Involvement of E3 Ligases and Deubiquitinases in the Control of HIF-α Subunit Abundance


Abstract
:1. Introduction
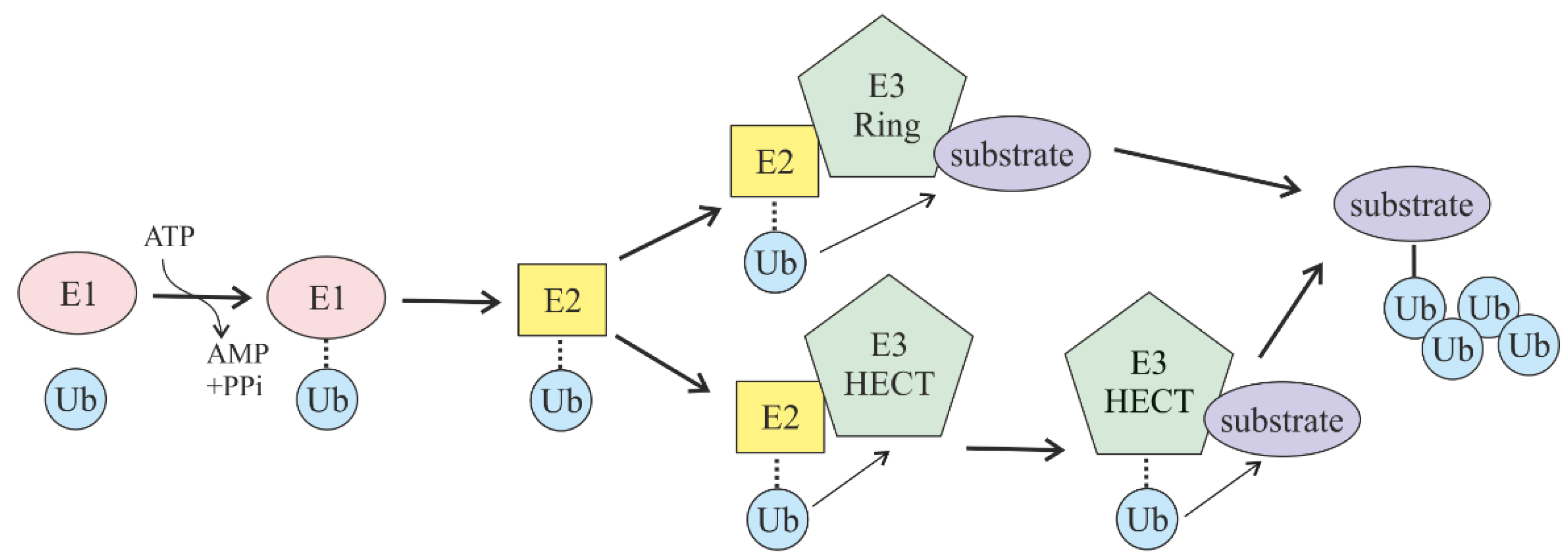
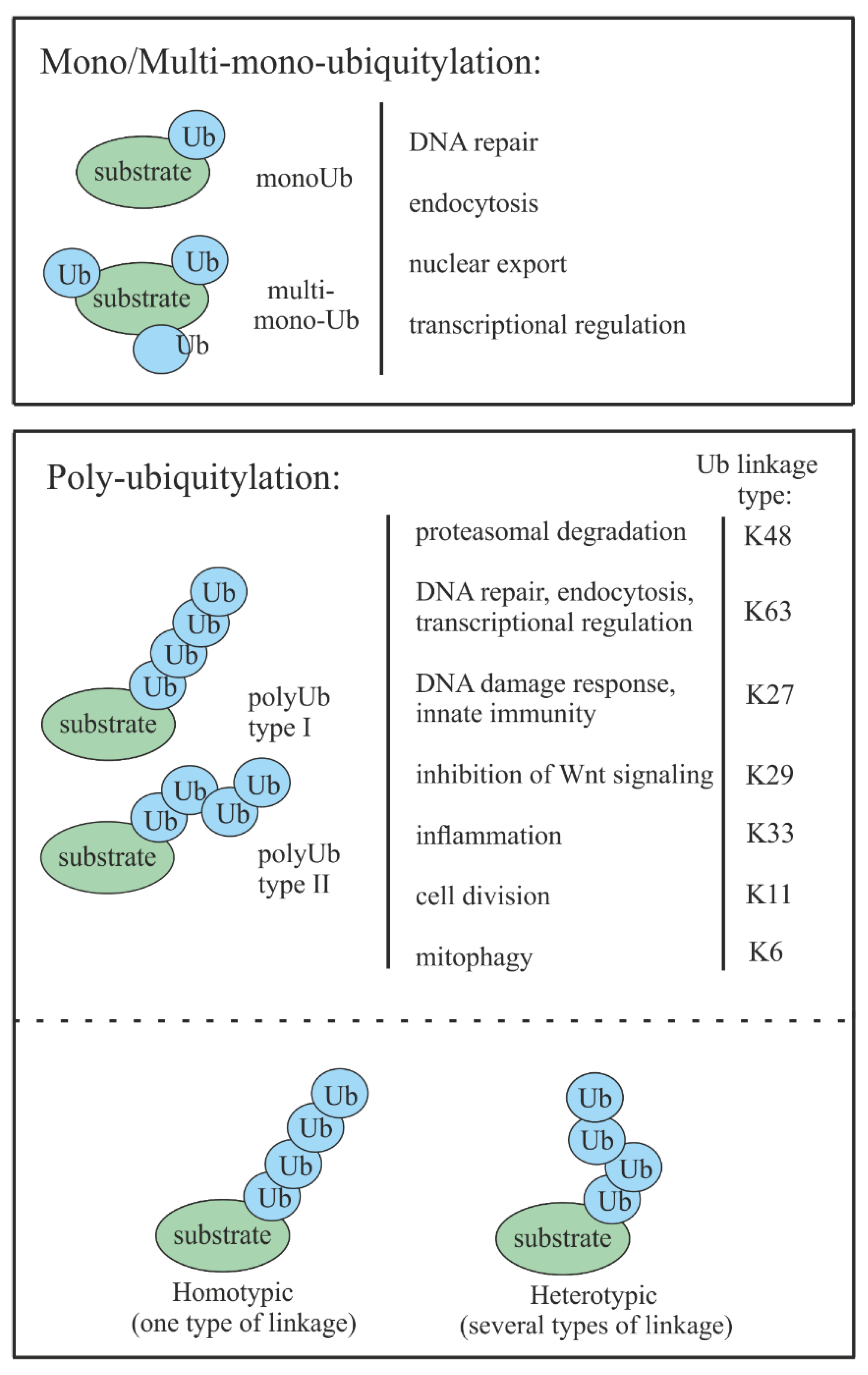
2. Ubiquitin-Mediated Proteasomal Degradation
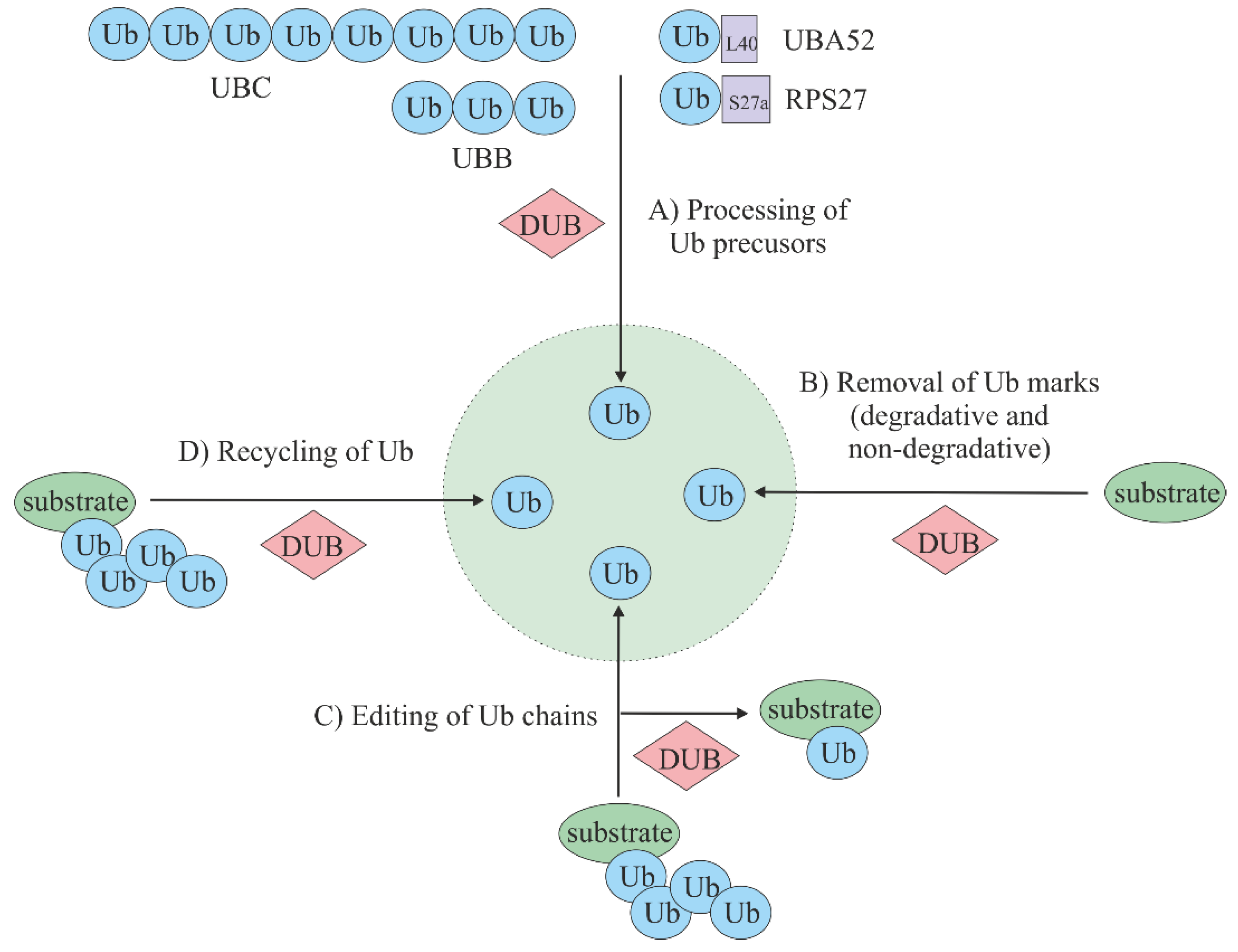
3. Deubiquitinating Enzymes (DUBs)
4. Hypoxia and Cancer
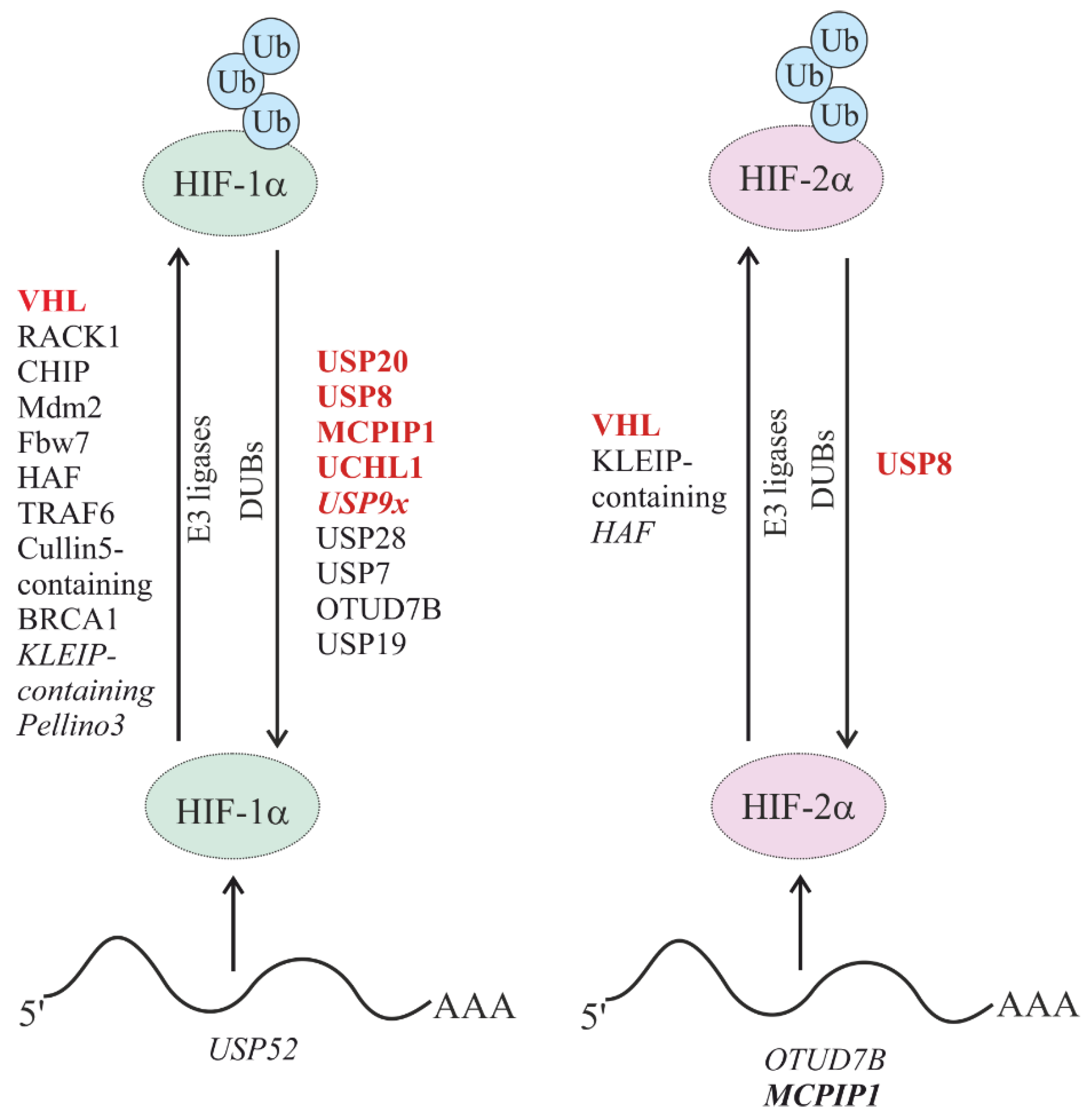
5. Degradation of HIFs
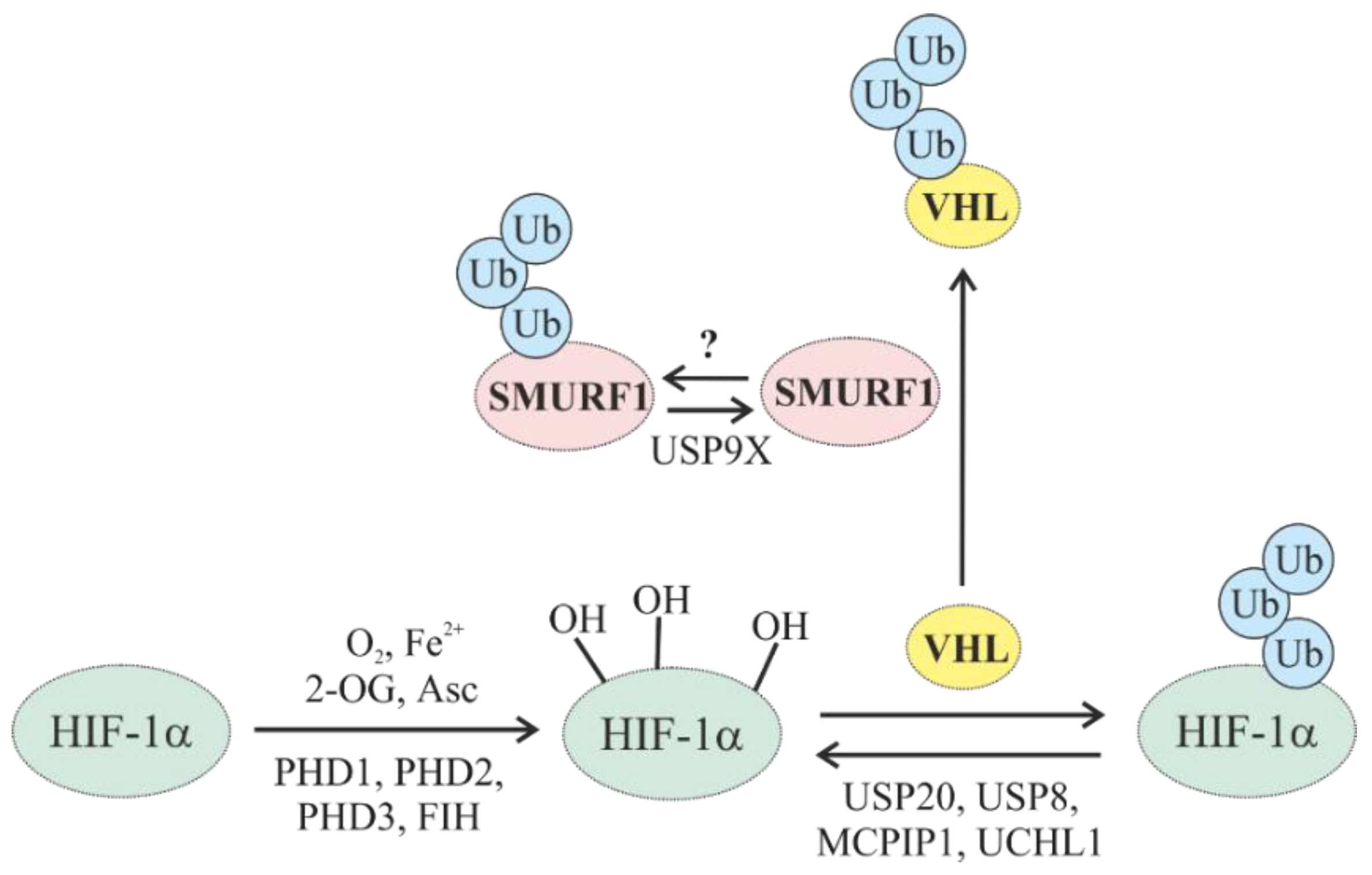
5.1. Oxygen-Dependent Degradation of HIFs
5.1.1. Oxygen-Dependent Degradation of HIF by E3 Ub Ligases
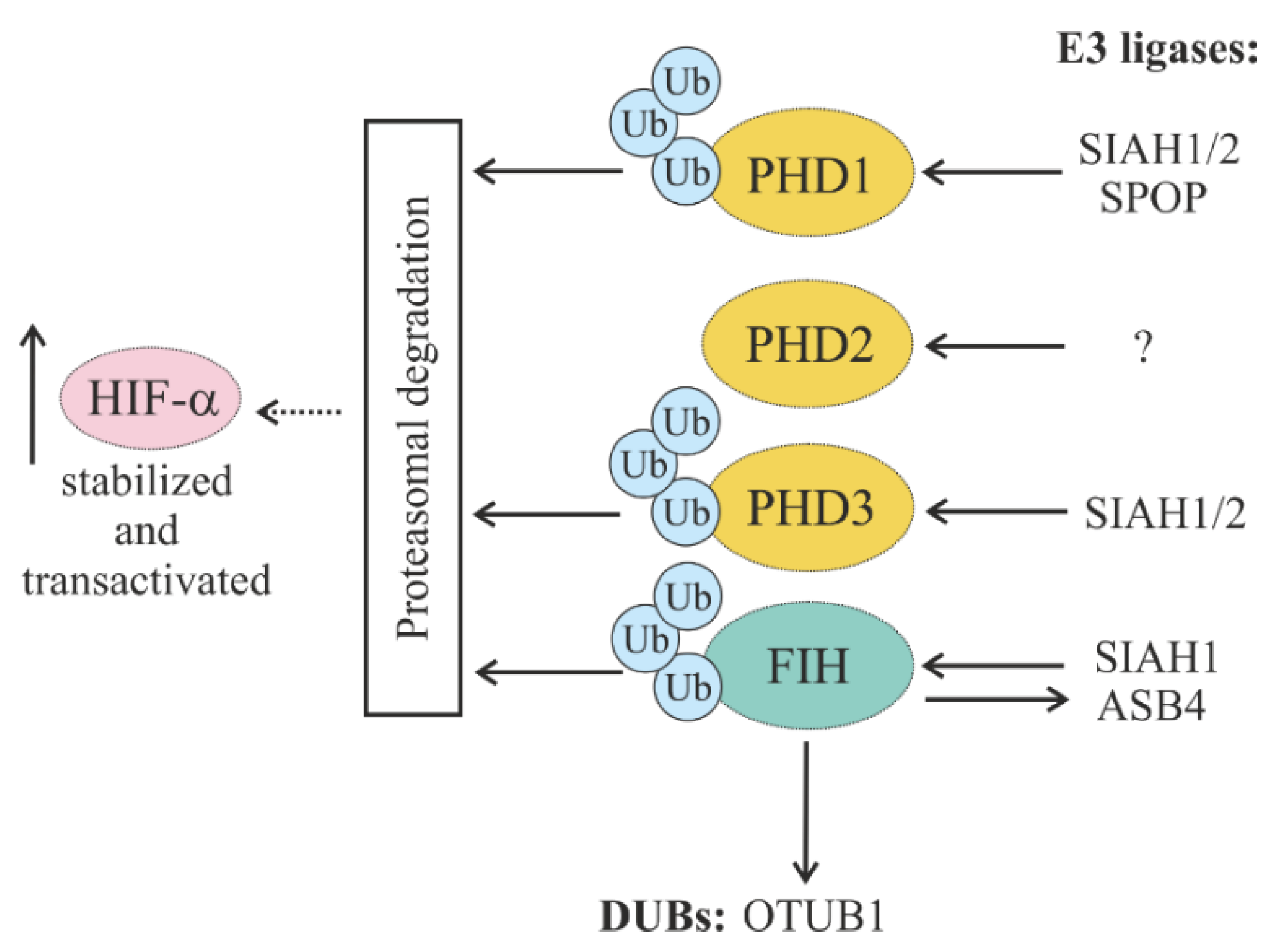
5.1.2. Regulation of HIF Hydroxylases (PHDs and FIH) through Ubiquitylation
5.1.3. Regulation of Oxygen-Dependent HIF Degradation by DUBs
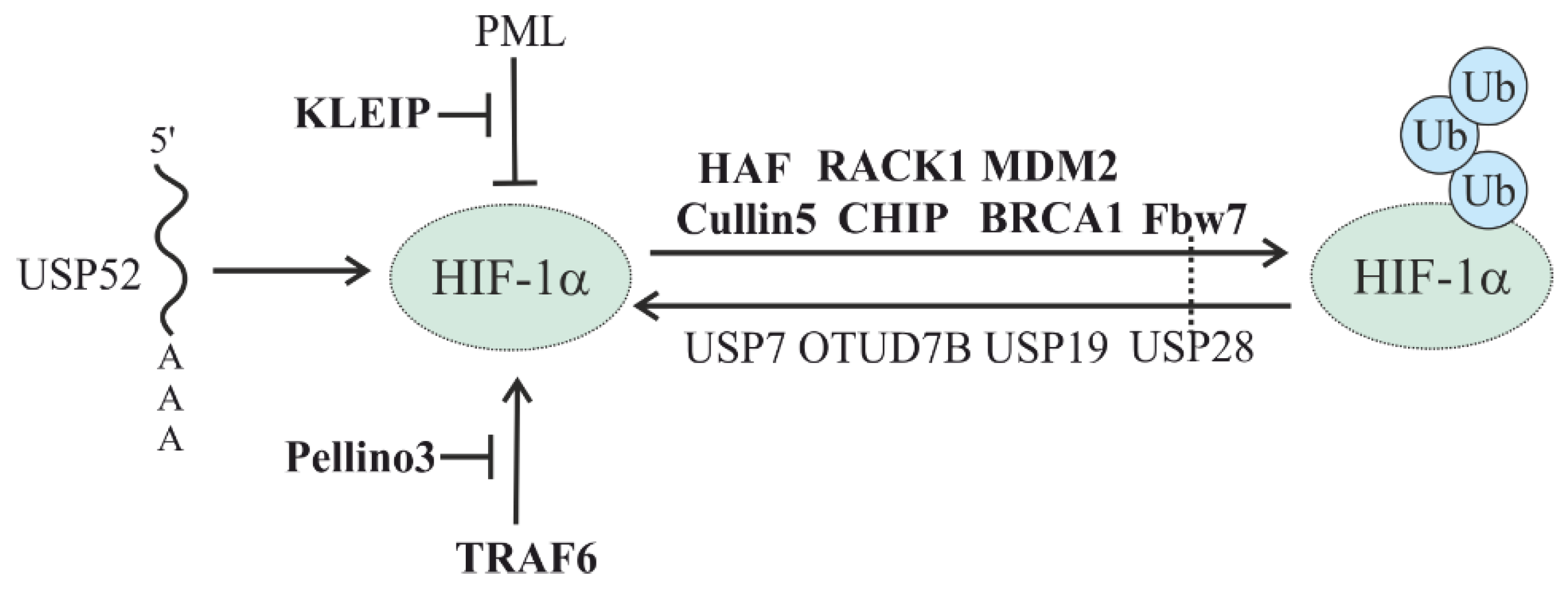
5.2. Oxygen-Independent Regulation of HIFs
5.2.1. HSP-Mediated HIF Degradation
5.2.2. Other E3 Ligases in the Oxygen-Independent Degradation of HIF
5.2.3. Oxygen-Independent Regulation of HIF Stability by DUBs
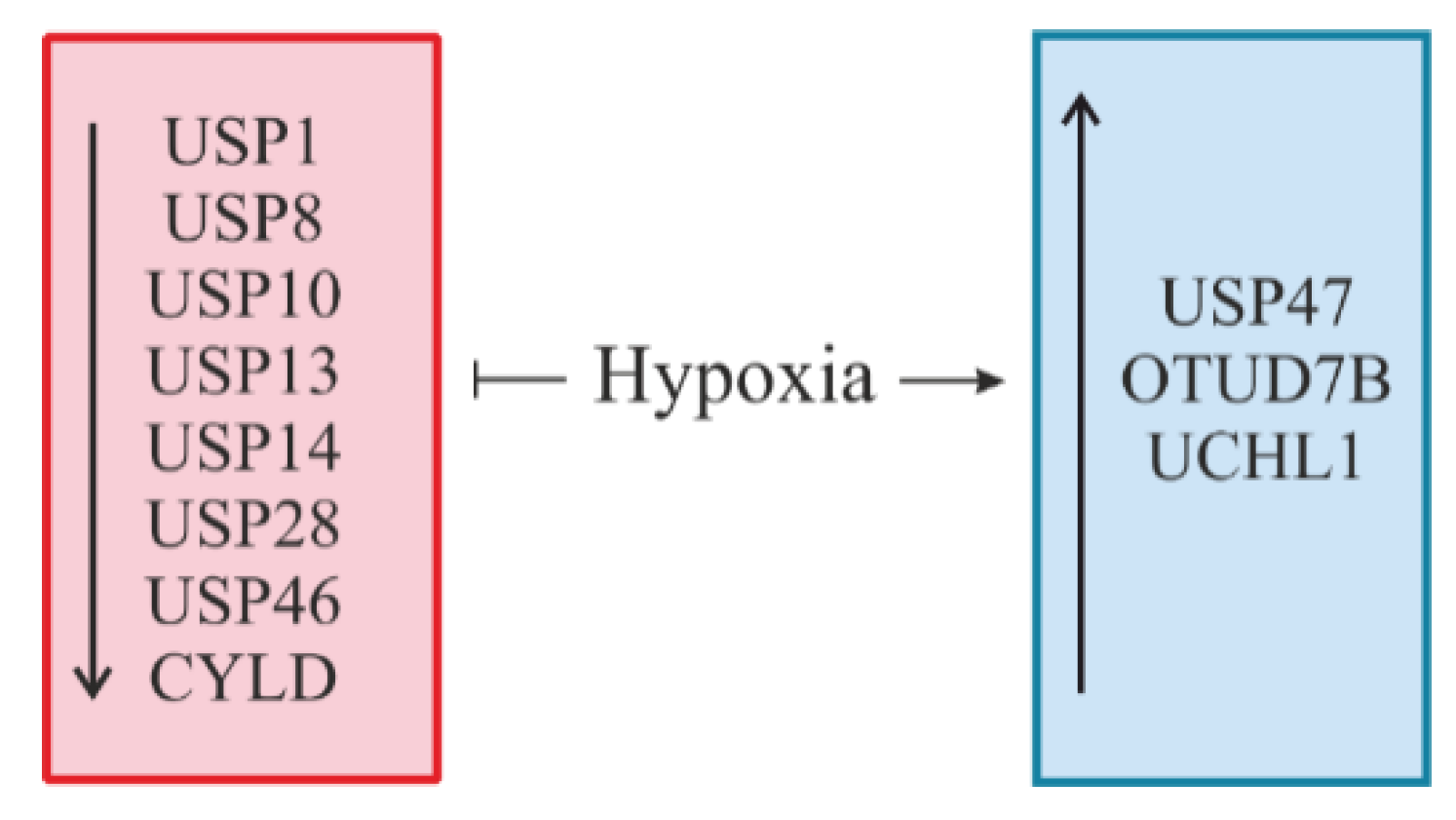
6. Hypoxia: A Novel Regulator of DUBs
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Best Pract. Res. Clin. Haematol. 2017, 30, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Holkova, B.; Grant, S. Proteasome inhibitors in mantle cell lymphoma. Best Pract. Res. Clin. Haematol. 2012, 25, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; Richardson, P.G.; Cavo, M.; Orlowski, R.Z.; San Miguel, J.F.; Palumbo, A.; Harousseau, J.L. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012, 120, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Ramakrishna, S. Deubiquitylation of deubiquitylases. Open Biol. 2017, 7, 170016. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- D’Arcy, P.; Wang, X.; Linder, S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol. Ther. 2015, 147, 32–54. [Google Scholar] [CrossRef] [Green Version]
- De Bie, P.; Ciechanover, A. Ubiquitination of E3 ligases: Self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death Differ. 2011, 18, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.D.; Hicke, L. Non-traditional functions of ubiquitin and ubiquitin-binding proteins. J. Biol. Chem. 2003, 278, 35857–35860. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Haglund, K.; Dikic, I. Ubiquitylation and cell signaling. EMBO J. 2005, 24, 3353–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haglund, K.; Sigismund, S.; Polo, S.; Szymkiewicz, I.; Di Fiore, P.P.; Dikic, I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat. Cell Biol. 2003, 5, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Kirkpatrick, D.; Jiang, X.; Gygi, S.; Sorkin, A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol. Cell 2006, 21, 737–748. [Google Scholar] [CrossRef]
- Pickart, C.M. Ubiquitin in chains. Trends Biochem. Sci. 2000, 25, 544–548. [Google Scholar] [CrossRef]
- Woelk, T.; Sigismund, S.; Penengo, L.; Polo, S. The ubiquitination code: A signalling problem. Cell Div. 2007, 2, 11. [Google Scholar] [CrossRef]
- Cao, J.; Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol. 2012, 2, 26. [Google Scholar] [CrossRef]
- Bach, I.; Ostendorff, H.P. Orchestrating nuclear functions: Ubiquitin sets the rhythm. Trends Biochem. Sci. 2003, 28, 189–195. [Google Scholar] [CrossRef]
- Huang, T.T.; D’Andrea, A.D. Regulation of DNA repair by ubiquitylation. Nat. Rev. Mol. Cell Biol. 2006, 7, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jeon, M.S.; Liao, L.; Yang, C.; Elly, C.; Yates, J.R., 3rd; Liu, Y.C. K33-linked polyubiquitination of T cell receptor-zeta regulates proteolysis-independent T cell signaling. Immunity 2010, 33, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Wickliffe, K.E.; Williamson, A.; Meyer, H.J.; Kelly, A.; Rape, M. K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol. 2011, 21, 656–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durcan, T.M.; Tang, M.Y.; Perusse, J.R.; Dashti, E.A.; Aguileta, M.A.; McLelland, G.L.; Gros, P.; Shaler, T.A.; Faubert, D.; Coulombe, B.; et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014, 33, 2473–2491. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Fraile, J.M.; Quesada, V.; Rodriguez, D.; Freije, J.M.; Lopez-Otin, C. Deubiquitinases in cancer: New functions and therapeutic options. Oncogene 2012, 31, 2373–2388. [Google Scholar] [CrossRef] [PubMed]
- Abdul Rehman, S.A.; Kristariyanto, Y.A.; Choi, S.Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- Heideker, J.; Wertz, I.E. DUBs, the regulation of cell identity and disease. Biochem. J. 2015, 465, 1–26. [Google Scholar] [CrossRef]
- Tian, Z.; D’Arcy, P.; Wang, X.; Ray, A.; Tai, Y.T.; Hu, Y.; Carrasco, R.D.; Richardson, P.; Linder, S.; Chauhan, D.; et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014, 123, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Poondla, N.; Chandrasekaran, A.P.; Kim, K.S.; Ramakrishna, S. Deubiquitinating enzymes as cancer biomarkers: New therapeutic opportunities? BMB Rep. 2019, 52, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Exp. 1998, 7, 205–213. [Google Scholar]
- Bardos, J.I.; Ashcroft, M. Hypoxia-inducible factor-1 and oncogenic signalling. Bioessays 2004, 26, 262–269. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Hirota, K.; Semenza, G.L. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit. Rev. Oncol. Hematol. 2006, 59, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Wigerup, C.; Pahlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diebold, I.; Djordjevic, T.; Hess, J.; Görlach, A. Rac-1 promotes pulmonary artery smooth muscle cell proliferation by upregulation of plasminogen activator inhibitor-1: Role of NFκB-dependent hypoxia-inducible factor-1α transcription. Thromb. Haemost. 2008, 100, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Hu, H.; Kshitiz; Ahmed, I.; Levchenko, A.; Semenza, G.L. Chaperone-mediated autophagy targets hypoxia-inducible factor-1alpha (HIF-1alpha) for lysosomal degradation. J. Biol. Chem. 2013, 288, 10703–10714. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kaelin, W.G., Jr. The EGLN-HIF O2-Sensing System: Multiple Inputs and Feedbacks. Mol. Cell 2017, 66, 772–779. [Google Scholar] [CrossRef]
- Robinson, C.M.; Ohh, M. The multifaceted von Hippel-Lindau tumour suppressor protein. FEBS Lett. 2014, 588, 2704–2711. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain: A hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Zhang, C.; Peng, Z.; Zhu, M.; Wang, P.; Du, X.; Li, X.; Liu, Y.; Jin, Y.; McNutt, M.A.; Yin, Y. USP9X destabilizes pVHL and promotes cell proliferation. Oncotarget 2016, 7, 60519–60534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.C.; Yang, H.; Fribourgh, J.L.; Wolfe, L.S.; Xiong, Y. Insights into Cullin-RING E3 ubiquitin ligase recruitment: Structure of the VHL-EloBC-Cul2 complex. Structure 2015, 23, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Kruizinga, R.C.; van Marion, D.M.; den Dunnen, W.F.; de Groot, J.C.; Hoving, E.W.; Oosting, S.F.; Timmer-Bosscha, H.; Derks, R.P.; Cornelissen, C.; van der Luijt, R.B.; et al. Difference in CXCR4 expression between sporadic and VHL-related hemangioblastoma. Fam. Cancer 2016, 15, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aufforth, R.D.; Ramakant, P.; Sadowski, S.M.; Mehta, A.; Trebska-McGowan, K.; Nilubol, N.; Pacak, K.; Kebebew, E. Pheochromocytoma Screening Initiation and Frequency in von Hippel-Lindau Syndrome. J. Clin. Endocrinol. Metab. 2015, 100, 4498–4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanharanta, S.; Shu, W.; Brenet, F.; Hakimi, A.A.; Heguy, A.; Viale, A.; Reuter, V.E.; Hsieh, J.J.; Scandura, J.M.; Massague, J. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat. Med. 2013, 19, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Na, X.; Wu, G.; Ryan, C.K.; Schoen, S.R.; di’Santagnese, P.A.; Messing, E.M. Overproduction of vascular endothelial growth factor related to von Hippel-Lindau tumor suppressor gene mutations and hypoxia-inducible factor-1 alpha expression in renal cell carcinomas. J. Urol. 2003, 170, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Krieg, M.; Marti, H.H.; Plate, K.H. Coexpression of erythropoietin and vascular endothelial growth factor in nervous system tumors associated with von Hippel-Lindau tumor suppressor gene loss of function. Blood 1998, 92, 3388–3393. [Google Scholar]
- Barth, S.; Nesper, J.; Hasgall, P.A.; Wirthner, R.; Nytko, K.J.; Edlich, F.; Katschinski, D.M.; Stiehl, D.P.; Wenger, R.H.; Camenisch, G. The peptidyl prolyl cis/trans isomerase FKBP38 determines hypoxia-inducible transcription factor prolyl-4-hydroxylase PHD2 protein stability. Mol. Cell Biol. 2007, 27, 3758–3768. [Google Scholar] [CrossRef]
- Nakayama, K.; Ronai, Z. Siah: New players in the cellular response to hypoxia. Cell Cycle 2004, 3, 1345–1347. [Google Scholar] [CrossRef]
- Fukuba, H.; Takahashi, T.; Jin, H.G.; Kohriyama, T.; Matsumoto, M. Abundance of aspargynyl-hydroxylase FIH is regulated by Siah-1 under normoxic conditions. Neurosci. Lett. 2008, 433, 209–214. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, S.; Dai, X.; Gan, W.; Nie, X.; Wei, W.; Hu, G.; Guo, J. Tumor suppressor SPOP ubiquitinates and degrades EglN2 to compromise growth of prostate cancer cells. Cancer Lett. 2017, 390, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, J.E., III; Wu, Y.; Smith, K.; Charles, P.; Powers, K.; Wang, H.; Patterson, C. ASB4 is a hydroxylation substrate of FIH and promotes vascular differentiation via an oxygen-dependent mechanism. Mol. Cell Biol. 2007, 27, 6407–6419. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, D.; Messing, E.M.; Wu, G. VHL protein-interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1alpha. EMBO Rep. 2005, 6, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Azenha, D.; Lopes, M.C.; Martins, T.C. Claspin functions in cell homeostasis-A link to cancer? DNA Repair 2017, 59, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, C.; Ji, J.; Jiang, J.; Shi, M.; Cai, Q.; Yu, Y.; Zhu, Z.; Zhang, J. Deubiquitinating enzyme USP20 is a positive regulator of Claspin and suppresses the malignant characteristics of gastric cancer cells. Int. J. Oncol. 2017, 50, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Luo, K.; Deng, M.; Li, Y.; Yin, P.; Gao, B.; Fang, Y.; Wu, P.; Liu, T.; Lou, Z. HERC2-USP20 axis regulates DNA damage checkpoint through Claspin. Nucleic Acids Res. 2014, 42, 13110–13121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Luo, K.; Zhao, F.; Yin, P.; Song, Y.; Deng, M.; Huang, J.; Chen, Y.; Li, L.; Lee, S.; et al. USP20 positively regulates tumorigenesis and chemoresistance through beta-catenin stabilization. Cell Death Differ. 2018, 25, 1855–1869. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Zhang, M.; Saad, Y.; Kolattukudy, P.E. Antidicer RNAse activity of monocyte chemotactic protein-induced protein-1 is critical for inducing angiogenesis. Am. J. Physiol. Cell Physiol. 2013, 305, C1021–C1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Lu, Y.X.; Cheng, D.; Zhang, K.; Zheng, J.; Liu, Y.; Wang, X.; Yuan, Y.F.; Tang, Y.D. Monocyte Chemoattractant Protein-Induced Protein 1 Targets Hypoxia-Inducible Factor 1alpha to Protect Against Hepatic Ischemia/Reperfusion Injury. Hepatology 2018, 68, 2359–2375. [Google Scholar] [CrossRef] [PubMed]
- Ligeza, J.; Marona, P.; Gach, N.; Lipert, B.; Miekus, K.; Wilk, W.; Jaszczynski, J.; Stelmach, A.; Loboda, A.; Dulak, J.; et al. MCPIP1 contributes to clear cell renal cell carcinomas development. Angiogenesis 2017, 20, 325–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Ning, H.; Gu, L.; Peng, H.; Wang, Q.; Hou, R.; Fu, M.; Hoft, D.F.; Liu, J. MCPIP1 Selectively Destabilizes Transcripts Associated with an Antiapoptotic Gene Expression Program in Breast Cancer Cells That Can Elicit Complete Tumor Regression. Cancer Res. 2016, 76, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Troilo, A.; Alexander, I.; Muehl, S.; Jaramillo, D.; Knobeloch, K.; Krek, W. HIF1α deubiquitination by USP8 is essential for ciliogenesis in normoxia. EMBO Rep. 2014, 15, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Alwan, H.A.; van Leeuwen, J.E. UBPY-mediated epidermal growth factor receptor (EGFR) de-ubiquitination promotes EGFR degradation. J. Biol. Chem. 2007, 282, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a Deubiquitinating Enzyme Essential for TGFß Signaling, Controls Smad4 Monoubiquitination. Cell 2009, 136, 123–135. [Google Scholar] [CrossRef]
- Nagai, H.; Noguchi, T.; Homma, K.; Katagiri, K.; Takeda, K.; Matsuzawa, A.; Ichijo, H. Ubiquitin-like sequence in ASK1 plays critical roles in the recognition and stabilization by USP9X and oxidative stress-induced cell death. Mol. Cell 2009, 36, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Perez-Mancera, P.A.; Rust, A.G.; van der Weyden, L.; Kristiansen, G.; Li, A.; Sarver, A.L.; Silverstein, K.A.; Grutzmann, R.; Aust, D.; Rummele, P.; et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 2012, 486, 266–270. [Google Scholar] [CrossRef]
- Schwickart, M.; Huang, X.; Lill, J.R.; Liu, J.; Ferrando, R.; French, D.M.; Maecker, H.; O’Rourke, K.; Bazan, F.; Eastham-Anderson, J.; et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010, 463, 103–107. [Google Scholar] [CrossRef]
- Goto, Y.; Zeng, L.; Yeom, C.J.; Zhu, Y.; Morinibu, A.; Shinomiya, K.; Kobayashi, M.; Hirota, K.; Itasaka, S.; Yoshimura, M.; et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1alpha. Nat. Commun. 2015, 6, 6153. [Google Scholar] [CrossRef]
- Castegna, A.; Aksenov, M.; Aksenova, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: Creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic. Biol. Med. 2002, 33, 562–571. [Google Scholar] [CrossRef]
- Di Domenico, F.; Coccia, R.; Cocciolo, A.; Murphy, M.P.; Cenini, G.; Head, E.; Butterfield, D.A.; Giorgi, A.; Schinina, M.E.; Mancuso, C.; et al. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochim. Biophys. Acta 2013, 1832, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Y.; Yang, M.; Zhao, M.; Luo, Q.; Yang, L.; Peng, H.; Wang, J.; Huang, S.K.; Zheng, Z.X.; Yuan, X.H.; et al. The de-ubiquitinase UCHL1 promotes gastric cancer metastasis via the Akt and Erk1/2 pathways. Tumour Biol. 2015, 36, 8379–8387. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Bedekovics, T.; Chesi, M.; Bergsagel, P.L.; Galardy, P.J. UCHL1 is a biomarker of aggressive multiple myeloma required for disease progression. Oncotarget 2015, 6, 40704–40718. [Google Scholar] [CrossRef] [PubMed]
- Abdelmaksoud-Dammak, R.; Saadallah-Kallel, A.; Miladi-Abdennadher, I.; Ayedi, L.; Khabir, A.; Sallemi-Boudawara, T.; Frikha, M.; Daoud, J.; Mokdad-Gargouri, R. CpG methylation of ubiquitin carboxyl-terminal hydrolase 1 (UCHL1) and P53 mutation pattern in sporadic colorectal cancer. Tumour Biol. 2016, 37, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, R.; Goto, Y.; Koyasu, S.; Kobayashi, M.; Morinibu, A.; Yoshimura, M.; Hiraoka, M.; Hammond, E.M.; Harada, H. UCHL1-HIF-1 axis-mediated antioxidant property of cancer cells as a therapeutic target for radiosensitization. Sci. Rep. 2017, 7, 6879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Fallon, L.; Lashuel, H.A.; Liu, Z.; Lansbury, P.T., Jr. The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell 2002, 111, 209–218. [Google Scholar] [CrossRef]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef]
- Ehrlich, E.S.; Wang, T.; Luo, K.; Xiao, Z.; Niewiadomska, A.M.; Martinez, T.; Xu, W.; Neckers, L.; Yu, X.F. Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2009, 106, 20330–20335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.V.; Hubbi, M.E.; Pan, F.; McDonald, K.R.; Mansharamani, M.; Cole, R.N.; Liu, J.O.; Semenza, G.L. Calcineurin promotes hypoxia-inducible factor 1alpha expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J. Biol. Chem. 2007, 282, 37064–37073. [Google Scholar] [CrossRef] [PubMed]
- Amir, S.; Wang, R.; Simons, J.W.; Mabjeesh, N.J. SEPT9_v1 up-regulates hypoxia-inducible factor 1 by preventing its RACK1-mediated degradation. J. Biol. Chem. 2009, 284, 11142–11151. [Google Scholar] [CrossRef]
- Paatero, I.; Jokilammi, A.; Heikkinen, P.T.; Iljin, K.; Kallioniemi, O.; Jones, F.E.; Jaakkola, P.M.; Elenius, K. Interaction with ErbB4 promotes hypoxia-inducible factor-1α signaling. J. Biol. Chem. 2012, 287, 9659–9671. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Liu, Y.V.; McDonald, K.R.; Wesley, J.B.; Zhang, H.; Semenza, G.L. Spermidine/spermine N(1)-acetyltransferase-1 binds to hypoxia-inducible factor-1alpha (HIF-1alpha) and RACK1 and promotes ubiquitination and degradation of HIF-1alpha. J. Biol. Chem. 2007, 282, 33358–33366. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Liu, Y.V.; McDonald, K.R.; Wesley, J.B.; Hubbi, M.E.; Byun, H.; Semenza, G.L. Spermidine/spermine-N1-acetyltransferase 2 is an essential component of the ubiquitin ligase complex that regulates hypoxia-inducible factor 1alpha. J. Biol. Chem. 2007, 282, 23572–23580. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Fernandes, R.; Ramalho, J.; Marques, C.; Shang, F.; Taylor, A.; Pereira, P. The chaperone-dependent ubiquitin ligase CHIP targets HIF-1alpha for degradation in the presence of methylglyoxal. PLoS ONE 2010, 5, e15062. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Zhong, J.; Chang, R.; Hu, H.; Pandey, A.; Semenza, G.L. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but Not HIF-2alpha. J. Biol. Chem. 2010, 285, 3651–3663. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.V.; Soares, A.R.; Ramalho, J.S.; Pereira, P.; Girao, H. K63 linked ubiquitin chain formation is a signal for HIF1A degradation by Chaperone-Mediated Autophagy. Sci. Rep. 2015, 5, 10210. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.V.; Fofo, H.; Bejarano, E.; Bento, C.F.; Ramalho, J.S.; Girao, H.; Pereira, P. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 2013, 9, 1349–1366. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Singh, A.R.; Durden, D.L. MDM2 regulates hypoxic hypoxia-inducible factor 1alpha stability in an E3 ligase, proteasome, and PTEN-phosphatidylinositol 3-kinase-AKT-dependent manner. J. Biol. Chem. 2014, 289, 22785–22797. [Google Scholar] [CrossRef]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44. [Google Scholar]
- Amelio, I.; Inoue, S.; Markert, E.K.; Levine, A.J.; Knight, R.A.; Mak, T.W.; Melino, G. TAp73 opposes tumor angiogenesis by promoting hypoxia-inducible factor 1alpha degradation. Proc. Natl. Acad. Sci. USA 2015, 112, 226–231. [Google Scholar] [CrossRef]
- Koh, M.Y.; Lemos, R., Jr.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1alpha- to HIF-2alpha-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Darnay, B.G.; Powis, G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1alpha, leading to its oxygen-independent degradation. Mol. Cell Biol. 2008, 28, 7081–7095. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Nguyen, V.; Lemos, R., Jr.; Darnay, B.G.; Kiriakova, G.; Abdelmelek, M.; Ho, T.H.; Karam, J.; Monzon, F.A.; Jonasch, E.; et al. Hypoxia-induced SUMOylation of E3 ligase HAF determines specific activation of HIF2 in clear-cell renal cell carcinoma. Cancer Res. 2015, 75, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, X.B.; Meng, Y.; Fan, L.; Li, M.; Fang, J. TRAF6 upregulates expression of HIF-1alpha and promotes tumor angiogenesis. Cancer Res. 2013, 73, 4950–4959. [Google Scholar] [CrossRef] [PubMed]
- Rezaeian, A.H.; Li, C.F.; Wu, C.Y.; Zhang, X.; Delacerda, J.; You, M.J.; Han, F.; Cai, Z.; Jeong, Y.S.; Jin, G.; et al. A hypoxia-responsive TRAF6-ATM-H2AX signalling axis promotes HIF1alpha activation, tumorigenesis and metastasis. Nat. Cell Biol. 2017, 19, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, B.; Humphries, F.; Hogan, A.E.; O’Shea, D.; Moynagh, P.N. The E3 ubiquitin ligase Pellino3 protects against obesity-induced inflammation and insulin resistance. Immunity 2014, 41, 973–987. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.C.; Lee, Y.R.; Huang, S.F.; Lin, Y.M.; Chen, T.Y.; Chung, H.C.; Tsai, C.H.; Chen, H.Y.; Chiang, C.T.; Lai, C.K.; et al. A Cullin3-KLHL20 Ubiquitin ligase-dependent pathway targets PML to potentiate HIF-1 signaling and prostate cancer progression. Cancer Cell 2011, 20, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Woik, N.; Dietz, C.T.; Schaker, K.; Kroll, J. Kelch-like ECT2-interacting protein KLEIP regulates late-stage pulmonary maturation via Hif-2alpha in mice. Dis. Model. Mech. 2014, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kim, H.J.; Rih, J.K.; Mattson, T.L.; Kim, K.W.; Cho, C.H.; Isaacs, J.S.; Bae, I. BRCA1 plays a role in the hypoxic response by regulating HIF-1alpha stability and by modulating vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 13047–13056. [Google Scholar] [CrossRef] [PubMed]
- Flugel, D.; Gorlach, A.; Michiels, C.; Kietzmann, T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol. Cell Biol. 2007, 27, 3253–3265. [Google Scholar] [CrossRef]
- Flugel, D.; Gorlach, A.; Kietzmann, T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood 2012, 119, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Cassavaugh, J.M.; Hale, S.A.; Wellman, T.L.; Howe, A.K.; Wong, C.; Lounsbury, K.M. Negative regulation of HIF-1alpha by an FBW7-mediated degradation pathway during hypoxia. J. Cell Biochem. 2011, 112, 3882–3890. [Google Scholar] [CrossRef] [PubMed]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 2007, 9, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Diefenbacher, M.E.; Popov, N.; Blake, S.M.; Schülein-Völk, C.; Nye, E.; Spencer-Dene, B.; Jaenicke, L.A.; Eilers, M.; Behrens, A. The deubiquitinase USP28 controls intestinal homeostasis and promotes Colorectal cancer. J. Clin. Investig. 2014, 124, 3407–3418. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Xu, Y.; Gong, M.; Cao, Y.; An, R. USP28 is a potential prognostic marker for bladder cancer. Tumour Biol. 2014, 35, 4017–4022. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.J.; Zhang, T.; Feng, X.J.; Chang, J.; Suo, F.Z.; Ma, J.L.; Liu, Y.J.; Liu, Y.; Zheng, Y.C.; Liu, H.M. USP28 contributes to the proliferation and metastasis of gastric cancer. J. Cell Biochem. 2018. [Google Scholar] [CrossRef]
- Richter, K.; Paakkola, T.; Mennerich, D.; Kubaichuk, K.; Konzack, A.; Kippari, H.A.; Kozlova, N.; Koivunen, P.; Haapasaari, K.M.; Jukkola-Vuorinen, A.; et al. USP28 Deficiency Promotes Breast and Liver Carcinogenesis as well as Tumor Angiogenesis in a HIF-independent Manner. Mol. Cancer Res. 2018, 16, 1000–1012. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef]
- Wu, H.T.; Kuo, Y.C.; Hung, J.J.; Huang, C.H.; Chen, W.Y.; Chou, T.Y.; Chen, Y.; Chen, Y.J.; Chen, Y.J.; Cheng, W.C.; et al. K63-polyubiquitinated HAUSP deubiquitinates HIF-1alpha and dictates H3K56 acetylation promoting hypoxia-induced tumour progression. Nat. Commun. 2016, 7, 13644. [Google Scholar] [CrossRef]
- Hassink, G.C.; Zhao, B.; Sompallae, R.; Altun, M.; Gastaldello, S.; Zinin, N.V.; Masucci, M.G.; Lindsten, K. The ER-resident ubiquitin-specific protease 19 participates in the UPR and rescues ERAD substrates. EMBO Rep. 2009, 10, 755–761. [Google Scholar] [CrossRef]
- Nakayama, K.; Frew, I.J.; Hagensen, M.; Skals, M.; Habelhah, H.; Bhoumik, A.; Kadoya, T.; Erdjument-Bromage, H.; Tempst, P.; Frappell, P.B.; et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell 2004, 117, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, Z.; Hou, F.; Harding, R.; Huang, X.; Dong, A.; Walker, J.R.; Tong, Y. The substrate binding domains of human SIAH E3 ubiquitin ligases are now crystal clear. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3095–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altun, M.; Zhao, B.; Velasco, K.; Liu, H.; Hassink, G.; Paschke, J.; Pereira, T.; Lindsten, K. Ubiquitin-specific protease 19 (USP19) regulates hypoxia-inducible factor 1alpha (HIF-1alpha) during hypoxia. J. Biol. Chem. 2012, 287, 1962–1969. [Google Scholar] [CrossRef] [PubMed]
- Bett, J.S.; Ibrahim, A.F.; Garg, A.K.; Kelly, V.; Pedrioli, P.; Rocha, S.; Hay, R.T. The P-body component USP52/PAN2 is a novel regulator of HIF1A mRNA stability. Biochem. J. 2013, 451, 185–194. [Google Scholar] [CrossRef]
- Bremm, A.; Moniz, S.; Mader, J.; Rocha, S.; Komander, D. Cezanne (OTUD7B) regulates HIF-1α homeostasis in a proteasome-independent manner. EMBO Rep. 2014, 15, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Luong le, A.; Fragiadaki, M.; Smith, J.; Boyle, J.; Lutz, J.; Dean, J.L.; Harten, S.; Ashcroft, M.; Walmsley, S.R.; Haskard, D.O.; et al. Cezanne regulates inflammatory responses to hypoxia in endothelial cells by targeting TRAF6 for deubiquitination. Circ. Res. 2013, 112, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Moniz, S.; Bandarra, D.; Biddlestone, J.; Campbell, K.J.; Komander, D.; Bremm, A.; Rocha, S. Cezanne regulates E2F1-dependent HIF2alpha expression. J. Cell Sci. 2015, 128, 3082–3093. [Google Scholar] [CrossRef] [PubMed]
- Minchenko, O.H.; Tsymbal, D.O.; Minchenko, D.O.; Riabovol, O.O.; Halkin, O.V.; Ratushna, O.O. IRE-1α regulates expression of ubiquitin specific peptidases during hypoxic response in U87 glioma cells. Endoplasm. Reticul. Stress Dis. 2016, 3, 50–62. [Google Scholar]
- Nijman, S.M.B.; Huang, T.T.; Dirac, A.M.G.; Brummelkamp, T.R.; Kerkhoven, R.M.; D’Andrea, A.D.; Bernards, R. The deubiquitinating enzyme USP1 regulates the fanconi anemia pathway. Mol. Cell 2005, 17, 331–339. [Google Scholar] [CrossRef]
- Huang, T.T.; Nijman, S.M.B.; Mirchandani, K.D.; Galardy, P.J.; Cohn, M.A.; Haas, W.; Gygi, S.P.; Ploegh, H.L.; Bernards, R.; D’Andrea, A.D. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 2006, 8, 339–347. [Google Scholar] [CrossRef]
- Lin, Z.; Yang, H.; Tan, C.; Li, J.; Liu, Z.; Quan, Q.; Kong, S.; Ye, J.; Gao, B.; Fang, D. USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep. 2013, 5, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 Regulates p53 Localization and Stability by Deubiquitinating p53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mines, M.A.; Goodwin, J.S.; Limbird, L.E.; Cui, F.F.; Fan, G.H. Deubiquitination of CXCR4 by USP14 is critical for both CXCL12-induced CXCR4 degradation and chemotaxis but not ERK ativation. J. Biol. Chem. 2009, 284, 5742–5752. [Google Scholar] [CrossRef] [PubMed]
- Nagai, A.; Kadowaki, H.; Maruyama, T.; Takeda, K.; Nishitoh, H.; Ichijo, H. USP14 inhibits ER-associated degradation via interaction with IRE1alpha. Biochem. Biophys. Res. Commun. 2009, 379, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, Y.; Wang, Y.; Liu, H.; Yang, Y.; Wang, Q. Effect of deubiquitinase USP8 on hypoxia/reoxygenationinduced inflammation by deubiquitination of TAK1 in renal tubular epithelial cells. Int. J. Mol. Med. 2018, 42, 3467–3476. [Google Scholar]
- Scortegagna, M.; Subtil, T.; Qi, J.; Kim, H.; Zhao, W.; Gu, W.; Kluger, H.; Ronai, Z.A. USP13 enzyme regulates Siah2 ligase stability and activity via noncatalytic ubiquitin-binding domains. J. Biol. Chem. 2011, 286, 27333–27341. [Google Scholar] [CrossRef]
- Geng, J.; Huang, X.; Li, Y.; Xu, X.; Li, S.; Jiang, D.; Liang, J.; Jiang, D.; Wang, C.; Dai, H. Down-regulation of USP13 mediates phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 124. [Google Scholar] [CrossRef]
- Li, Q.; Kluz, T.; Sun, H.; Costa, M. Mechanisms of c-myc degradation by nickel compounds and hypoxia. PLoS ONE 2009, 4, e8531. [Google Scholar] [CrossRef]
- Wen, Y.A.; Stevens, P.D.; Gasser, M.L.; Andrei, R.; Gao, T. Downregulation of PHLPP expression contributes to hypoxia-induced resistance to chemotherapy in colon cancer cells. Mol. Cell Biol. 2013, 33, 4594–4605. [Google Scholar] [CrossRef]
- Guo, J.; Shinriki, S.; Su, Y.; Nakamura, T.; Hayashi, M.; Tsuda, Y.; Murakami, Y.; Tasaki, M.; Hide, T.; Takezaki, T.; et al. Hypoxia suppresses cylindromatosis (CYLD) expression to promote inflammation in glioblastoma: Possible link to acquired resistance to anti-VEGF therapy. Oncotarget 2014, 5, 6353–6364. [Google Scholar] [CrossRef]
- An, J.; Mo, D.; Liu, H.; Veena, M.S.; Srivatsan, E.S.; Massoumi, R.; Rettig, M.B. Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-kappaB activation. Cancer Cell 2008, 14, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Liu, Y.; Xu, X.; Zhang, W.; Yu, T.; Jia, J.; Liu, C. Deubiquitinase USP47/UBP64E Regulates beta-Catenin Ubiquitination and Degradation and Plays a Positive Role in Wnt Signaling. Mol. Cell Biol. 2015, 35, 3301–3311. [Google Scholar] [CrossRef]
- Choi, B.J.; Park, S.A.; Lee, S.Y.; Cha, Y.N.; Surh, Y.J. Hypoxia induces epithelial-mesenchymal transition in colorectal cancer cells through ubiquitin-specific protease 47-mediated stabilization of Snail: A potential role of Sox9. Sci. Rep. 2017, 7, 15918. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ying, W.; Wang, W.; Li, W.; Feng, X. HIF1α and HIF2α mediated UCHL1 upregulation in hypoxia-induced neuronal injury following neuronal hypoxic ischemic encephalopathy. Int. J. Clin. Exp. Pathol. 2016, 9, 2677–2685. [Google Scholar]
- Scholz, C.C.; Rodriguez, J.; Pickel, C.; Burr, S.; Fabrizio, J.A.; Nolan, K.A.; Spielmann, P.; Cavadas, M.A.; Crifo, B.; Halligan, D.N.; et al. FIH Regulates Cellular Metabolism through Hydroxylation of the Deubiquitinase OTUB1. PLoS Biol. 2016, 14, e1002347. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhai, B.; Koivunen, P.; Shin, S.J.; Lu, G.; Liu, J.; Geisen, C.; Chakraborty, A.A.; Moslehi, J.J.; Smalley, D.M.; et al. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev. 2014, 28, 1429–1444. [Google Scholar] [CrossRef] [Green Version]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef]
- Confalonieri, S.; Quarto, M.; Goisis, G.; Nuciforo, P.; Donzelli, M.; Jodice, G.; Pelosi, G.; Viale, G.; Pece, S.; Di Fiore, P.P. Alterations of ubiquitin ligases in human cancer and their association with the natural history of the tumor. Oncogene 2009, 28, 2959–2968. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.K.; Li, Q.; Xu, L.H.; Hu, L.J.; Liao, W.G.; Zhang, X.R.; Liu, Z.M.; Wu, D.; Zeng, M.S. Expression and clinical significance of SIAH in laryngeal squamous cell carcinoma. Med. Oncol. 2013, 30, 485. [Google Scholar] [CrossRef]
- Kim, C.J.; Cho, Y.G.; Park, C.H.; Jeong, S.W.; Nam, S.W.; Kim, S.Y.; Lee, S.H.; Yoo, N.J.; Lee, J.Y.; Park, W.S. Inactivating mutations of the Siah-1 gene in gastric cancer. Oncogene 2004, 23, 8591–8596. [Google Scholar] [CrossRef] [Green Version]
- Yoshibayashi, H.; Okabe, H.; Satoh, S.; Hida, K.; Kawashima, K.; Hamasu, S.; Nomura, A.; Hasegawa, S.; Ikai, I.; Sakai, Y. SIAH1 causes growth arrest and apoptosis in hepatoma cells through beta-catenin degradation-dependent and -independent mechanisms. Oncol. Rep. 2007, 17, 549–556. [Google Scholar] [PubMed]
- Chan, P.; Moller, A.; Liu, M.C.; Sceneay, J.E.; Wong, C.S.; Waddell, N.; Huang, K.T.; Dobrovic, A.; Millar, E.K.; O’Toole, S.A.; et al. The expression of the ubiquitin ligase SIAH2 (seven in absentia homolog 2) is mediated through gene copy number in breast cancer and is associated with a basal-like phenotype and p53 expression. Breast Cancer Res. 2011, 13, R19. [Google Scholar] [CrossRef] [PubMed]
- Brauckhoff, A.; Malz, M.; Tschaharganeh, D.; Malek, N.; Weber, A.; Riener, M.O.; Soll, C.; Samarin, J.; Bissinger, M.; Schmidt, J.; et al. Nuclear expression of the ubiquitin ligase seven in absentia homolog (SIAH)-1 induces proliferation and migration of liver cancer cells. J. Hepatol. 2011, 55, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ci, W.; Karmakar, S.; Chen, K.; Dhar, R.; Fan, Z.; Guo, Z.; Zhang, J.; Ke, Y.; Wang, L.; et al. SPOP promotes tumorigenesis by acting as a key regulatory hub in kidney cancer. Cancer Cell 2014, 25, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Bao, Y.C.; Ji, X.X.; Chen, B.; Deng, Q.F.; Zhou, S.W. SPOP promotes SIRT2 degradation and suppresses non-small cell lung cancer cell growth. Biochem. Biophys. Res. Commun. 2017, 483, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; He, B.; Xu, L.; Barbieri, C.E.; Eedunuri, V.K.; Chew, S.A.; Zimmermann, M.; Bond, R.; Shou, J.; Li, C.; et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc. Natl. Acad. Sci. USA 2013, 110, 6997–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, S.; Lee, S.Y.; Lee, J.; Jeong, C.H.; Farrand, L.; Lim, S.; Reddy, K.; Kim, J.Y.; Lee, M.H.; Lee, H.J.; et al. USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin. Cancer Res. 2013, 19, 3894–3904. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Zhao, C.; Wei, N.; Wu, X.; Cui, J.; Xing, Y. High Expression of Ubiquitin-Specific Protease 8 (USP8) Is Associated with Poor Prognosis in Patients with Cervical Squamous Cell Carcinoma. Med. Sci. Monit. 2018, 24, 4934–4943. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yao, D.; Zhang, P.; Ding, W.; Zhang, X.; Zhang, C.; Gong, S.; Zhang, Y.; Wang, J.; Sun, T.; et al. Deubiquitinase USP9X promotes cell migration, invasion and inhibits apoptosis of human pancreatic cancer. Oncol. Rep. 2017, 38, 3531–3537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Xie, W.; Song, X.; Wu, K.; Xiao, L.; Liu, Y.; Zhang, L. Aberrant expression of deubiquitylating enzyme USP9X predicts poor prognosis in gastric cancer. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 687–692. [Google Scholar] [CrossRef]
- Li, X.; Song, N.; Liu, L.; Liu, X.; Ding, X.; Song, X.; Yang, S.; Shan, L.; Zhou, X.; Su, D.; et al. USP9X regulates centrosome duplication and promotes breast carcinogenesis. Nat. Commun. 2017, 8, 14866. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.M.; Carvalho, J.; Spencer-Dene, B.; Mitter, R.; Frith, D.; Snijders, A.P.; Wood, S.A.; Behrens, A. The deubiquitinase USP9X regulates FBW7 stability and suppresses colorectal cancer. J. Clin. Investig. 2018, 128, 1326–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marona, P.; Gorka, J.; Mazurek, Z.; Wilk, W.; Rys, J.; Majka, M.; Jura, J.; Miekus, K. MCPIP1 Downregulation in Clear Cell Renal Cell Carcinoma Promotes Vascularization and Metastatic Progression. Cancer Res. 2017, 77, 4905–4920. [Google Scholar] [CrossRef]
- Chen, G.; Gharib, T.G.; Huang, C.C.; Thomas, D.G.; Shedden, K.A.; Taylor, J.M.; Kardia, S.L.; Misek, D.E.; Giordano, T.J.; Iannettoni, M.D.; et al. Proteomic analysis of lung adenocarcinoma: Identification of a highly expressed set of proteins in tumors. Clin. Cancer Res. 2002, 8, 2298–2305. [Google Scholar] [PubMed]
- Jin, C.; Yu, W.; Lou, X.; Zhou, F.; Han, X.; Zhao, N.; Lin, B. UCHL1 Is a Putative Tumor Suppressor in Ovarian Cancer Cells and Contributes to Cisplatin Resistance. J. Cancer 2013, 4, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhu, M.; Yang, X.; Wang, Y.; Qin, B.; Cui, C.; Chen, H.; Sang, A. Inhibition of RACK1 ameliorates choroidal neovascularization formation in vitro and in vivo. Exp. Mol. Pathol. 2016, 100, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Deng, Y.Z.; Zhao, J.S.; Ji, X.D.; Shi, J.; Feng, Y.X.; Li, G.; Li, J.J.; Zhu, D.; Koeffler, H.P.; et al. RACK1 promotes non-small-cell lung cancer tumorigenicity through activating sonic hedgehog signaling pathway. J. Biol. Chem. 2012, 287, 7845–7858. [Google Scholar] [CrossRef]
- Fei, L.; Ma, Y.; Zhang, M.; Liu, X.; Luo, Y.; Wang, C.; Zhang, H.; Zhang, W.; Han, Y. RACK1 promotes lung cancer cell growth via an MCM7/RACK1/ Akt signaling complex. Oncotarget 2017, 8, 40501–40513. [Google Scholar] [CrossRef]
- Zhong, X.; Li, M.; Nie, B.; Wu, F.; Zhang, L.; Wang, E.; Han, Y. Overexpressions of RACK1 and CD147 associated with poor prognosis in stage T1 pulmonary adenocarcinoma. Ann. Surg. Oncol. 2013, 20, 1044–1052. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, W.; Wang, J.; Feng, J.; Wang, Q.; Jin, J.; Lv, M.; Li, X.; Li, Y.; Ma, Y.; et al. Receptor for activated C kinase 1 promotes hepatocellular carcinoma growth by enhancing mitogen-activated protein kinase kinase 7 activity. Hepatology 2013, 57, 140–151. [Google Scholar] [CrossRef]
- Cao, X.X.; Xu, J.D.; Liu, X.L.; Xu, J.W.; Wang, W.J.; Li, Q.Q.; Chen, Q.; Xu, Z.D.; Liu, X.P. RACK1: A superior independent predictor for poor clinical outcome in breast cancer. Int. J. Cancer 2010, 127, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Jiang, B.; Ma, J.; Ma, Z.; Wan, X.; Liu, H.; Chen, Z.; Cheng, Q.; Chen, R. Forced downregulation of RACK1 inhibits glioma development by suppressing Src/Akt signaling activity. Oncol. Rep. 2013, 30, 2195–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.Z.; Yao, F.; Li, J.J.; Mao, Z.F.; Hu, P.T.; Long, L.Y.; Li, G.; Ji, X.D.; Shi, S.; Guan, D.X.; et al. RACK1 suppresses gastric tumorigenesis by stabilizing the beta-catenin destruction complex. Gastroenterology 2012, 142, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.L.; Kim, M.; Huang, S.M.; Lee, H.J.; Kim, J.M. Expression of carboxyl terminus of Hsp70-interacting protein (CHIP) indicates poor prognosis in human gallbladder carcinoma. Oncol. Lett. 2013, 5, 813–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.; Luo, K.J.; Hu, Y.; Yang, H.; Fu, J.H. Metastatic lymph node CHIP expression is a potential prognostic marker for resected esophageal squamous cell carcinoma patients. Ann. Surg. Oncol. 2013, 20, 1668–1675. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zang, J.; Dai, H.J.; Li, F.; Guo, F. Ubiquitin ligase CHIP functions as an oncogene and activates the AKT signaling pathway in prostate cancer. Int. J. Oncol. 2018, 53, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Patani, N.; Jiang, W.; Newbold, R.; Mokbel, K. Prognostic implications of carboxyl-terminus of Hsc70 interacting protein and lysyl-oxidase expression in human breast cancer. J. Carcinog. 2010, 9, 9. [Google Scholar] [CrossRef]
- Wang, S.; Wu, X.; Zhang, J.; Chen, Y.; Xu, J.; Xia, X.; He, S.; Qiang, F.; Li, A.; Shu, Y.; et al. CHIP functions as a novel suppressor of tumour angiogenesis with prognostic significance in human gastric cancer. Gut 2013, 62, 496–508. [Google Scholar] [CrossRef]
- Wang, T.; Yang, J.; Xu, J.; Li, J.; Cao, Z.; Zhou, L.; You, L.; Shu, H.; Lu, Z.; Li, H.; et al. CHIP is a novel tumor suppressor in pancreatic cancer through targeting EGFR. Oncotarget 2014, 5, 1969–1986. [Google Scholar] [CrossRef]
- Wang, Y.; Ren, F.; Wang, Y.; Feng, Y.; Wang, D.; Jia, B.; Qiu, Y.; Wang, S.; Yu, J.; Sung, J.J.; et al. CHIP/Stub1 functions as a tumor suppressor and represses NF-kappaB-mediated signaling in colorectal cancer. Carcinogenesis 2014, 35, 983–991. [Google Scholar] [CrossRef]
- Qian, T.; Wang, J.; Wang, Q.; Liu, Y.; Yu, S.; Wang, Z.; Sun, D.; Wang, S. CHIP involves in non-small cell lung cancer prognosis through VEGF pathway. Biomed. Pharmacother. 2016, 83, 271–276. [Google Scholar]
- Zhang, L.; Hill, R.P. Hypoxia enhances metastatic efficiency by up-regulating Mdm2 in KHT cells and increasing resistance to apoptosis. Cancer Res. 2004, 64, 4180–4189. [Google Scholar] [CrossRef] [PubMed]
- Mairinger, F.D.; Walter, R.F.; Ting, S.; Vollbrecht, C.; Kollmeier, J.; Griff, S.; Hager, T.; Mairinger, T.; Christoph, D.C.; Theegarten, D.; et al. Mdm2 protein expression is strongly associated with survival in malignant pleural mesothelioma. Future Oncol. 2014, 10, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, D.D.; Wu, Y.P.; Su, D.; Zhou, T.Y.; Gai, R.H.; Fu, Y.Y.; Zheng, L.; He, Q.J.; Zhu, H.; et al. MDM2 promotes epithelial-mesenchymal transition and metastasis of ovarian cancer SKOV3 cells. Br. J. Cancer 2017, 117, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Akhoondi, S.; Sun, D.; von der, L.N.; Apostolidou, S.; Klotz, K.; Maljukova, A.; Cepeda, D.; Fiegl, H.; Dafou, D.; Marth, C.; et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007, 67, 9006–9012. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, M.; Delugin, M.; Abraldes, I.; Allain, N.; Belaud-Rotureau, M.A.; Turmo, M.; Prigent, C.; Loiseau, H.; Bikfalvi, A.; Javerzat, S. FBXW7/hCDC4 controls glioma cell proliferation in vitro and is a prognostic marker for survival in glioblastoma patients. Cell Div. 2007, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yang, C.; Fan, P.; Xiao, J.; Zhang, W.; Zhan, S.; Liu, T.; Wang, D.; Wu, H. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J. Biol. Chem. 2017, 292, 6269–6280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Li, Y.; Li, J.; Ma, Y.; Dai, W.; Mo, S.; Xu, Y.; Li, X.; Cai, S. FBW7 suppresses metastasis of colorectal cancer by inhibiting HIF1alpha/CEACAM5 functional axis. Int. J. Biol. Sci. 2018, 14, 726–735. [Google Scholar] [CrossRef]
- Imaizumi, T.; Kuramoto, T.; Matsunaga, K.; Shichijo, S.; Yutani, S.; Shigemori, M.; Oizumi, K.; Itoh, K. Expression of the tumor-rejection antigen SART1 in brain tumors. Int. J. Cancer 1999, 83, 760–764. [Google Scholar] [CrossRef]
- Kawamoto, M.; Shichijo, S.; Imai, Y.; Imaizumi, T.; Koga, T.; Yanaga, H.; Itoh, K. Expression of the SART-1 tumor rejection antigen in breast cancer. Int. J. Cancer 1999, 80, 64–67. [Google Scholar] [CrossRef]
- Sasatomi, T.; Yamana, H.; Shichijo, S.; Tanaka, S.; Okamura, T.; Ogata, Y.; Itoh, K.; Shirouzu, K. Expression of the SART1 tumor-rejection antigens in colorectal cancers. Dis. Colon Rectum 2000, 43, 1754–1758. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Hao, A.; Cui, H.; Wu, W.; Yang, H.; Hu, B.; Li, P. TRAF6 expression is associated with poorer prognosis and high recurrence in urothelial bladder cancer. Oncol. Lett. 2017, 14, 2432–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, F.; Zhang, L.; Qiu, W.; Yi, X. TRAF6 promotes the invasion and metastasis and predicts a poor prognosis in gastric cancer. Pathol. Res. Pract. 2016, 212, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Z.; Zhang, G.; Zhu, Q.; Zeng, H.; Wang, T.; Gao, F.; Qi, Z.; Zhang, J.; Wang, R. High TRAF6 Expression Is Associated with Esophageal Carcinoma Recurrence and Prompts Cancer Cell Invasion. Oncol. Res. 2017, 25, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Wang, D.; Wu, W.; Jin, D.; Kuang, T.; Ni, X.; Zhang, L.; Lou, W. TRAF6 is over-expressed in pancreatic cancer and promotes the tumorigenicity of pancreatic cancer cells. Med. Oncol. 2014, 31, 260. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Zhang, X.; Zeng, W.; Su, J.; Yang, K.; Lu, L.; Lim, C.B.; Tang, W.; Wu, L.; Zhao, S.; et al. TRAF6 regulates melanoma invasion and metastasis through ubiquitination of Basigin. Oncotarget 2016, 7, 7179–7192. [Google Scholar] [CrossRef]
- Fay, M.J.; Longo, K.A.; Karathanasis, G.A.; Shope, D.M.; Mandernach, C.J.; Leong, J.R.; Hicks, A.; Pherson, K.; Husain, A. Analysis of CUL-5 expression in breast epithelial cells, breast cancer cell lines, normal tissues and tumor tissues. Mol. Cancer 2003, 2, 40. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, L.; Hou, D.; Ouyang, Y.; Guo, X.; Wang, Y.; Li, J.; Gong, K. MicroRNA-19a regulates the proliferation, migration and invasion of human gastric cancer cells by targeting CUL5. Arch. Biochem. Biophys. 2019, 662, 93–100. [Google Scholar] [CrossRef]
- Devor, E.J.; Schickling, B.M.; Reyes, H.D.; Warrier, A.; Lindsay, B.; Goodheart, M.J.; Santillan, D.A.; Leslie, K.K. Cullin-5, a ubiquitin ligase scaffold protein, is significantly underexpressed in endometrial adenocarcinomas and is a target of miR-182. Oncol. Rep. 2016, 35, 2461–2465. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.M.; Wang, X.B.; Chen, M.M.; Liu, T.; Li, Y.X.; Jia, W.H.; Liu, M.; Li, X.; Tang, H. MicroRNA-19a and -19b regulate cervical carcinoma cell proliferation and invasion by targeting CUL5. Cancer Lett. 2012, 322, 148–158. [Google Scholar] [CrossRef]
- Ma, C.; Qi, Y.; Shao, L.; Liu, M.; Li, X.; Tang, H. Downregulation of miR-7 upregulates Cullin 5 (CUL5) to facilitate G1/S transition in human hepatocellular carcinoma cells. IUBMB Life 2013, 65, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Chu, A.; Turker, M.S.; Glazer, P.M. Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Mol. Cell Biol. 2011, 31, 3339–3350. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Paul, S. The breast cancer susceptibility genes (BRCA) in breast and ovarian cancers. Front. Biosci. 2014, 19, 605–618. [Google Scholar] [CrossRef]
- Hu, T.; Zhang, J.; Sha, B.; Li, M.; Wang, L.; Zhang, Y.; Liu, X.; Dong, Z.; Liu, Z.; Li, P.; et al. Targeting the overexpressed USP7 inhibits esophageal squamous cell carcinoma cell growth by inducing NOXA-mediated apoptosis. Mol. Carcinog. 2019, 58, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Ma, S.; Shan, L.; Wang, Y.; Wang, Y.; Cao, C.; Liu, B.; Yang, C.; Wang, L.; Tian, S.; et al. Ubiquitin-specific protease 7 sustains DNA damage response and promotes cervical carcinogenesis. J. Clin. Investig. 2018, 128, 4280–4296. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Q.; Wang, Y.; Zhuang, H.; Chen, B. Clinical Significance of Ubiquitin Specific Protease 7 (USP7) in Predicting Prognosis of Hepatocellular Carcinoma and its Functional Mechanisms. Med. Sci. Monit. 2018, 24, 1742–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Bedard, N.; Chevalier, S.; Wing, S.S. Identification of distinctive patterns of USP19-mediated growth regulation in normal and malignant cells. PLoS ONE 2011, 6, e15936. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Tu, H.Q.; Chang, Y.; Tan, B.; Wang, G.; Zhou, J.; Wang, L.; Mu, R.; Zhang, W.N. USP19 deubiquitinates HDAC1/2 to regulate DNA damage repair and control chromosomal stability. Oncotarget 2017, 8, 2197–2208. [Google Scholar] [CrossRef]
- Yang, S.; Liu, L.; Cao, C.; Song, N.; Wang, Y.; Ma, S.; Zhang, Q.; Yu, N.; Ding, X.; Yang, F.; et al. USP52 acts as a deubiquitinase and promotes histone chaperone ASF1A stabilization. Nat. Commun. 2018, 9, 1285. [Google Scholar] [CrossRef]
- Wang, J.H.; Wei, W.; Guo, Z.X.; Shi, M.; Guo, R.P. Decreased Cezanne expression is associated with the progression and poor prognosis in hepatocellular carcinoma. J. Transl. Med. 2015, 13, 41. [Google Scholar] [CrossRef]
- Pang, Z.; Cui, L.; Ding, N.; Zhu, L.; Qu, X.; Dong, W.; Du, J.; Liu, Q. Expressions of insulin-like growth factor receptor-1 and cezanne-1 in lung adenocarcinoma. Med. Oncol. 2017, 34, 78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, H.; Yang, L.; Zhang, Y.; Wang, P.; Huang, G.; Zheng, J.; Ren, H.; Qin, S. OTUD7B and NIK expression in non-small cell lung cancer: Association with clinicopathological features and prognostic implications. Pathol. Res. Pract. 2016, 212, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jie, Z.; Joo, D.; Ordureau, A.; Liu, P.; Gan, W.; Guo, J.; Zhang, J.; North, B.J.; Dai, X.; et al. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature 2017, 545, 365–369. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Involvement in HIF Regulation | Involvement in Cancer | References |
|---|---|---|---|
| Ub E3 Ligases | |||
| VHL-containing | Ubiquitylates hydroxylated HIF-α for proteasomal degradation |
| [53,54,55,147] |
| SIAH 1/2 | Ubiquitylate PHD1/3, leading to HIF-1α stabilization; SIAH1 facilitates FIH degradation via the proteasomal pathway |
| [60,148,149,150,151,152,153] |
| SPOP | Ubiquitylates PHD1, promoting its proteasomal degradation |
| [61,154,155,156] |
| DUBs | |||
| USP8 | Reverses the VHL-mediated degradation of HIF-1α and HIF-2α |
| [72,74,135,157,158] |
| USP9X | Affects the ubiquitylation of HIF-1α indirectly by reducing VHL via deubiquitylation of the E3 ligase SMURF1, which targets VHL |
| [51,159,160,161,162] |
| USP20 | Counteracts the VHL-mediated ubiquitylation of HIF-1α |
| [63,65,66,67] |
| MCPIP1 | Deubiquitylates HIF-1α; suppresses the levels of HIF-1α and SIRT-1 miR repressors |
| [68,69,70,71,163] |
| UCHL1 | Abrogates VHL-mediated ubiquitylation of HIF-1α |
| [79,82,83,84,164,165] |
| Enzymes | Involvement in HIF Regulation | Involvement in Cancer | References |
|---|---|---|---|
| Ub E3 Ligases | |||
| RACK1 | Competes with HSP90 for binding to HIF-1α to drive HIF-1α degradation |
| [87,166,167,168,169,170,171,172,173] |
| CHIP | Promotes HIF-1α but not HIF-2α degradation via both proteasomal or lysosomal machinery |
| [46,94,95,96,97,174,175,176,177,178,179,180,181] |
| MDM2 | Regulates HIF-1α stability directly due to E3 ligase activity or indirectly by forming a ternary complex, which is degraded in a p53-dependent manner |
| [98,99,182,183,184] |
| Fbw7 | Recruited to GSK-3-phosphorylated HIF-1α for proteasomal degradation. |
| [110,111,112,185,186,187,188] |
| HAF | Selectively degrades HIF-1α and promotes HIF-2α transactivation during hypoxia |
| [101,102,189,190,191] |
| TRAF6 | Increases HIF-1α polylysine-63 ubiquitylation, protecting it from proteasomal degradation; TRAF6-ATM-H2AX signaling axis promotes HIF1α stabilization and activation |
| [104,192,193,194,195,196] |
| Cullin 5-containing | Involved in HSP90-mediated regulation of HIF-1α stability |
| [88,197,198,199,200,201] |
| KLEIP-containing | By reducing levels of PML, leads to a activation of mTOR, which promotes HIF-1α signaling; stabilizes mRNA expression of HIF-2α |
| [107,108] |
| BRCA1 | Regulates stability of HIF-1α (NRM) |
| [109,202,203] |
| Pellino3 | Ubiquitylates TRAF6 with polylysine-63 to block its interaction with HIF-1α, making it more prone to proteasomal degradation | [106] | |
| DUBs | |||
| USP7 | Deubiquitylation of HIF-1α |
| [119,204,205,206] |
| USP19 | Stabilizes HIF-1α and interacts with SIAH1/2 and PHD1/3 regulators |
| [121,122,207,208] |
| USP28 | Counteracts Fbw7-mediated HIF-1α ubiquitylation |
| [111,114,115,116,117] |
| USP52 | Stabilizes HIF-1α mRNA |
| [124,209] |
| OTUD7B | Stabilizes HIF-1α and E2F1 transcription factor to control the expression of HIF-2α mRNA; affects HIF-2α at the transcript level |
| [70,125,127,210,211,212,213] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubaichuk, K.; Kietzmann, T. Involvement of E3 Ligases and Deubiquitinases in the Control of HIF-α Subunit Abundance. Cells 2019, 8, 598. https://doi.org/10.3390/cells8060598
Kubaichuk K, Kietzmann T. Involvement of E3 Ligases and Deubiquitinases in the Control of HIF-α Subunit Abundance. Cells. 2019; 8(6):598. https://doi.org/10.3390/cells8060598
Chicago/Turabian StyleKubaichuk, Kateryna, and Thomas Kietzmann. 2019. "Involvement of E3 Ligases and Deubiquitinases in the Control of HIF-α Subunit Abundance" Cells 8, no. 6: 598. https://doi.org/10.3390/cells8060598
APA StyleKubaichuk, K., & Kietzmann, T. (2019). Involvement of E3 Ligases and Deubiquitinases in the Control of HIF-α Subunit Abundance. Cells, 8(6), 598. https://doi.org/10.3390/cells8060598