2-Oxonanonoidal Antibiotic Actinolactomycin Inhibits Cancer Progression by Suppressing HIF-1α

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture, Cell Proliferation, Apoptosis Assay and Hypoxia Condition
2.2. Luciferase Assay
2.3. Western Blot and Immunoprecipitation
2.4. Pulse-Labeling
2.5. Real Time-PCR
2.6. Animal Experiment
2.7. Statistical Analysis
3. Results
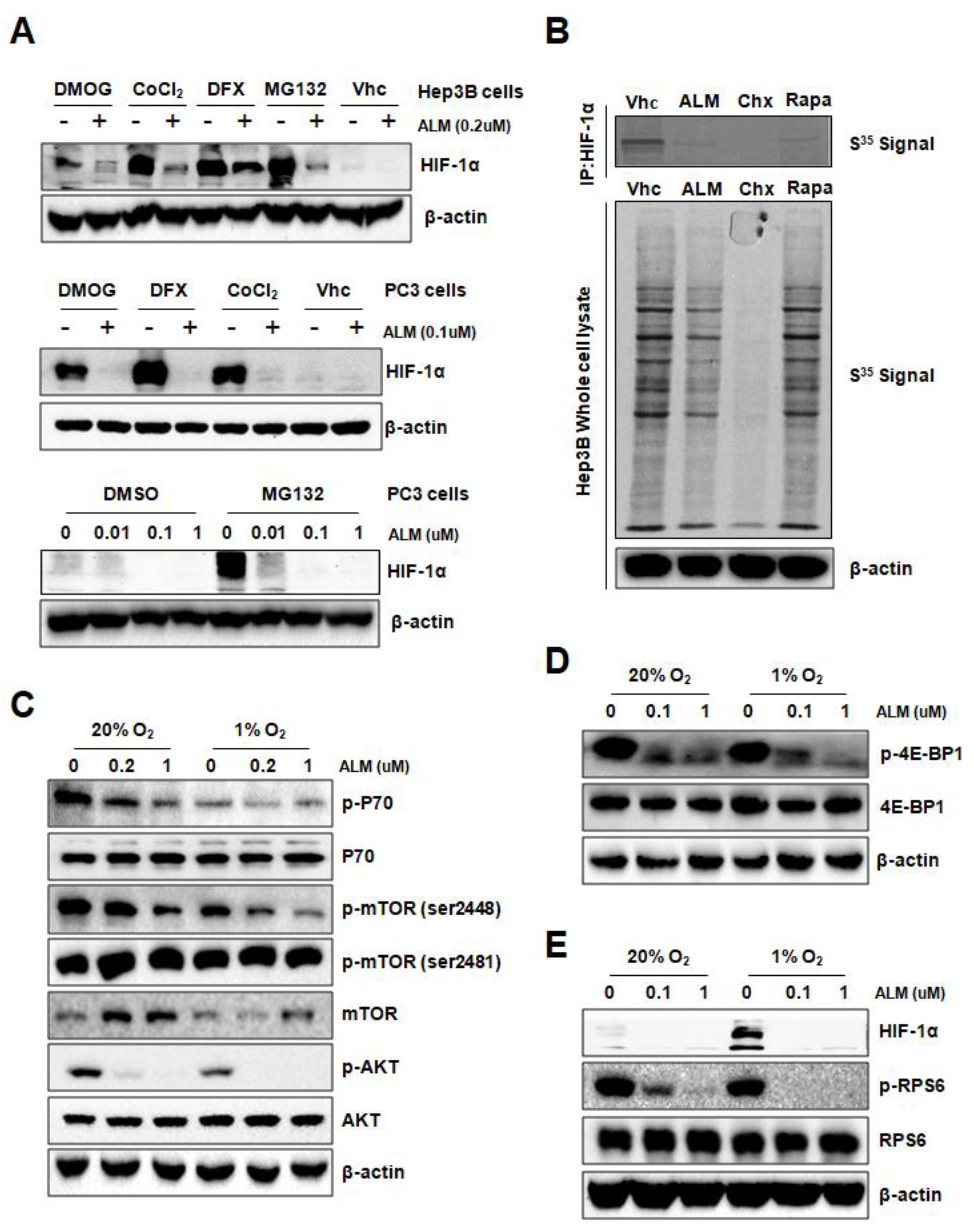
3.1. ALM Inhibits HIF-1α Transactivity and Protein Expression
3.2. ALM Inhibits HIF-1α Translation by Down-Regulating AKT and mTOR Activity
3.3. ALM Induced HIF-1α-Dependent Growth Inhibition and Apoptosis in PC3 Cells
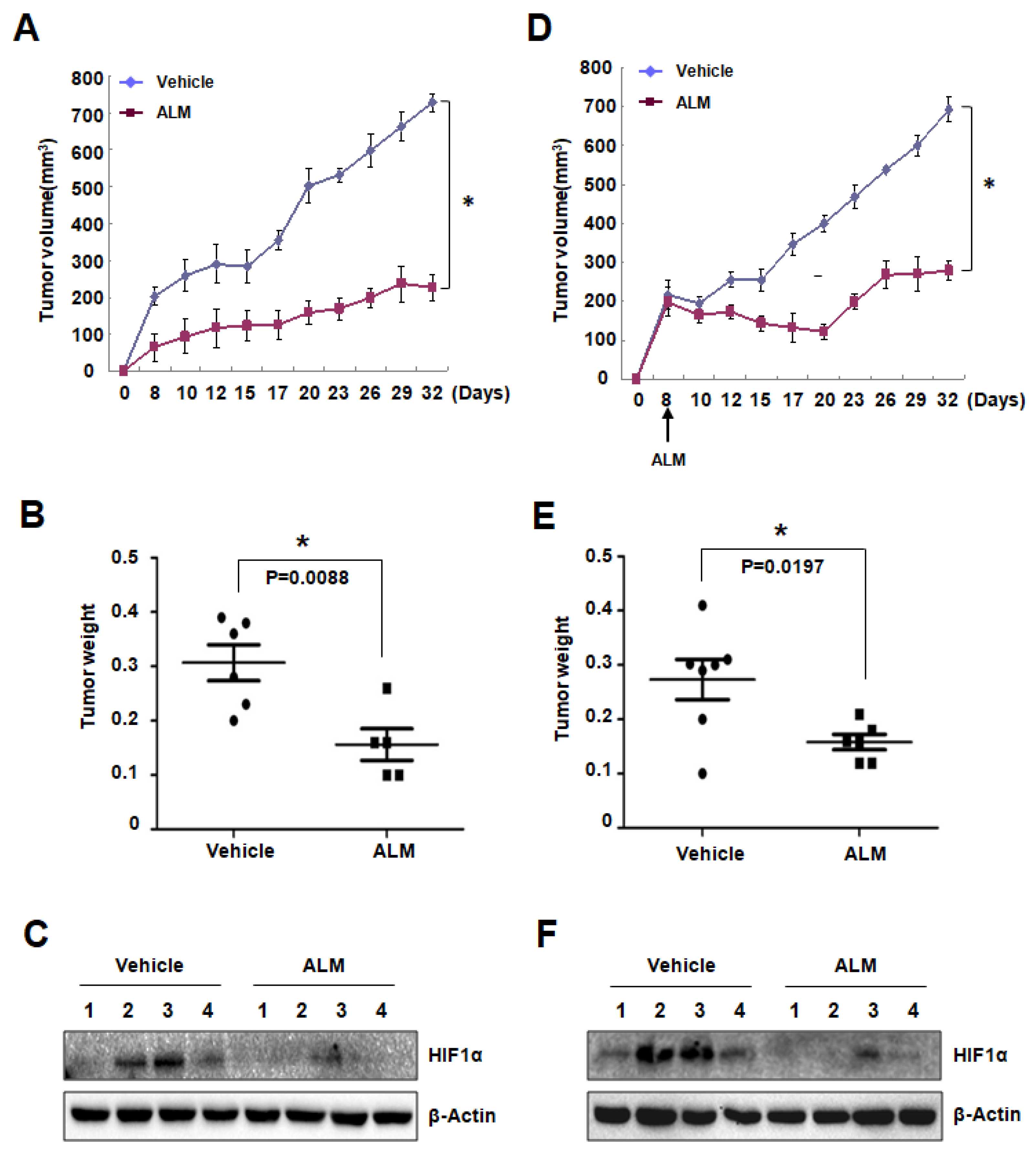
3.4. ALM Treatment Inhibited PC3 Xenograft Growth
3.5. Low Dose of ALM Enhances the Inhibitory Effect of mTOR Inhibitors on Cell Growth
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Semenza, G.L. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Et Biophys. Acta 2016, 1863, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Antiangiogenic therapy and tumor progression. Cancer Cell 2004, 5, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V. Hypoxia-inducible factor: Achilles’ heel of antiangiogenic cancer therapy (review). Int. J. Oncol. 2001, 19, 257–262. [Google Scholar] [CrossRef]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef]
- Koshiji, M.; To, K.K.; Hammer, S.; Kumamoto, K.; Harris, A.L.; Modrich, P.; Huang, L.E. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol. Cell 2005, 17, 793–803. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M.; et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017, 4, 19–24. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Et Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Giaccia, A.; Siim, B.G.; Johnson, R.S. HIF-1 as a target for drug development. Nat. Rev. Drug Discov. 2003, 2, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Wigerup, C.; Pahlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [Green Version]
- Guillemin, K.; Krasnow, M.A. The hypoxic response: Huffing and HIFing. Cell 1997, 89, 9–12. [Google Scholar] [CrossRef]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef]
- Bunn, H.F.; Poyton, R.O. Oxygen sensing and molecular adaptation to hypoxia. Physiol. Rev. 1996, 76, 839–885. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. The sweet side of HIF. Kidney Int. 2010, 78, 10–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef]
- Semenza, G.L. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov. Today 2007, 12, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.S.; Martelli, A.M.; Cocco, L.; Libra, M.; Nicoletti, F.; Abrams, S.L.; McCubrey, J.A. The therapeutic potential of mTOR inhibitors in breast cancer. Br. J. Clin. Pharmacol. 2016, 82, 1189–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobday, T.J.; Qin, R.; Reidy-Lagunes, D.; Moore, M.J.; Strosberg, J.; Kaubisch, A.; Shah, M.; Kindler, H.L.; Lenz, H.J.; Chen, H.; et al. Multicenter Phase II Trial of Temsirolimus and Bevacizumab in Pancreatic Neuroendocrine Tumors. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.L.; Segelov, E.; Singh, S. Everolimus in the management of metastatic neuroendocrine tumours. Ther. Adv. Gastroenterol. 2017, 10, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, S.S.; Case, S. Everolimus (RAD001) in the treatment of advanced renal cell carcinoma: A review. Oncol. 2010, 15, 236–245. [Google Scholar] [CrossRef]
- Krueger, D.A.; Care, M.M.; Holland, K.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.A.; Byars, A.; Sahmoud, T.; Franz, D.N. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. New Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Franz, D.N.; Agricola, K.; Mays, M.; Tudor, C.; Care, M.M.; Holland-Bouley, K.; Berkowitz, N.; Miao, S.; Peyrard, S.; Krueger, D.A. Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann. Neurol. 2015, 78, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Ito, T.; Jensen, R.T. Everolimus in the treatment of neuroendocrine tumors: Efficacy, side-effects, resistance, and factors affecting its place in the treatment sequence. Expert Opin. Pharmacother. 2018, 19, 909–928. [Google Scholar] [CrossRef] [PubMed]
- Morviducci, L.; Rota, F.; Rizza, L.; Di Giacinto, P.; Ramponi, S.; Nardone, M.R.; Tubili, C.; Lenzi, A.; Zuppi, P.; Baldelli, R. Everolimus is a new anti-cancer molecule: Metabolic side effects as lipid disorders and hyperglycemia. Diabetes Res. Clin. Pract. 2018, 143, 428–431. [Google Scholar] [CrossRef]
- Houde, V.P.; Brule, S.; Festuccia, W.T.; Blanchard, P.G.; Bellmann, K.; Deshaies, Y.; Marette, A. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes 2010, 59, 1338–1348. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, J.; Hu, L.; Yang, Z.; Suo, C.; Wang, Y.J.; Gao, P.; Cui, C.; Sun, L. 2-Oxonanonoidal Antibiotic Actinolactomycin Inhibits Cancer Progression by Suppressing HIF-1α. Cells 2019, 8, 439. https://doi.org/10.3390/cells8050439
Cheng J, Hu L, Yang Z, Suo C, Wang YJ, Gao P, Cui C, Sun L. 2-Oxonanonoidal Antibiotic Actinolactomycin Inhibits Cancer Progression by Suppressing HIF-1α. Cells. 2019; 8(5):439. https://doi.org/10.3390/cells8050439
Chicago/Turabian StyleCheng, Jiadong, Lan Hu, Zheng Yang, Caixia Suo, Yueyang Jack Wang, Ping Gao, Chengbin Cui, and Linchong Sun. 2019. "2-Oxonanonoidal Antibiotic Actinolactomycin Inhibits Cancer Progression by Suppressing HIF-1α" Cells 8, no. 5: 439. https://doi.org/10.3390/cells8050439
APA StyleCheng, J., Hu, L., Yang, Z., Suo, C., Wang, Y. J., Gao, P., Cui, C., & Sun, L. (2019). 2-Oxonanonoidal Antibiotic Actinolactomycin Inhibits Cancer Progression by Suppressing HIF-1α. Cells, 8(5), 439. https://doi.org/10.3390/cells8050439