The Role of YAP and TAZ in Angiogenesis and Vascular Mimicry



Abstract
:1. Introduction
1.1. Angiogenesis and Its Roles in Development and Pathogenesis
1.1.1. Angiogenesis
1.1.2. Angiogenesis in Development
1.1.3. Angiogenesis in Pathogenesis
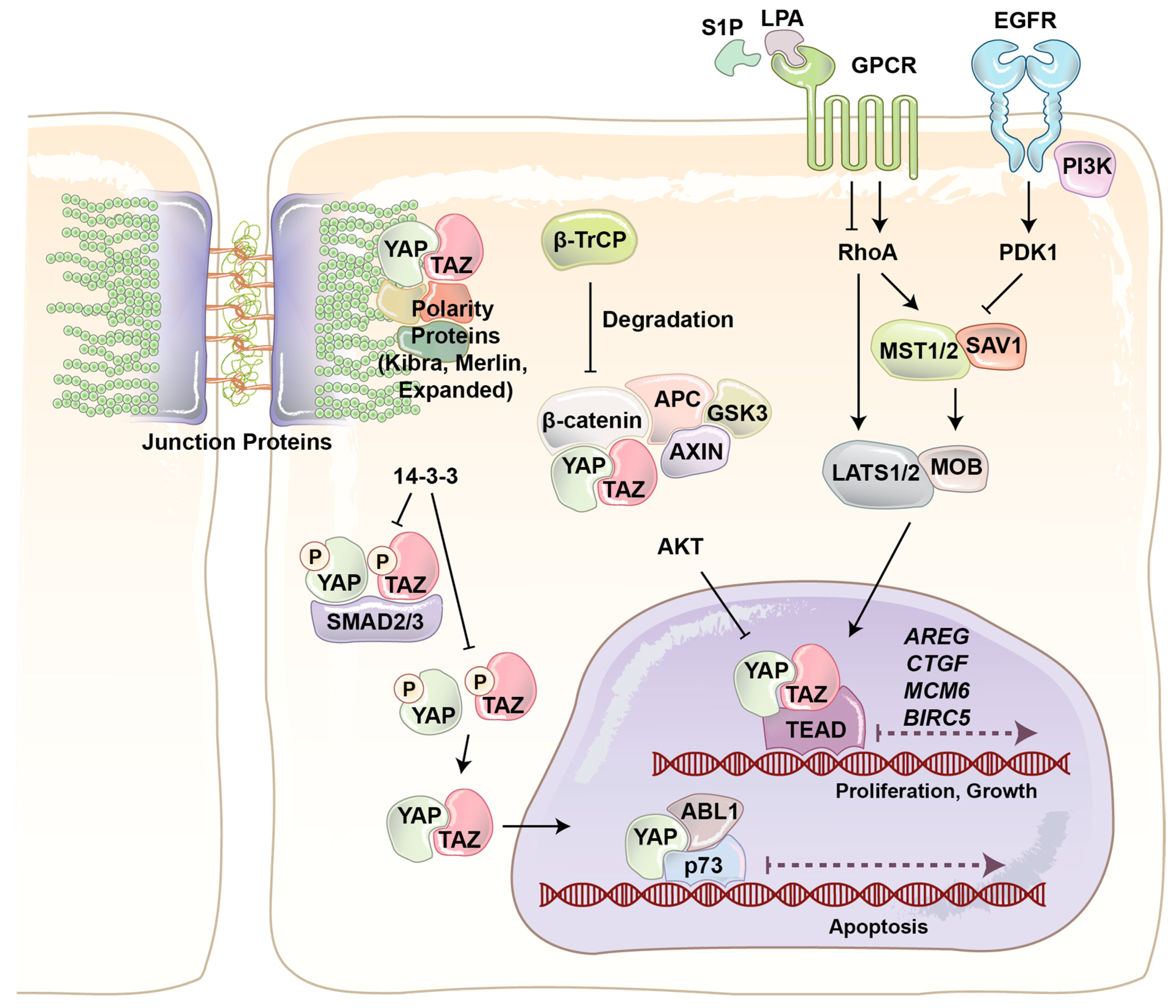
1.2. Yes-Associated Protein (YAP) and Paralog Transcription Activator with PDZ Binding Motif (TAZ)
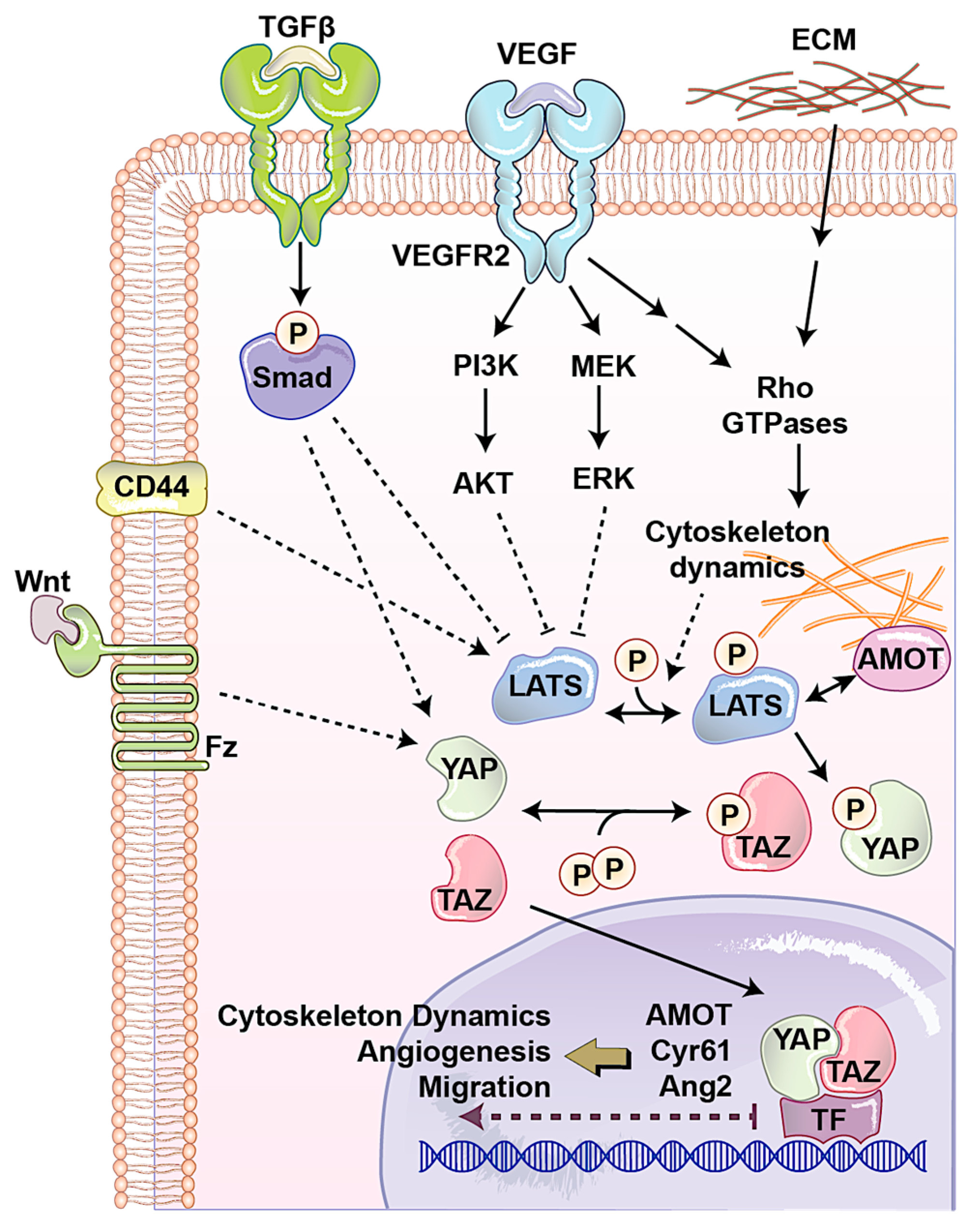
2. Roles of YAP/TAZ in the Regulation of Endothelial Function during Angiogenesis
2.1. Angiomotin (AMOT) Family
2.2. CD44
2.3. Extracellular Matrix (ECM)
2.4. Transforming Growth Factor-β (TGFβ)/Bone Morphogenic Protein (BMP)
2.5. WNT Pathway
2.6. PROX1 Transcriptional Programing—Key Regulators of Blood Vessel and Lymphatic Endothelial Cell Trans-Differentiation
3. Roles of YAP/TAZ in Developmental Vasculogenesis and Angiogenesis
4. Roles of YAP/TAZ in Pathological Angiogenesis
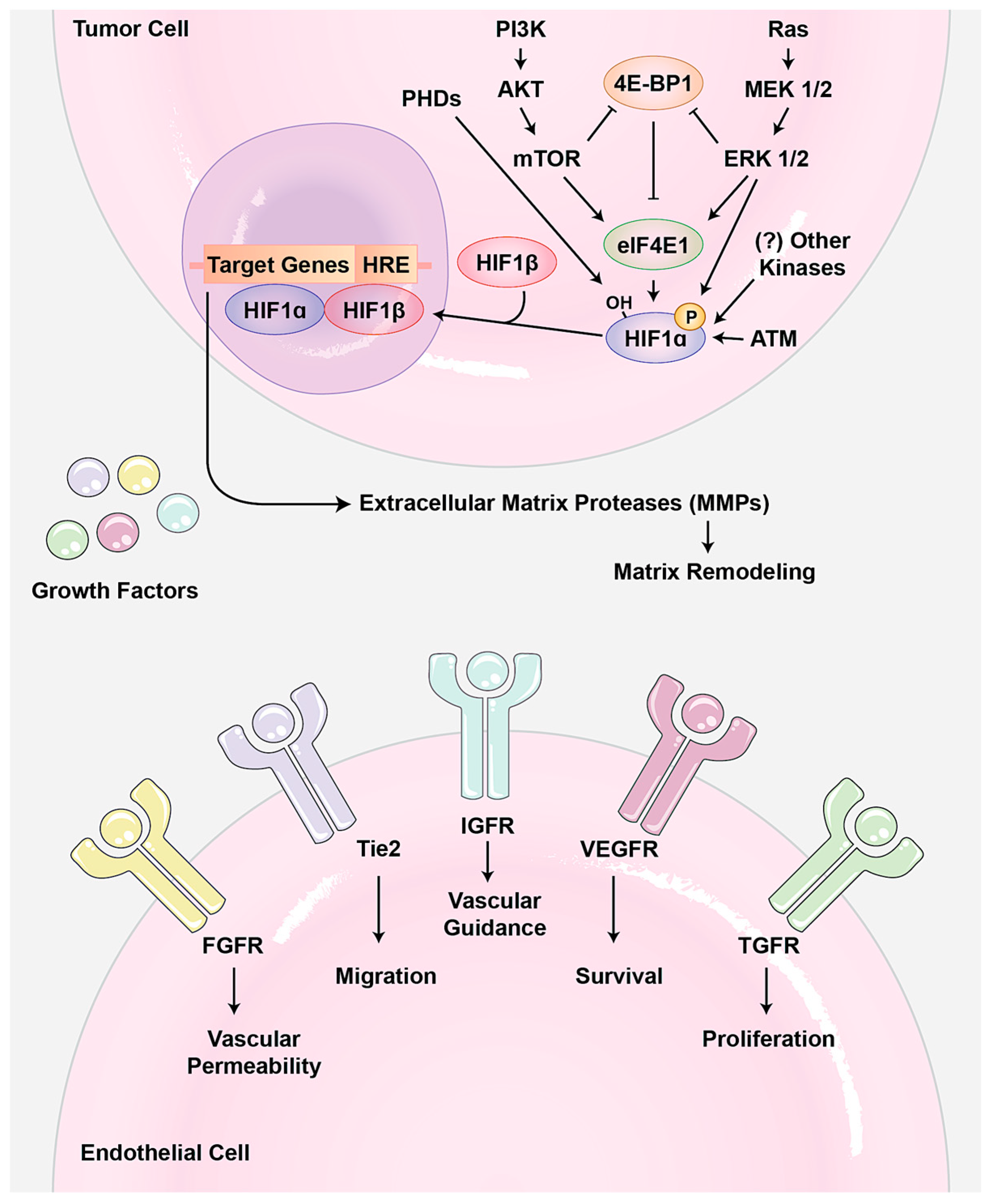
5. Roles of YAP/TAZ in Tumor Angiogenesis and Vascular Mimicry
6. Conclusion and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Djonov, V.; Baum, O.; Burri, P.H. Vascular remodeling by intussusceptive angiogenesis. Cell Tissue Res. 2003, 314, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Caduff, J.H.; Fischer, L.C.; Burri, P.H. Scanning electron microscope study of the developing microvasculature in the postnatal rat lung. Anat. Rec. 1986, 216, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Otrock, Z.K.; Mahfouz, R.A.; Makarem, J.A.; Shamseddine, A.I. Understanding the biology of angiogenesis: Review of the most important molecular mechanisms. Blood Cellsmoleculesand Dis. 2007, 39, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Deveza, L.; Choi, J.; Yang, F.J. Therapeutic angiogenesis for treating cardiovascular diseases. Theranostics 2012, 2, 801. [Google Scholar] [CrossRef]
- Couffinhal, T.; Dufourcq, P.; Daret, D.; Duplaa, C. The mechanisms of angiogenesis. Medical and therapeutic applications. La Revue de Medecine Interne 2001, 22, 1064–1082. [Google Scholar] [CrossRef]
- Muñoz-Chápuli, R. Evolution of angiogenesis. Int. J. Dev. Biol. 2011, 55, 345–351. [Google Scholar] [CrossRef]
- Breier, G. Angiogenesis in embryonic development—A review. Placenta 2000, 21, S11–S15. [Google Scholar] [CrossRef] [PubMed]
- Klagsbrun, M.; D’Amore, P.A. Vascular endothelial growth factor and its receptors. Cytokine Growth Factor Rev. 1996, 3, 259–270. [Google Scholar] [CrossRef]
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef]
- Lobov, I.; Renard, R.; Papadopoulos, N.; Gale, N.; Thurston, G.; Yancopoulos, G.; Wiegand, S.J. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc. Natl. Acad. Sci. USA 2007, 104, 3219–3224. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Lenard, A.; Belting, H.-G.; Affolter, M. Cell behaviors and dynamics during angiogenesis. Co. Biol. 2016, 143, 2249–2260. [Google Scholar]
- Flanagan, J.G.; Vanderhaeghen, P. The ephrins and Eph receptors in neural development. Annu. Rev. Neurosci. 1998, 21, 309–345. [Google Scholar] [CrossRef]
- Creamer, D.; Sullivan, D.; Bicknell, R.; Barker, J. Angiogenesis in psoriasis. Angiogenesis 2002, 5, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Tuo, J.; Wang, Y.; Cheng, R.; Li, Y.; Chen, M.; Qiu, F.; Qian, H.; Shen, D.; Penalva, R.; Xu, H. Wnt signaling in age-related macular degeneration: Human macular tissue and mouse model. J. Transl. Med. 2015, 13, 330. [Google Scholar] [CrossRef] [PubMed]
- Fallah, A.; Sadeghinia, A.; Kahroba, H.; Samadi, A.; Heidari, H.R.; Bradaran, B.; Zeinali, S.; Molavi, O. Therapeutic targeting of angiogenesis molecular pathways in angiogenesis-dependent diseases. J. Cell. Physiol. 2019, 110, 775–785. [Google Scholar] [CrossRef]
- Zetter, P.B.R. Angiogenesis and tumor metastasis. Annu. Rev. Med. 1998, 49, 407–424. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Lawler, J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J. Cell. Mol. Med. 2002, 6, 1–12. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef]
- Moya, I.M.; Halder, G. Hippo–YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2018, 20, 211–226. [Google Scholar] [CrossRef]
- Maugeri-Saccà, M.; Barba, M.; Pizzuti, L.; Vici, P.; Di Lauro, L.; Dattilo, R.; Vitale, I.; Bartucci, M.; Mottolese, M.; De Maria, R. The Hippo transducers TAZ and YAP in breast cancer: Oncogenic activities and clinical implications. J. Exp. Clin. Cancer Res. 2015, 17, e14. [Google Scholar] [CrossRef]
- Hansen, C.G.; Moroishi, T.; Guan, K.-L. YAP and TAZ: A nexus for Hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef]
- Zhang, K.; Qi, H.-X.; Hu, Z.-M.; Chang, Y.-N.; Shi, Z.-M.; Han, X.-H.; Han, Y.-W.; Zhang, R.-X.; Zhang, Z.; Chen, T. YAP and TAZ take center stage in cancer. Biochemistry 2015, 54, 6555–6566. [Google Scholar] [CrossRef]
- Csibi, A.; Blenis, J. Hippo-YAP and mTOR pathways collaborate to regulate organ size. Nat. Cell Biol. 2012, 14, 1244–1245. [Google Scholar] [CrossRef]
- Tumaneng, K.; Russell, R.C.; Guan, K.-L. Organ size control by Hippo and TOR pathways. Curr. Biol. 2012, 22, R368–R379. [Google Scholar] [CrossRef]
- Hong, W.; Guan, K.-L. The YAP and TAZ transcription co-activators: Key downstream effectors of the mammalian Hippo pathway. Semin. Cell Dev. Biol. 2012, 23, 785–793. [Google Scholar] [CrossRef]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.R.; Schiripo, T.A.; Haber, D.A.; Hariharan, I.K. salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef]
- Lai, Z.-C.; Wei, X.; Shimizu, T.; Ramos, E.; Rohrbaugh, M.; Nikolaidis, N.; Ho, L.-L.; Li, Y. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell 2005, 120, 675–685. [Google Scholar] [CrossRef]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Saucedo, L.J.; Edgar, B.A. Filling out the Hippo pathway. Nat. Rev. Mol. Cell Biol. 2007, 8, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed]
- Callus, B.A.; Verhagen, A.M.; Vaux, D.L. Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. Febs J. 2006, 273, 4264–4276. [Google Scholar] [CrossRef]
- Praskova, M.; Xia, F.; Avruch, J. MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr. Biol. 2008, 18, 311–321. [Google Scholar] [CrossRef]
- Chan, E.H.Y.; Nousiainen, M.; Chalamalasetty, R.B.; Schäfer, A.; Nigg, E.A.; Silljé, H.H.W. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 2005, 24, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Guan, K.-L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.-Y.; Lei, Q.; Guan, K.-L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-C.; Graves, H.K.; Moya, I.M.; Tao, C.; Hamaratoglu, F.; Gladden, A.B.; Halder, G. Differential regulation of the Hippo pathway by adherens junctions and apical–basal cell polarity modules. Proc. Natl. Acad. Sci. USA 2015, 112, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef]
- van Rensburg, H.J.J.; Lai, D.; Azad, T.; Hao, Y.; Yang, X. TAZ enhances mammary cell proliferation in 3D culture through transcriptional regulation of IRS1. Cell. Signal. 2018, 52, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Kim, N.-G.; Gumbiner, B.M. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl. Acad. Sci. USA 2013, 110, 2569–2574. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Montminy, T.; Azad, T.; Lightbody, E.; Hao, Y.; SenGupta, S.; Asselin, E.; Nicol, C.; Yang, X. PI3K Positively Regulates YAP and TAZ in Mammary Tumorigenesis Through Multiple Signaling Pathways. Mol. Cancer Res. 2018, 16, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Glienke, J.; Schmitt, A.O.; Pilarsky, C.; Hinzmann, B.; Weiß, B.; Rosenthal, A.; Thierauch, K.H. Differential gene expression by endothelial cells in distinct angiogenic states. Eur. J. Biochem. 2000, 267, 2820–2830. [Google Scholar] [CrossRef]
- Auerbach, R.; Lewis, R.; Shinners, B.; Kubai, L.; Akhtar, N. Angiogenesis assays: A critical overview. Clin. Chem. 2003, 49, 32–40. [Google Scholar] [CrossRef]
- Auerbach, R.; Akhtar, N.; Lewis, R.L.; Shinners, B.L. Angiogenesis assays: Problems and pitfalls. Cancer Metastasis Rev. 2000, 19, 167–172. [Google Scholar] [CrossRef]
- Staton, C.A.; Lewis, C.; Bicknell, R. Angiogenesis Assays: A Critical Appraisal of Current Techniques; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Goodwin, A.M. In vitro assays of angiogenesis for assessment of angiogenic and anti-angiogenic agents. Microvasc. Res. 2007, 74, 172–183. [Google Scholar] [CrossRef]
- Cui, X.; Morales, R.-T.T.; Qian, W.; Wang, H.; Gagner, J.-P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials 2018, 161, 164–178. [Google Scholar] [CrossRef]
- Song, L.; Ahmed, M.F.; Li, Y.; Zeng, C.; Li, Y. Vascular Differentiation from Pluripotent Stem Cells in 3-D Auxetic Scaffolds. J. Tissue Eng. Regen. Med. 2018, 12, 1679–1689. [Google Scholar] [CrossRef]
- Aase, K.; Ernkvist, M.; Ebarasi, L.; Jakobsson, L.; Majumdar, A.; Yi, C.; Birot, O.; Ming, Y.; Kvanta, A.; Edholm, D. Angiomotin regulates endothelial cell migration during embryonic angiogenesis. Genes Dev. 2007, 21, 2055–2068. [Google Scholar] [CrossRef]
- Zheng, Y.; Vertuani, S.; Nystrom, S.; Audebert, S.; Meijer, I.; Tegnebratt, T.; Borg, J.-P.; Uhlén, P.; Majumdar, A.; Holmgren, L. Angiomotin-like protein 1 controls endothelial polarity and junction stability during sprouting angiogenesis. Circ. Res. 2009, 105, 260–270. [Google Scholar] [CrossRef]
- Skouloudaki, K.; Walz, G. YAP1 recruits c-Abl to protect angiomotin-like 1 from Nedd4-mediated degradation. PLoS ONE 2012, 7, e35735. [Google Scholar] [CrossRef]
- Dai, X.; She, P.; Chi, F.; Feng, Y.; Liu, H.; Jin, D.; Zhao, Y.; Guo, X.; Jiang, D.; Guan, K.-L.; Zhong, T.P.; Zhao, B. Phosphorylation of angiomotin by Lats1/2 kinases inhibits F-actin binding, cell migration and angiogenesis. J. Biol. Chem. 2013, 288, 34041–34051. [Google Scholar] [CrossRef]
- Hong, S.A.; Son, M.W.; Cho, J.; Jang, S.H.; Lee, H.J.; Lee, J.H.; Cho, H.D.; Oh, M.H.; Lee, M.S. Low angiomotin-p130 with concomitant high Yes-associated protein 1 expression is associated with adverse prognosis of advanced gastric cancer. Apmis 2017, 125, 996–1006. [Google Scholar] [CrossRef]
- Singleton, P.A.; Salgia, R.; Moreno-Vinasco, L.; Moitra, J.; Sammani, S.; Mirzapoiazova, T.; Garcia, J.G.N. CD44 Regulates Hepatocyte Growth Factor-mediated Vascular Integrity: ROLE OF c-Met, Tiam1/Rac1, DYNAMIN 2, AND CORTACTIN. J. Biol. Chem. 2007, 282, 30643–30657. [Google Scholar] [CrossRef] [PubMed]
- Savani, R.C.; Cao, G.; Pooler, P.M.; Zaman, A.; Zhou, Z.; DeLisser, H.M. Differential involvement of the hyaluronan (HA) receptors CD44 and receptor for HA-mediated motility in endothelial cell function and angiogenesis. J. Biol. Chem. 2001, 276, 36770–36778. [Google Scholar] [CrossRef]
- Flynn, K.M.; Michaud, M.; Canosa, S.; Madri, J.A. CD44 regulates vascular endothelial barrier integrity via a PECAM-1 dependent mechanism. Angiogenesis 2013, 16, 689–705. [Google Scholar] [CrossRef]
- Stamenkovic, I.; Yu, Q. Merlin, a “magic” linker between the extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival. Curr. Protein Pept. Sci. 2010, 11, 471–484. [Google Scholar] [CrossRef]
- Badouel, C.; McNeill, H. SnapShot: The hippo signaling pathway. Cell 2011, 145, 484–484.e1. [Google Scholar] [CrossRef]
- Tsuneki, M.; Madri, J.A. Adhesion molecule-mediated hippo pathway modulates hemangioendothelioma cell behavior. Mol. Cell. Biol. 2014, 34, 4485–4499. [Google Scholar] [CrossRef]
- Tsuneki, M.; Madri, J.A. CD44 regulation of endothelial cell proliferation and apoptosis via modulation of CD31 and VE-cadherin expression. J. Biol. Chem. 2014, 289, 5357–5370. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Stamenkovic, I.; Yu, Q. CD44 attenuates activation of the hippo signaling pathway and is a prime therapeutic target for glioblastoma. Cancer Res. 2010, 70, 2455–2464. [Google Scholar] [CrossRef]
- Wada, K.-I.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Yamamoto, K.; Agarwala, S.; Terai, K.; Fukui, H.; Fukuhara, S.; Ando, K.; Miyazaki, T.; Yokota, Y.; Schmelzer, E. Flow-dependent endothelial YAP regulation contributes to vessel maintenance. Dev. Cell 2017, 40, 523–536.e6. [Google Scholar] [CrossRef] [PubMed]
- Gegenfurtner, F.A.; Jahn, B.; Wagner, H.; Ziegenhain, C.; Enard, W.; Geistlinger, L.; Rädler, J.O.; Vollmar, A.M.; Zahler, S. Micropatterning as a tool to identify regulatory triggers and kinetics of actin-mediated endothelial mechanosensing. J. Cell Sci. 2018, 131, 212886. [Google Scholar] [CrossRef]
- Dickson, M.C.; Martin, J.S.; Cousins, F.M.; Kulkarni, A.B.; Karlsson, S.; Akhurst, R.J. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 1995, 121, 1845–1854. [Google Scholar]
- Oshima, M.; Oshima, H.; Taketo, M.M. TGF-β receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 1996, 179, 297–302. [Google Scholar] [CrossRef]
- Larsson, J.; Goumans, M.J.; Sjöstrand, L.J.; van Rooijen, M.A.; Ward, D.; Levéen, P.; Xu, X.; ten Dijke, P.; Mummery, C.L.; Karlsson, S. Abnormal angiogenesis but intact hematopoietic potential in TGF-β type I receptor-deficient mice. EMBO J. 2001, 20, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Pefani, D.E.; Pankova, D.; Abraham, A.G.; Grawenda, A.M.; Vlahov, N.; Scrace, S. TGF-β Targets the Hippo Pathway Scaffold RASSF1A to Facilitate YAP/SMAD2 Nuclear Translocation. Mol. Cell 2016, 63, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Cheng, H.; Gao, R.; Mu, C.; Chen, L.; Wu, S.; Chen, Q.; Zhu, Y. Zyxin-Siah2–Lats2 axis mediates cooperation between Hippo and TGF-β signalling pathways. Nat. Commun. 2016, 7, 11123. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Tweedie, E.; Conley, B.; Ames, J.; FitzSimons, M.; Brooks, P.; Liaw, L.; Vary, C.P.H. BMP9 crosstalk with the hippo pathway regulates endothelial cell matricellular and chemokine responses. PLoS ONE 2015, 10, e0122892. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Matkar, P.N.; Quan, A.; Mantella, L.-E.; Teoh, H.; Al-Omran, M.; Verma, S. Investigation of TGFβ1-induced long noncoding RNAs in endothelial cells. Int. J. Vasc. Med. 2016, 2016, 2459687. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Hot, B.; Valnohova, J.; Arthofer, E.; Simon, K.; Shin, J.; Uhlén, M.; Kostenis, E.; Mulder, J.; Schulte, G. FZD10-Gα13 signalling axis points to a role of FZD10 in CNS angiogenesis. Cell. Signal. 2017, 32, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M. Alternative Wnt signaling activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef]
- Min, J.-K.; Park, H.; Choi, H.-J.; Kim, Y.; Pyun, B.-J.; Agrawal, V.; Song, B.-W.; Jeon, J.; Maeng, Y.-S.; Rho, S.-S. The WNT antagonist Dickkopf2 promotes angiogenesis in rodent and human endothelial cells. J. Clin. Investig. 2011, 121, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Podgrabinska, S.; Braun, P.; Velasco, P.; Kloos, B.; Pepper, M.S.; Jackson, D.G.; Skobe, M. Molecular characterization of lymphatic endothelial cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16069–16074. [Google Scholar] [CrossRef] [PubMed]
- Reneman, R.S.; Arts, T.; Hoeks, A.P.G. Wall shear stress–an important determinant of endothelial cell function and structure–in the arterial system in vivo. J. Vasc. Res. 2006, 3, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Diaz, M.F.; Price, K.M.; Ozuna, J.A.; Zhang, S.; Sevick-Muraca, E.M.; Hagan, J.P.; Wenzel, P.L. Fluid shear stress activates YAP1 to promote cancer cell motility. Nat. Commun. 2017, 8, 14122. [Google Scholar] [CrossRef]
- Ivanov, K.I.; Agalarov, Y.; Valmu, L.; Samuilova, O.; Liebl, J.; Houhou, N.; Maby-El Hajjami, H.; Norrmén, C.; Jaquet, M.; Miura, N.; et al. Phosphorylation regulates FOXC2-mediated transcription in lymphatic endothelial cells. Mol. Cell. Biol. 2013, 33, 3749–3761. [Google Scholar] [CrossRef] [PubMed]
- Sabine, A.; Bovay, E.; Demir, C.S.; Kimura, W.; Jaquet, M.; Agalarov, Y.; Zangger, N.; Scallan, J.P.; Graber, W.; Gulpinar, E. FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J. Clin. Investig. 2015, 125, 3861–3877. [Google Scholar] [CrossRef]
- Johnson, N.C.; Dillard, M.E.; Baluk, P.; McDonald, D.M.; Harvey, N.L.; Frase, S.L.; Oliver, G. Lymphatic endothelial cell identity is reversible and its maintenance requires Prox1 activity. Genes Dev. 2008, 22, 3282–3291. [Google Scholar] [CrossRef]
- Cho, H.; Kim, J.; Ahn, J.H.; Hong, Y.-K.; Mäkinen, T.; Lim, D.-S.; Koh, G.Y. YAP and TAZ Negatively Regulate Prox1 During Developmental and Pathologic Lymphangiogenesis. Circ. Res. 2019, 124, 225–242. [Google Scholar] [CrossRef]
- Patan, S. Vasculogenesis and angiogenesis. Cancer Treat. Res. 2004, 117, 3–32. [Google Scholar]
- Morin-Kensicki, E.M.; Boone, B.N.; Howell, M.; Stonebraker, J.R.; Teed, J.; Alb, J.G.; Magnuson, T.R.; O’Neal, W.; Milgram, S.L. Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol. Cell. Biol. 2006, 26, 77–87. [Google Scholar] [CrossRef]
- Nagasawa-Masuda, A.; Terai, K. Yap/Taz transcriptional activity is essential for vascular regression via Ctgf expression and actin polymerization. PLoS ONE 2017, 12, e0174633. [Google Scholar] [CrossRef]
- Brigstock, D.R. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis 2002, 5, 153–165. [Google Scholar] [CrossRef]
- Chaqour, B.; Goppelt-Struebe, M. Mechanical regulation of the Cyr61/CCN1 and CTGF/CCN2 proteins. Fed. Eur. Biochem. Soc. J. 2006, 273, 3639–3649. [Google Scholar] [CrossRef]
- Jiang, W.G.; Watkins, G.; Fodstad, O.; Douglas-Jones, A.; Mokbel, K.; Mansel, R.E. Differential expression of the CCN family members Cyr61, CTGF and Nov in human breast cancer. Endocr. Relat. Cancer 2004, 11, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.; Ho, K.C.; Hao, Y.; Yang, X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011, 71, 2728–2738. [Google Scholar] [CrossRef]
- Neto, F.; Klaus-Bergmann, A.; Ong, Y.T.; Alt, S.; Vion, A.-C.; Szymborska, A.; Carvalho, J.R.; Hollfinger, I.; Bartels-Klein, E.; Franco, C.A. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. eLife 2018, 7, e31037. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-J.; Zhang, H.; Park, H.; Choi, K.-S.; Lee, H.-W.; Agrawal, V.; Kim, Y.-M.; Kwon, Y.-G. Yes-associated protein regulates endothelial cell contact-mediated expression of angiopoietin-2. Nat. Commun. 2015, 6, 6943. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, M.; Fan, J.; Odaka, Y.; Liu, N.; Hassan, A.; Duan, X.; Stump, P.; Byerly, L.; Donaldson, M.; Hao, J. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc. Natl. Acad. Sci. USA 2017, 114, 10918–10923. [Google Scholar] [CrossRef]
- Olsson, A.-K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling? In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669. [Google Scholar] [CrossRef]
- Azad, T.; Nouri, K.; van Rensburg Janse, H.J.; Hao, Y.; Yang, X. Monitoring Hippo Signaling Pathway Activity Using a Luciferase-based Large Tumor Suppressor (LATS) Biosensor. J. Vis. Exp. 2018, E58416. [Google Scholar] [CrossRef]
- Azad, T.; van Rensburg, H.J.J.; Lightbody, E.D.; Neveu, B.; Champagne, A.; Ghaffari, A.; Kay, V.R.; Hao, Y.; Shen, H.; Yeung, B. A LATS biosensor screen identifies VEGFR as a regulator of the Hippo pathway in angiogenesis. Nat. Commun. 2018, 9, 1061. [Google Scholar] [CrossRef]
- Wang, X.; Valls, A.F.; Schermann, G.; Shen, Y.; Moya, I.M.; Castro, L.; Urban, S.; Solecki, G.M.; Winkler, F.; Riedemann, L. YAP/TAZ orchestrate VEGF signaling during developmental angiogenesis. Dev. Cell 2017, 42, 462–478.e7. [Google Scholar] [CrossRef]
- He, J.; Bao, Q.; Zhang, Y.; Liu, M.; Lv, H.; Liu, Y.; Yao, L.; Li, B.; Zhang, C.; He, S. Yes-associated protein promotes angiogenesis via signal transducer and activator of transcription 3 in endothelial cells. Circ. Res. 2018, 122, 591–605. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef]
- Paiva, A.E.; Lousado, L.; Guerra, D.A.; Azevedo, P.O.; Sena, I.F.; Andreotti, J.P.; Santos, G.S.; Gonçalves, R.; Mintz, A.; Birbrair, A. Pericytes in the premetastatic niche. Cancer Res. 2018, 78, 2779–2786. [Google Scholar] [CrossRef]
- Kato, K.; Diéguez-Hurtado, R.; Hong, S.P.; Kato-Azuma, S.; Adams, S.; Stehling, M.; Trappmann, B.; Wrana, J.L.; Koh, G.Y.; Adams, R.H. Pulmonary pericytes regulate lung morphogenesis. Nat. Commun. 2018, 9, 2448. [Google Scholar] [CrossRef]
- Tímár, J.; Döme, B.; Fazekas, K.; Janovics, Á.; Paku, S. Angiogenesis-dependent diseases and angiogenesis therapy. Pathol. Oncol. Res. 2001, 7, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Barreto, S.; Gonzalez-Vazquez, A.; Cameron, A.R.; Cavanagh, B.; Murray, D.J.; O’Brien, F.J. Identification of the mechanisms by which age alters the mechanosensitivity of mesenchymal stromal cells on substrates of differing stiffness: Implications for osteogenesis and angiogenesis. Acta Biomater. 2017, 53, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Schwartz, J.B.; Mustapic, M.; Lobach, I.V.; Daneman, R.; Abner, E.L.; Jicha, G.A. Altered cargo proteins of human plasma endothelial cell–derived exosomes in atherosclerotic cerebrovascular disease. FASEB J. 2017, 31, 3689–3694. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Yuan, W.; Su, Z.; Lin, C.; Huang, T.; Chen, Y.; Wang, J. Yes-associated protein mediates angiotensin II-induced vascular smooth muscle cell phenotypic modulation and hypertensive vascular remodelling. Cell Prolif. 2018, 51, e12517. [Google Scholar] [CrossRef]
- Bharadwaj, A.S.; Appukuttan, B.; Wilmarth, P.A.; Pan, Y.; Stempel, A.J.; Chipps, T.J.; Benedetti, E.E.; Zamora, D.O.; Choi, D.; David, L.L. Role of the retinal vascular endothelial cell in ocular disease. Prog. Retin. Eye Res. 2013, 32, 102–180. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Shi, H.; Zhu, R.; Li, L.; Qin, B.; Kang, L.; Chen, H.; Guan, H. Inhibition of YAP ameliorates choroidal neovascularization via inhibiting endothelial cell proliferation. Mol. Vis. 2018, 24, 83–93. [Google Scholar]
- Kim, J.; Kim, Y.H.; Kim, J.; Bae, H.; Lee, D.-H.; Kim, K.H.; Hong, S.P.; Jang, S.P.; Kubota, Y.; Kwon, Y.-G. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Investig. 2017, 127, 3441–3461. [Google Scholar] [CrossRef]
- Zhu, M.; Liu, X.; Wang, Y.; Chen, L.; Wang, L.; Qin, X.; Xu, J.; Li, L.; Tu, Y.; Zhou, T. YAP via interacting with STAT3 regulates VEGF-induced angiogenesis in human retinal microvascular endothelial cells. Exp. Cell Res. 2018, 373, 155–163. [Google Scholar] [CrossRef]
- Hao, G.-M.; Lv, T.-T.; Wu, Y.; Wang, H.-L.; Xing, W.; Wang, Y.; Li, C.; Zhang, Z.-J.; Wang, Z.-L.; Wang, W. The Hippo signaling pathway: A potential therapeutic target is reversed by a Chinese patent drug in rats with diabetic retinopathy. BMC Complementary Altern. Med. 2017, 17, 187. [Google Scholar] [CrossRef]
- Um, J.; Yu, J.; Park, K.S. Substance P accelerates wound healing in type 2 diabetic mice through endothelial progenitor cell mobilization and Yes-associated protein activation. Mol. Med. Rep. 2017, 15, 3035–3040. [Google Scholar] [CrossRef]
- Xu, X.-M.; Xu, T.-M.; Wei, Y.-B.; Gao, X.-X.; Sun, J.-C.; Wang, Y.; Kong, Q.-J.; Shi, J.-G. Low-Intensity Pulsed Ultrasound Treatment Accelerates Angiogenesis by Activating YAP/TAZ in Human Umbilical Vein Endothelial Cells. Ultrasound Med. Biol. 2018, 44, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Mao, Y.; Luo, W.; Wu, W.; Xu, H.; Wang, X.L.; Shen, Y.H. Palmitic acid dysregulates the Hippo-YAP pathway and inhibits angiogenesis by inducing mitochondrial damage and activating the cytosolic DNA sensor cGAS-STING-IRF3 signaling. J. Biol. Chem. 2017, 292, 15002–15015. [Google Scholar] [CrossRef]
- Mammoto, A.; Muyleart, M.; Kadlec, A.; Gutterman, D.; Mammoto, T. YAP1-TEAD1 signaling controls angiogenesis and mitochondrial biogenesis through PGC1α. Microvasc. Res. 2018, 119, 73–83. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Mao, L.; Xiong, J.; Wen, J.; Wang, Y.; Geng, D.; Liu, Y. TAZ Expression on Endothelial Cells Is Closely Related to Blood Vascular Density and VEGFR2 Expression in Astrocytomas. J. Neuropathol. Exp. Neurol. 2019, 78, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Küffer, S.; Cheung, K.C.P.; Jiang, X.; Trümper, L.; Wulf, G.G.; Ströbel, P. CD31 expression determines redox status and chemoresistance in human angiosarcomas. Clin. Cancer Res. 2018, 24, 460–473. [Google Scholar] [CrossRef]
- Marti, P.; Stein, C.; Blumer, T.; Abraham, Y.; Dill, M.T.; Pikiolek, M.; Orsini, V.; Jurisic, G.; Megel, P.; Makowska, Z. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology 2015, 62, 1497–1510. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Tian, Y.; Zhang, B.; Zhang, X.; Shi, H.; Liang, Z.; Wu, P.; Li, R.; You, B.; Yang, L. YAP signaling in gastric cancer-derived mesenchymal stem cells is critical for its promoting role in cancer progression. Int. J. Oncol. 2017, 51, 1055–1066. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Delgado-Bellido, D.; Serrano-Saenz, S.; Fernández-Cortés, M.; Oliver, F.J. Vasculogenic mimicry signaling revisited: Focus on non-vascular VE-cadherin. Mol. Cancer 2017, 16, 65. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Chao, J.T.; Chien, D.S.; Chu, Y.W. Tumor cell vascular mimicry: Novel targeting opportunity in melanoma. Pharmacol. Ther. 2016, 159, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, M.J.; Seftor, E.A.; Hess, A.R.; Seftor, R.E. Vasculogenic mimicry and tumour-cell plasticity: Lessons from melanoma. Nat. Rev. Cancer 2003, 3, 411. [Google Scholar] [CrossRef]
- Shirakawa, K.; Kobayashi, H.; Heike, Y.; Kawamoto, S.; Brechbiel, M.W.; Kasumi, F.; Iwanaga, T.; Konishi, F.; Terada, M.; Wakasugi, H. Hemodynamics in vasculogenic mimicry and angiogenesis of inflammatory breast cancer xenograft. Cancer Res. 2002, 62, 560–566. [Google Scholar]
- Williamson, S.C.; Metcalf, R.L.; Trapani, F.; Mohan, S.; Antonello, J.; Abbott, B.; Leong, H.S.; Chester, C.P.; Simms, N.; Polanski, R. Vasculogenic mimicry in small cell lung cancer. Nat. Commun. 2016, 7, 13322. [Google Scholar] [CrossRef]
- Sood, A.K.; Seftor, E.A.; Fletcher, M.S.; Gardner, L.M.; Heidger, P.M.; Buller, R.E.; Seftor, R.E.; Hendrix, M.J. Molecular determinants of ovarian cancer plasticity. Am. J. Pathol. 2001, 158, 1279–1288. [Google Scholar] [CrossRef]
- Cai, X.S.; Jia, Y.W.; Mei, J.; Tang, R.Y. Tumor blood vessels formation in osteosarcoma: Vasculogenesis mimicry. Chin. Med. J. 2004, 117, 94–98. [Google Scholar]
- Li, M.; Gu, Y.; Zhang, Z.; Zhang, S.; Zhang, D.; Saleem, A.F.; Zhao, X.; Sun, B. Vasculogenic mimicry: A new prognostic sign of gastric adenocarcinoma. Pathol. Oncol. Res. 2010, 16, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Streeter, E.H.; Harris, A.L. Angiogenesis in bladder cancer—Prognostic marker and target for future therapy. Surg. Oncol. 2002, 11, 85–100. [Google Scholar] [CrossRef]
- Sun, T.; Sun, B.C.; Zhao, X.L.; Zhao, N.; Dong, X.Y.; Che, N.; Yao, Z.; Ma, Y.M.; Gu, Q.; Zong, W.K. Promotion of tumor cell metastasis and vasculogenic mimicry by way of transcription coactivation by Bcl-2 and Twist1: A study of hepatocellular carcinoma. Hepatology 2011, 54, 1690–1706. [Google Scholar] [CrossRef] [PubMed]
- Baeten, C.I.; Hillen, F.; Pauwels, P.; de Bruine, A.P.; Baeten, C.G. Prognostic role of vasculogenic mimicry in colorectal cancer. Dis. Colon Rectum 2009, 52, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Bora-Singhal, N.; Nguyen, J.; Schaal, C.; Perumal, D.; Singh, S.; Coppola, D.; Chellappan, S. YAP1 regulates OCT4 activity and SOX2 expression to facilitate self-renewal and vascular mimicry of stem-like cells. Stem Cells 2015, 33, 1705–1718. [Google Scholar] [CrossRef]
- Wei, H.; Wang, F.; Wang, Y.; Li, T.; Xiu, P.; Zhong, J.; Sun, X.; Li, J. Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex. Cancer Sci. 2017, 108, 478–487. [Google Scholar] [CrossRef]
- van Rensburg, H.J.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo pathway component TAZ promotes immune evasion in human cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201–207. [Google Scholar] [CrossRef]
- Zhao, Y.; Adjei, A.A. Targeting angiogenesis in cancer therapy: Moving beyond vascular endothelial growth factor. Oncologist 2015, 20, 660–673. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Main Finding | Ref. |
|---|---|
| Angiomotin is a negative regulator of YAP | [44] |
| Angiomotin-like 1 degradation by Nedd4 is regulated by YAP through c-ABL | [60] |
| YAP/TAZ are the main mediators of mechanotransduction in endothelial cells | [72] |
| BMP9 crosstalk with the Hippo pathway regulates endothelial cell matricellular response | [80] |
| mRNAs upregulated in ECs in response to TGFβ1 treatment are involved in hippo signaling | [81] |
| Flow-dependent endothelial YAP regulation contributes to vessel maintenance | [73] |
| YAP/TAZ negatively regulate prox1 during developmental and pathologic lymphangiogenesis | [92] |
| YAP disruption in mice causes defects in yolk sac vasculogenesis and chorioallantoic fusion | [94] |
| YAP/TAZ activity is essential for vascular regression via Ctgf and actin polymerization | [95] |
| Adherens junction and endothelial cell distribution in angiogenesis is regulated by YAP/TAZ | [100] |
| YAP regulates angiopoietin-2 expression in ECs | [101] |
| Vascular tip cell migration is regulated by YAP/TAZ-CDC42 signaling pathway | [102] |
| VEGFR is a regulator of YAP/TAZ in the Hippo pathway in angiogenesis through PI3K/MAPK pathways | [106] |
| VEGF activates YAP/TAZ via its effects on actin cytoskeleton | [107] |
| YAP promotes angiogenesis via Stat3 | [108] |
| YAP mediates angiotensin II-induced vascular smooth muscle cell phenotypic modulation and hypertensive vascular remodelling | [115] |
| YAP inhibition ameliorates choroidal neovascularization | [117] |
| YAP/TAZ regulates vascular barrier maturation | [118] |
| YAP via interacting with STAT3 regulates VEGF-induced angiogenesis in retina | [119] |
| Substance P accelerates wound healing in type 2 diabetic mice through YAP activation | [121] |
| Ultrasound treatment accelerates angiogenesis by activating YAP/TAZ | [122] |
| Palmitic acid inhibits angiogenesis through YAP suppression | [123] |
| YAP1-TEAD1 controls angiogenesis and mitochondrial biogenesis through PGC1α. | [124] |
| Blood vascular density and VEGFR2 expression in astrocytomas is regulated by TAZ | [126] |
| Cell proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma is regulated by YAP | [128] |
| YAP regulates OCT4 activity and SOX2 expression to facilitate vascular mimicry | [142] |
| Verteporfin suppresses vasculogenic mimicry of pancreatic ductal adenocarcinoma | [143] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azad, T.; Ghahremani, M.; Yang, X. The Role of YAP and TAZ in Angiogenesis and Vascular Mimicry. Cells 2019, 8, 407. https://doi.org/10.3390/cells8050407
Azad T, Ghahremani M, Yang X. The Role of YAP and TAZ in Angiogenesis and Vascular Mimicry. Cells. 2019; 8(5):407. https://doi.org/10.3390/cells8050407
Chicago/Turabian StyleAzad, Taha, Mina Ghahremani, and Xiaolong Yang. 2019. "The Role of YAP and TAZ in Angiogenesis and Vascular Mimicry" Cells 8, no. 5: 407. https://doi.org/10.3390/cells8050407
APA StyleAzad, T., Ghahremani, M., & Yang, X. (2019). The Role of YAP and TAZ in Angiogenesis and Vascular Mimicry. Cells, 8(5), 407. https://doi.org/10.3390/cells8050407