interaction. Cell Chem. Boil. 2019, 26, 378–389.e13. [Google Scholar] [CrossRef]
- Zhou, Z.; Hu, T.; Xu, Z.; Lin, Z.; Zhang, Z.; Feng, T.; Zhu, L.; Rong, Y.; Shen, H.; Luk, J.M.; et al. Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. Faseb J. 2015, 29, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.A.; Sessions, R.B.; Shoemark, D.K.; Williams, C.; Ebrahimighaei, R.; McNeill, M.C.; Crump, M.P.; McKay, T.R.; Harris, G.; Newby, A.C.; et al. Antiproliferative and Antimigratory Effects of a Novel YAP–TEAD Interaction Inhibitor Identified Using in Silico Molecular Docking. J. Med. Chem. 2019, 62, 1291–1305. [Google Scholar] [CrossRef]
- Crawford, J.J.; Bronner, S.M.; Zbieg, J.R. Hippo Pathway Inhibition by Blocking the Yap/Taz-Tead Interface: A Patent Review. Expert Opin. Ther. Pat. 2018, 28, 868–873. [Google Scholar] [CrossRef]
- Noland, C.L.; Gierke, S.; Schnier, P.D.; Murray, J.; Sandoval, W.N.; Sagolla, M.; Dey, A.; Hannoush, R.N.; Fairbrother, W.J.; Cunningham, C.N. Palmitoylation of TEAD Transcription Factors Is Required for Their Stability and Function in Hippo Pathway Signaling. Structure 2016, 24, 179–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.; Han, X.; Zheng, B.; DeRan, M.; Yu, J.; Jarugumilli, G.K.; Deng, H.; Pan, D.; Luo, X.; Wu, X. Autopalmitoylation of Tead Proteins Regulates Transcriptional Output of the Hippo Pathway. Nat. Chem. Biol. 2016, 12, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M. Selective migration of terminally differentiating cells from the basal layer of cultured human epidermis. J. Cell Boil. 1984, 98, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Watt, F.M.; Green, H. Stratification and terminal differentiation of cultured epidermal cells. Nat. Cell Boil. 1982, 295, 434–436. [Google Scholar] [CrossRef]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rognoni, E.; Walko, G. The Roles of YAP/TAZ and the Hippo Pathway in Healthy and Diseased Skin. Cells 2019, 8, 411. https://doi.org/10.3390/cells8050411
Rognoni E, Walko G. The Roles of YAP/TAZ and the Hippo Pathway in Healthy and Diseased Skin. Cells. 2019; 8(5):411. https://doi.org/10.3390/cells8050411
Chicago/Turabian StyleRognoni, Emanuel, and Gernot Walko. 2019. "The Roles of YAP/TAZ and the Hippo Pathway in Healthy and Diseased Skin" Cells 8, no. 5: 411. https://doi.org/10.3390/cells8050411
APA StyleRognoni, E., & Walko, G. (2019). The Roles of YAP/TAZ and the Hippo Pathway in Healthy and Diseased Skin. Cells, 8(5), 411. https://doi.org/10.3390/cells8050411