The B-Side of Cancer Immunity: The Underrated Tune



Abstract
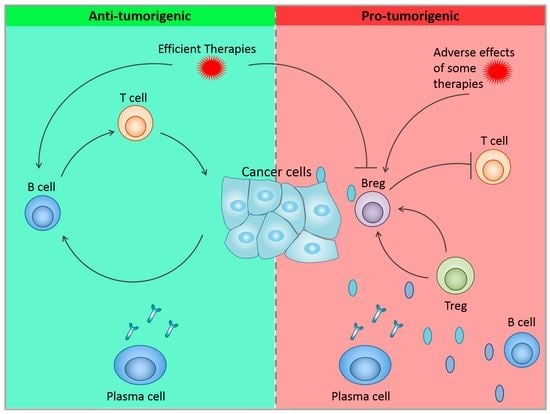
:
1. Introduction
2. Role of B Cells in Pro- and Anti-Tumor Immunity
2.1. Anti-Tumor Activity of B Cells
2.1.1. Antibody Production
2.1.2. Other Functions of B Cells
2.2. Pro-Tumor Activity of B Cells
2.2.1. Conventional B Cells
2.2.2. Regulatory B Cells
3. Tumor Microenvironment Factors Influencing B Cells Functions
3.1. Influence of Different TME Cells on B Cell Functions
3.1.1. Immune Cells Component Influencing B Cells
3.1.2. Direct Action of Tumor Cells on B Cells
3.2. Immune Checkpoint Stimulation on B Cells
3.3. Effect of Hypoxia on B Cells
4. Role of B Cells in Cancer Therapy
4.1. B Cells in Therapy: Implication in Resistance and Correlation with Response
4.2. Therapies Activating B Cells
4.3. Therapies Depleting/Inhibiting B Cells
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Packard, T.A.; Cambier, J.C. B lymphocyte antigen receptor signaling: Initiation, amplification, and regulation. F1000Prime Rep. 2013, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Allman, D.; Pillai, S. Peripheral B cell subsets. Curr. Opin. Immunol. 2008, 20, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Zubler, R.H. Naive and memory B cells in T-cell-dependent and T-independent responses. Springer Semin. Immun. 2001, 23, 405–419. [Google Scholar] [CrossRef]
- Mesin, L.; Ersching, J.; Victora, G.D. Germinal Center B Cell Dynamics. Immunity 2016, 45, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Lu, L.; Xia, Y.; Dai, F.; Wang, Y.; Bao, Y.; Lundy, S.K.; Ito, F.; Pan, Q.; Zhang, X.; et al. Antitumor effector B cells directly kill tumor cells via the Fas/FasL pathway and are regulated by IL-10. Eur. J. Immunol. 2015, 45, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Zhang, Y.; Rosenblatt, J.D. B cell regulation of the anti-tumor response and role in carcinogenesis. J. Immunother. Cancer 2016, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Pages, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Coronella-Wood, J.A.; Hersh, E.M. Naturally occurring B-cell responses to breast cancer. Cancer Immunol. Immunother. 2003, 52, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef]
- Milne, K.; Kobel, M.; Kalloger, S.E.; Barnes, R.O.; Gao, D.; Gilks, C.B.; Watson, P.H.; Nelson, B.H. Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PLoS ONE 2009, 4, e6412. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwe, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749. [Google Scholar] [CrossRef]
- Dieu-Nosjean, M.C.; Goc, J.; Giraldo, N.A.; Sautes-Fridman, C.; Fridman, W.H. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014, 35, 571–580. [Google Scholar] [CrossRef]
- Wouters, M.C.A.; Nelson, B.H. Prognostic Significance of Tumor-Infiltrating B Cells and Plasma Cells in Human Cancer. Clin. Cancer Res. 2018, 24, 6125–6135. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.; Katayama, H.; Ostrin, E.J.; Hanash, S.M. The Emerging Role of B Cells in Tumor Immunity. Cancer Res. 2016, 76, 5597–5601. [Google Scholar] [CrossRef]
- Andreu, P.; Johansson, M.; Affara, N.I.; Pucci, F.; Tan, T.; Junankar, S.; Korets, L.; Lam, J.; Tawfik, D.; DeNardo, D.G.; et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell 2010, 17, 121–134. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Korets, L.V.; Coussens, L.M. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 2005, 7, 411–423. [Google Scholar] [CrossRef]
- Yuen, G.J.; Demissie, E.; Pillai, S. B lymphocytes and cancer: A love-hate relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef]
- Li, Q.; Teitz-Tennenbaum, S.; Donald, E.J.; Li, M.; Chang, A.E. In vivo sensitized and in vitro activated B cells mediate tumor regression in cancer adoptive immunotherapy. J. Immunol. 2009, 183, 3195–3203. [Google Scholar] [CrossRef]
- Kemp, T.J.; Moore, J.M.; Griffith, T.S. Human B cells express functional TRAIL/Apo-2 ligand after CpG-containing oligodeoxynucleotide stimulation. J. Immunol. 2004, 173, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Ammirante, M.; Luo, J.L.; Grivennikov, S.; Nedospasov, S.; Karin, M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010, 464, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Olkhanud, P.B.; Damdinsuren, B.; Bodogai, M.; Gress, R.E.; Sen, R.; Wejksza, K.; Malchinkhuu, E.; Wersto, R.P.; Biragyn, A. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res. 2011, 71, 3505–3515. [Google Scholar] [CrossRef]
- Lindner, S.; Dahlke, K.; Sontheimer, K.; Hagn, M.; Kaltenmeier, C.; Barth, T.F.; Beyer, T.; Reister, F.; Fabricius, D.; Lotfi, R.; et al. Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer Res. 2013, 73, 2468–2479. [Google Scholar] [CrossRef]
- Jahrsdorfer, B.; Blackwell, S.E.; Wooldridge, J.E.; Huang, J.; Andreski, M.W.; Jacobus, L.S.; Taylor, C.M.; Weiner, G.J. B-chronic lymphocytic leukemia cells and other B cells can produce granzyme B and gain cytotoxic potential after interleukin-21-based activation. Blood 2006, 108, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Das, S.; Handler, J.S.; Hajdu, C.H.; Coffre, M.; Koralov, S.B.; Bar-Sagi, D. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Dis. 2016, 6, 247–255. [Google Scholar] [CrossRef]
- Da Gama Duarte, J.; Peyper, J.M.; Blackburn, J.M. B cells and antibody production in melanoma. Mamm. Genome. 2018, 29, 790–805. [Google Scholar] [CrossRef]
- Reuschenbach, M.; von Knebel Doeberitz, M.; Wentzensen, N. A systematic review of humoral immune responses against tumor antigens. Cancer Immunol. Immunother. 2009, 58, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, M.; Hanagiri, T.; Yasuda, M.; Kuroda, K.; Shigematsu, Y.; Baba, T.; Fukuyama, T.; Nagata, Y.; So, T.; Ichiki, Y.; et al. Antitumor effect of antibody against a SEREX-defined antigen (UOEH-LC-1) on lung cancer xenotransplanted into severe combined immunodeficiency mice. Cancer Res. 2007, 67, 8351–8357. [Google Scholar] [CrossRef] [PubMed]
- Carmi, Y.; Spitzer, M.H.; Linde, I.L.; Burt, B.M.; Prestwood, T.R.; Perlman, N.; Davidson, M.G.; Kenkel, J.A.; Segal, E.; Pusapati, G.V.; et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature 2015, 521, 99–104. [Google Scholar] [CrossRef]
- Lu, L.; Weng, C.; Mao, H.; Fang, X.; Liu, X.; Wu, Y.; Cao, X.; Li, B.; Chen, X.; Gan, Q.; et al. IL-17A promotes migration and tumor killing capability of B cells in esophageal squamous cell carcinoma. Oncotarget 2016, 7, 21853–21864. [Google Scholar] [CrossRef]
- Rubtsov, A.V.; Rubtsova, K.; Kappler, J.W.; Jacobelli, J.; Friedman, R.S.; Marrack, P. CD11c-Expressing B Cells Are Located at the T Cell/B Cell Border in Spleen and Are Potent APCs. J. Immunol. 2015, 195, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pinto, D. B cells as antigen presenting cells. Cell Immunol. 2005, 238, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Bruno, T.C.; Ebner, P.J.; Moore, B.L.; Squalls, O.G.; Waugh, K.A.; Eruslanov, E.B.; Singhal, S.; Mitchell, J.D.; Franklin, W.A.; Merrick, D.T.; et al. Antigen-Presenting Intratumoral B Cells Affect CD4(+) TIL Phenotypes in Non-Small Cell Lung Cancer Patients. Cancer Immunol. Res. 2017, 5, 898–907. [Google Scholar] [CrossRef]
- Pucci, F.; Garris, C.; Lai, C.P.; Newton, A.; Pfirschke, C.; Engblom, C.; Alvarez, D.; Sprachman, M.; Evavold, C.; Magnuson, A.; et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science 2016, 352, 242–246. [Google Scholar] [CrossRef]
- Yang, C.; Lee, H.; Pal, S.; Jove, V.; Deng, J.; Zhang, W.; Hoon, D.S.; Wakabayashi, M.; Forman, S.; Yu, H. B cells promote tumor progression via STAT3 regulated-angiogenesis. PLoS ONE 2013, 8, e64159. [Google Scholar] [CrossRef]
- Iwata, Y.; Matsushita, T.; Horikawa, M.; Dilillo, D.J.; Yanaba, K.; Venturi, G.M.; Szabolcs, P.M.; Bernstein, S.H.; Magro, C.M.; Williams, A.D.; et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 2011, 117, 530–541. [Google Scholar] [CrossRef]
- Lv, Y.; Wang, H.; Liu, Z. The Role of Regulatory B Cells in Patients with Acute Myeloid Leukemia. Med. Sci. Monit. 2019, 25, 3026–3031. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Lao, X.M.; Chen, M.M.; Liu, R.X.; Wei, Y.; Ouyang, F.Z.; Chen, D.P.; Zhao, X.Y.; Zhao, Q.; Li, X.F.; et al. PD-1hi Identifies a Novel Regulatory B-cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Dis. 2016, 6, 546–559. [Google Scholar] [CrossRef]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Schioppa, T.; Moore, R.; Thompson, R.G.; Rosser, E.C.; Kulbe, H.; Nedospasov, S.; Mauri, C.; Coussens, L.M.; Balkwill, F.R. B regulatory cells and the tumor-promoting actions of TNF-alpha during squamous carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 10662–10667. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Bar-Sagi, D. BTK signaling drives CD1d(hi)CD5(+) regulatory B-cell differentiation to promote pancreatic carcinogenesis. Oncogene 2019. [Google Scholar] [CrossRef]
- Shao, Y.; Lo, C.M.; Ling, C.C.; Liu, X.B.; Ng, K.T.; Chu, A.C.; Ma, Y.Y.; Li, C.X.; Fan, S.T.; Man, K. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett. 2014, 355, 264–272. [Google Scholar] [CrossRef]
- Zhao, D.M.; Thornton, A.M.; DiPaolo, R.J.; Shevach, E.M. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood 2006, 107, 3925–3932. [Google Scholar] [CrossRef]
- Shen, M.; Wang, J.; Yu, W.; Zhang, C.; Liu, M.; Wang, K.; Yang, L.; Wei, F.; Wang, S.E.; Sun, Q.; et al. A novel MDSC-induced PD-1(-)PD-L1(+) B-cell subset in breast tumor microenvironment possesses immuno-suppressive properties. Oncoimmunology 2018, 7, e1413520. [Google Scholar] [CrossRef]
- Wang, Y.; Schafer, C.C.; Hough, K.P.; Tousif, S.; Duncan, S.R.; Kearney, J.F.; Ponnazhagan, S.; Hsu, H.C.; Deshane, J.S. Myeloid-Derived Suppressor Cells Impair B Cell Responses in Lung Cancer through IL-7 and STAT5. J. Immunol. 2018, 201, 278–295. [Google Scholar] [CrossRef]
- Lee, S.; Margolin, K. Cytokines in cancer immunotherapy. Cancers 2011, 3, 3856–3893. [Google Scholar] [CrossRef] [PubMed]
- Wejksza, K.; Lee-Chang, C.; Bodogai, M.; Bonzo, J.; Gonzalez, F.J.; Lehrmann, E.; Becker, K.; Biragyn, A. Cancer-produced metabolites of 5-lipoxygenase induce tumor-evoked regulatory B cells via peroxisome proliferator-activated receptor alpha. J. Immunol. 2013, 190, 2575–2584. [Google Scholar] [CrossRef]
- Pimenta, E.M.; De, S.; Weiss, R.; Feng, D.; Hall, K.; Kilic, S.; Bhanot, G.; Ganesan, S.; Ran, S.; Barnes, B.J. IRF5 is a novel regulator of CXCL13 expression in breast cancer that regulates CXCR5(+) B- and T-cell trafficking to tumor-conditioned media. Immunol. Cell Biol. 2015, 93, 486–499. [Google Scholar] [CrossRef]
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075. [Google Scholar] [CrossRef]
- Somasundaram, R.; Zhang, G.; Fukunaga-Kalabis, M.; Perego, M.; Krepler, C.; Xu, X.; Wagner, C.; Hristova, D.; Zhang, J.; Tian, T.; et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat. Commun. 2017, 8, 607. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhang, Q.; Cheng, Y.; Chen, X.; Wang, G.; Shi, M.; Zhang, T.; Cao, Y.; Pan, H.; Zhang, L.; et al. Tumor-derived exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by promoting TIM-1(+) regulatory B cell expansion. J. Immunother. Cancer 2018, 6, 145. [Google Scholar] [CrossRef]
- Han, S.; Feng, S.; Ren, M.; Ma, E.; Wang, X.; Xu, L.; Xu, M. Glioma cell-derived placental growth factor induces regulatory B cells. Int. J. Biochem. Cell Biol. 2014, 57, 63–68. [Google Scholar] [CrossRef]
- Escors, D.; Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Garcia-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal. Transduct. Target Ther. 2018, 3, 26. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Minato, N.; Nakano, T.; Honjo, T. Immunological studies on PD-1 deficient mice: Implication of PD-1 as a negative regulator for B cell responses. Int. Immunol. 1998, 10, 1563–1572. [Google Scholar] [CrossRef]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef]
- Haas, K.M. Programmed cell death 1 suppresses B-1b cell expansion and long-lived IgG production in response to T cell-independent type 2 antigens. J. Immunol. 2011, 187, 5183–5195. [Google Scholar] [CrossRef]
- Haro, M.A.; Littrell, C.A.; Yin, Z.; Huang, X.; Haas, K.M. PD-1 Suppresses Development of Humoral Responses That Protect against Tn-Bearing Tumors. Cancer Immunol. Res. 2016, 4, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef]
- Gnanaprakasam, J.N.R.; Sherman, J.W.; Wang, R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 2017, 35, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, S.E.; O’Neill, L.A. HIF1alpha and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Caro-Maldonado, A.; Wang, R.; Nichols, A.G.; Kuraoka, M.; Milasta, S.; Sun, L.D.; Gavin, A.L.; Abel, E.D.; Kelsoe, G.; Green, D.R.; et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J. Immunol. 2014, 192, 3626–3636. [Google Scholar] [CrossRef]
- Shin, D.H.; Lin, H.; Zheng, H.; Kim, K.S.; Kim, J.Y.; Chun, Y.S.; Park, J.W.; Nam, J.H.; Kim, W.K.; Zhang, Y.H.; et al. HIF-1alpha-mediated upregulation of TASK-2 K(+) channels augments Ca(2)(+) signaling in mouse B cells under hypoxia. J. Immunol. 2014, 193, 4924–4933. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Grotsch, B.; Luo, Y.; Knaup, K.X.; Wiesener, M.S.; Chen, X.X.; Jantsch, J.; Fillatreau, S.; Schett, G.; Bozec, A. Hypoxia-inducible factor-1alpha is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 2018, 9, 251. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Spata, M.; Bayne, L.J.; Buza, E.L.; Durham, A.C.; Allman, D.; Vonderheide, R.H.; Simon, M.C. Hif1a Deletion Reveals Pro-Neoplastic Function of B Cells in Pancreatic Neoplasia. Cancer Dis. 2016, 6, 256–269. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E.; et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015, 521, 94–98. [Google Scholar] [CrossRef]
- Affara, N.I.; Ruffell, B.; Medler, T.R.; Gunderson, A.J.; Johansson, M.; Bornstein, S.; Bergsland, E.; Steinhoff, M.; Li, Y.; Gong, Q.; et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell 2014, 25, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, R.; Yang, Y.; Shi, C.; Shen, Y.; Lu, C.; Chen, Y.; Zhou, W.; Lin, A.; Yu, L.; et al. Specific Decrease in B-Cell-Derived Extracellular Vesicles Enhances Post-Chemotherapeutic CD8(+) T Cell Responses. Immunity 2019. [Google Scholar] [CrossRef]
- Griffin, M.; Scotto, D.; Josephs, D.H.; Mele, S.; Crescioli, S.; Bax, H.J.; Pellizzari, G.; Wynne, M.D.; Nakamura, M.; Hoffmann, R.M.; et al. BRAF inhibitors: Resistance and the promise of combination treatments for melanoma. Oncotarget 2017, 8, 78174–78192. [Google Scholar] [CrossRef] [PubMed]
- Wierz, M.; Pierson, S.; Guyonnet, L.; Viry, E.; Lequeux, A.; Oudin, A.; Niclou, S.P.; Ollert, M.; Berchem, G.; Janji, B.; et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood 2018, 131, 1617–1621. [Google Scholar] [CrossRef]
- Yuan, J.; Gnjatic, S.; Li, H.; Powel, S.; Gallardo, H.F.; Ritter, E.; Ku, G.Y.; Jungbluth, A.A.; Segal, N.H.; Rasalan, T.S.; et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc. Natl. Acad. Sci. USA 2008, 105, 20410–20415. [Google Scholar] [CrossRef]
- DeFalco, J.; Harbell, M.; Manning-Bog, A.; Baia, G.; Scholz, A.; Millare, B.; Sumi, M.; Zhang, D.; Chu, F.; Dowd, C.; et al. Non-progressing cancer patients have persistent B cell responses expressing shared antibody paratopes that target public tumor antigens. Clin. Immunol. 2018, 187, 37–45. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998.e1020–1013.e1020. [Google Scholar] [CrossRef] [PubMed]
- Thibult, M.L.; Mamessier, E.; Gertner-Dardenne, J.; Pastor, S.; Just-Landi, S.; Xerri, L.; Chetaille, B.; Olive, D. PD-1 is a novel regulator of human B-cell activation. Int. Immunol. 2013, 25, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, H.M.; Brouwer, M.; Linsley, P.S.; van Lier, R.A. Activated T cells can induce high levels of CTLA-4 expression on B cells. J. Immunol. 1995, 155, 1776–1783. [Google Scholar]
- Kisielow, M.; Kisielow, J.; Capoferri-Sollami, G.; Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 2005, 35, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Lino, A.C.; Dang, V.D.; Lampropoulou, V.; Welle, A.; Joedicke, J.; Pohar, J.; Simon, Q.; Thalmensi, J.; Baures, A.; Fluhler, V.; et al. LAG-3 Inhibitory Receptor Expression Identifies Immunosuppressive Natural Regulatory Plasma Cells. Immunity 2018, 49, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Winer, A.; Bodor, J.N.; Borghaei, H. Identifying and managing the adverse effects of immune checkpoint blockade. J. Thorac. Dis. 2018, 10, S480–S489. [Google Scholar] [CrossRef]
- Das, R.; Bar, N.; Ferreira, M.; Newman, A.M.; Zhang, L.; Bailur, J.K.; Bacchiocchi, A.; Kluger, H.; Wei, W.; Halaban, R.; et al. Early B cell changes predict autoimmunity following combination immune checkpoint blockade. J. Clin. Investig. 2018, 128, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Da Gama Duarte, J.; Parakh, S.; Andrews, M.C.; Woods, K.; Pasam, A.; Tutuka, C.; Ostrouska, S.; Blackburn, J.M.; Behren, A.; Cebon, J. Autoantibodies May Predict Immune-Related Toxicity: Results from a Phase I Study of Intralesional Bacillus Calmette-Guerin followed by Ipilimumab in Patients with Advanced Metastatic Melanoma. Front. Immunol. 2018, 9, 411. [Google Scholar] [CrossRef]
- Gowen, M.F.; Giles, K.M.; Simpson, D.; Tchack, J.; Zhou, H.; Moran, U.; Dawood, Z.; Pavlick, A.C.; Hu, S.; Wilson, M.A.; et al. Baseline antibody profiles predict toxicity in melanoma patients treated with immune checkpoint inhibitors. J. Trans. Med. 2018, 16, 82. [Google Scholar] [CrossRef]
- Ahmadi, T.; Flies, A.; Efebera, Y.; Sherr, D.H. CD40 Ligand-activated, antigen-specific B cells are comparable to mature dendritic cells in presenting protein antigens and major histocompatibility complex class I- and class II-binding peptides. Immunology 2008, 124, 129–140. [Google Scholar] [CrossRef]
- Carpenter, E.L.; Mick, R.; Ruter, J.; Vonderheide, R.H. Activation of human B cells by the agonist CD40 antibody CP-870,893 and augmentation with simultaneous toll-like receptor 9 stimulation. J. Trans. Med. 2009, 7, 93. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Draube, A.; Liebig, T.M.; Rothe, A.; Kochanek, M.; von Bergwelt-Baildon, M.S. The immunosuppressive factors IL-10, TGF-beta, and VEGF do not affect the antigen-presenting function of CD40-activated B cells. J. Exp. Clin. Cancer Res. 2012, 31, 47. [Google Scholar] [CrossRef]
- Gonzalez, N.K.; Wennhold, K.; Balkow, S.; Kondo, E.; Bolck, B.; Weber, T.; Garcia-Marquez, M.; Grabbe, S.; Bloch, W.; von Bergwelt-Baildon, M.; et al. In vitro and in vivo imaging of initial B-T-cell interactions in the setting of B-cell based cancer immunotherapy. Oncoimmunology 2015, 4, e1038684. [Google Scholar] [CrossRef]
- Rossetti, R.A.M.; Lorenzi, N.P.C.; Yokochi, K.; Rosa, M.; Benevides, L.; Margarido, P.F.R.; Baracat, E.C.; Carvalho, J.P.; Villa, L.L.; Lepique, A.P. B lymphocytes can be activated to act as antigen presenting cells to promote anti-tumor responses. PLoS ONE 2018, 13, e0199034. [Google Scholar] [CrossRef]
- Wennhold, K.; Weber, T.M.; Klein-Gonzalez, N.; Thelen, M.; Garcia-Marquez, M.; Chakupurakal, G.; Fiedler, A.; Schlosser, H.A.; Fischer, R.; Theurich, S.; et al. CD40-activated B cells induce anti-tumor immunity in vivo. Oncotarget 2017, 8, 27740–27753. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lao, X.; Pan, Q.; Ning, N.; Yet, J.; Xu, Y.; Li, S.; Chang, A.E. Adoptive transfer of tumor reactive B cells confers host T-cell immunity and tumor regression. Clin. Cancer Res. 2011, 17, 4987–4995. [Google Scholar] [CrossRef]
- Sorenmo, K.U.; Krick, E.; Coughlin, C.M.; Overley, B.; Gregor, T.P.; Vonderheide, R.H.; Mason, N.J. CD40-activated B cell cancer vaccine improves second clinical remission and survival in privately owned dogs with non-Hodgkin’s lymphoma. PLoS ONE 2011, 6, e24167. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, R.; Morello, S.; Forte, G.; Montinaro, A.; De Vita, G.; Luciano, A.; Palma, G.; Arra, C.; Maiolino, P.; Adcock, I.M.; et al. B cells contribute to the antitumor activity of CpG-oligodeoxynucleotide in a mouse model of metastatic lung carcinoma. Am. J. Respir. Crit. Care Med. 2011, 183, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Yuan, S.; Pennati, A.; Murphy, J.; Wu, J.H.; Lawson, D.; Galipeau, J. Engineered fusokine GIFT4 licenses the ability of B cells to trigger a tumoricidal T-cell response. Cancer Res. 2014, 74, 4133–4144. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhou, M.; Ren, H.; Hu, H.M.; Lu, L.; Cao, M.; Wang, L.X. Tumor-derived autophagosomes (DRibbles) induce B cell activation in a TLR2-MyD88 dependent manner. PLoS ONE 2013, 8, e53564. [Google Scholar] [CrossRef]
- Ren, H.; Zhao, S.; Li, W.; Dong, H.; Zhou, M.; Cao, M.; Hu, H.M.; Wang, L.X. Therapeutic antitumor efficacy of B cells loaded with tumor-derived autophagasomes vaccine (DRibbles). J. Immunother. 2014, 37, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, D.; Choi, G.; de Bruyn, M.; Wiersma, V.R.; Bremer, E. Antibody-Based Cancer Therapy: Successful Agents and Novel Approaches. Int. Rev. Cell Mol. Biol. 2017, 331, 289–383. [Google Scholar] [CrossRef]
- Scott, A.M.; Allison, J.P.; Wolchok, J.D. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012, 12, 14. [Google Scholar]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Pavoni, E.; Monteriu, G.; Santapaola, D.; Petronzelli, F.; Anastasi, A.M.; Pelliccia, A.; D’Alessio, V.; De Santis, R.; Minenkova, O. Tumor-infiltrating B lymphocytes as an efficient source of highly specific immunoglobulins recognizing tumor cells. BMC Biotechnol. 2007, 7, 70. [Google Scholar] [CrossRef]
- Kotlan, B.; Simsa, P.; Teillaud, J.L.; Fridman, W.H.; Toth, J.; McKnight, M.; Glassy, M.C. Novel ganglioside antigen identified by B cells in human medullary breast carcinomas: The proof of principle concerning the tumor-infiltrating B lymphocytes. J. Immunol. 2005, 175, 2278–2285. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Komura, D.; Konishi, H.; Suzuki, R.; Yamamoto, A.; Kakiuchi, M.; Sato, R.; Ushiku, T.; Yamamoto, S.; Tatsuno, K.; et al. Immunogenetic Profiling for Gastric Cancers Identifies Sulfated Glycosaminoglycans as Major and Functional B Cell Antigens in Human Malignancies. Cell Rep. 2017, 20, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Feng, Q.; Wang, C.; Zeng, X.; Du, Y.; Lin, L.; Wu, J.; Fu, L.; Yang, K.; Xu, X.; et al. Characterization of the B Cell Receptor Repertoire in the Intestinal Mucosa and of Tumor-Infiltrating Lymphocytes in Colorectal Adenoma and Carcinoma. J. Immunol. 2017, 198, 3719–3728. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H. Sequencing the functional antibody repertoire—Diagnostic and therapeutic discovery. Nat. Rev. Rheumatol. 2015, 11, 171–182. [Google Scholar] [CrossRef]
- Barbera-Guillem, E.; Nelson, M.B.; Barr, B.; Nyhus, J.K.; May, K.F., Jr.; Feng, L.; Sampsel, J.W. B lymphocyte pathology in human colorectal cancer. Experimental and clinical therapeutic effects of partial B cell depletion. Cancer Immunol. Immunother. 2000, 48, 541–549. [Google Scholar] [CrossRef]
- Winkler, J.K.; Schiller, M.; Bender, C.; Enk, A.H.; Hassel, J.C. Rituximab as a therapeutic option for patients with advanced melanoma. Cancer Immunol. Immunother. 2018, 67, 917–924. [Google Scholar] [CrossRef]
- Pinc, A.; Somasundaram, R.; Wagner, C.; Hormann, M.; Karanikas, G.; Jalili, A.; Bauer, W.; Brunner, P.; Grabmeier-Pfistershammer, K.; Gschaider, M.; et al. Targeting CD20 in melanoma patients at high risk of disease recurrence. Mol. Ther. 2012, 20, 1056–1062. [Google Scholar] [CrossRef]
- Theurich, S.; Schlaak, M.; Steguweit, H.; Heukamp, L.C.; Wennhold, K.; Kurschat, P.; Rabenhorst, A.; Hartmann, K.; Schlosser, H.; Shimabukuro-Vornhagen, A.; et al. Targeting Tumor-Infiltrating B Cells in Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2016, 34, e110–e116. [Google Scholar] [CrossRef] [PubMed]
- DiLillo, D.J.; Yanaba, K.; Tedder, T.F. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: Therapeutic B cell depletion enhances B16 melanoma growth in mice. J. Immunol. 2010, 184, 4006–4016. [Google Scholar] [CrossRef] [PubMed]
- Aklilu, M.; Stadler, W.M.; Markiewicz, M.; Vogelzang, N.J.; Mahowald, M.; Johnson, M.; Gajewski, T.F. Depletion of normal B cells with rituximab as an adjunct to IL-2 therapy for renal cell carcinoma and melanoma. Ann. Oncol. 2004, 15, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Bodogai, M.; Lee Chang, C.; Wejksza, K.; Lai, J.; Merino, M.; Wersto, R.P.; Gress, R.E.; Chan, A.C.; Hesdorffer, C.; Biragyn, A. Anti-CD20 antibody promotes cancer escape via enrichment of tumor-evoked regulatory B cells expressing low levels of CD20 and CD137L. Cancer Res. 2013, 73, 2127–2138. [Google Scholar] [CrossRef]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treat Rev. 2017, 58, 41–50. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Dis. 2016, 6, 270–285. [Google Scholar] [CrossRef]
- Lee-Chang, C.; Bodogai, M.; Martin-Montalvo, A.; Wejksza, K.; Sanghvi, M.; Moaddel, R.; de Cabo, R.; Biragyn, A. Inhibition of breast cancer metastasis by resveratrol-mediated inactivation of tumor-evoked regulatory B cells. J. Immunol. 2013, 191, 4141–4151. [Google Scholar] [CrossRef]
- Song, S.S.; Yuan, P.F.; Li, P.P.; Wu, H.X.; Ni, W.J.; Lu, J.T.; Wei, W. Protective Effects of Total Glucosides of Paeony on N-nitrosodiethylamine-induced Hepatocellular Carcinoma in Rats via Down-regulation of Regulatory B Cells. Immunol. Investig. 2015, 44, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cheng, Q.; Tang, K.; Sun, Y.; Zhang, K.; Zhang, Y.; Luo, S.; Zhang, H.; Ye, D.; Huang, B. Lipid mediator lipoxin A4 inhibits tumor growth by targeting IL-10-producing regulatory B (Breg) cells. Cancer Lett. 2015, 364, 118–124. [Google Scholar] [CrossRef] [PubMed]




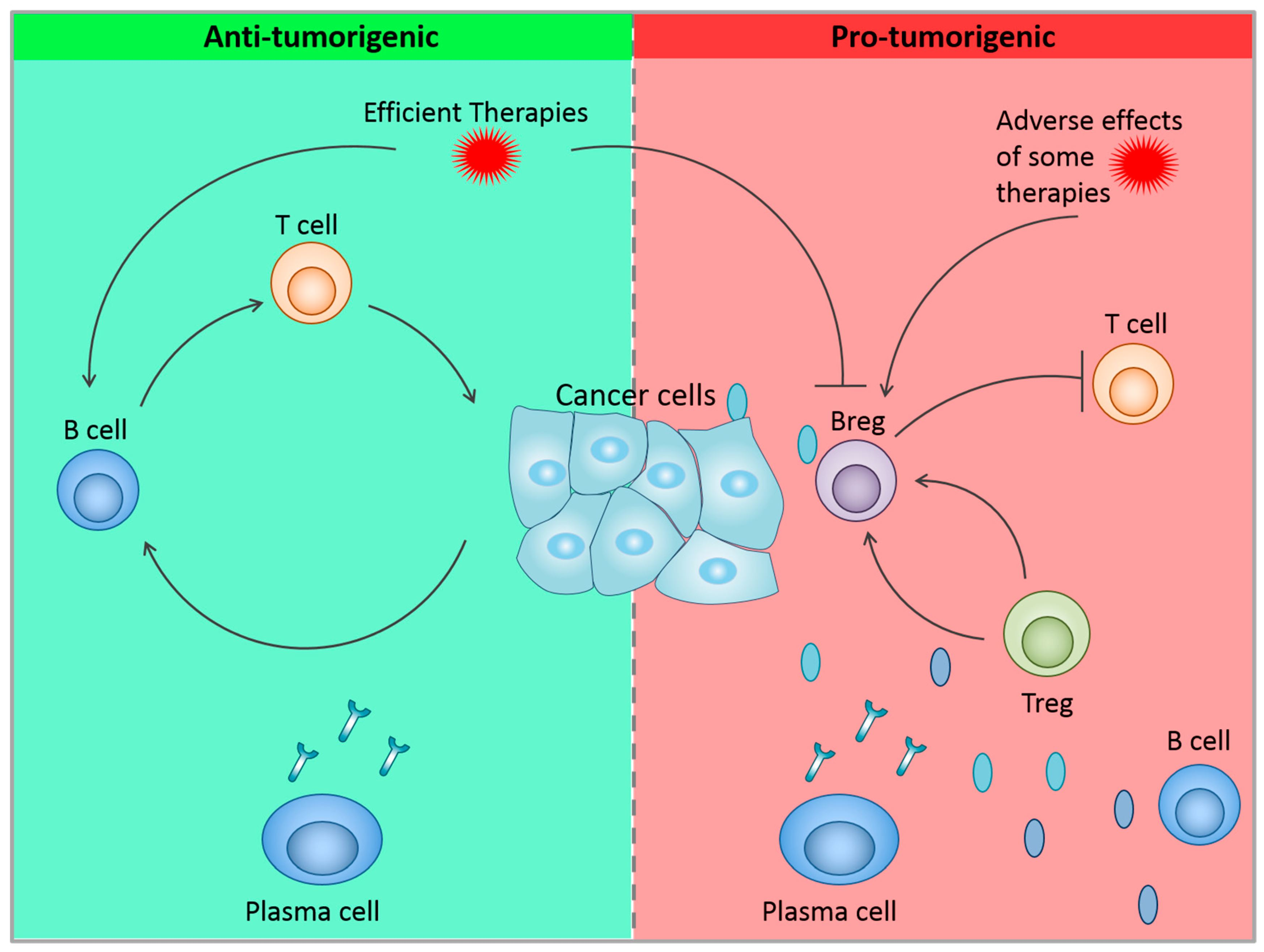{kind=link}
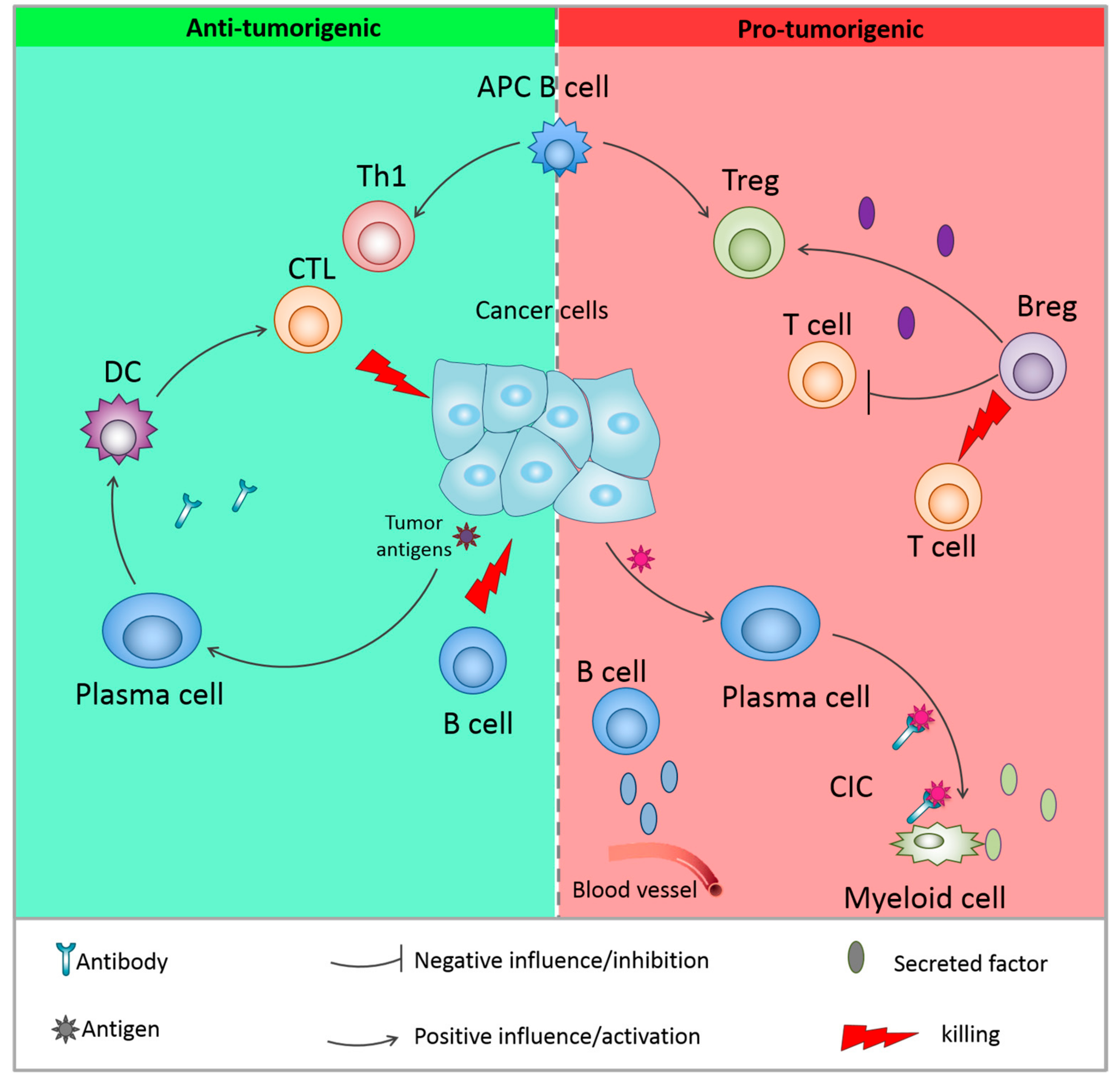{kind=link}
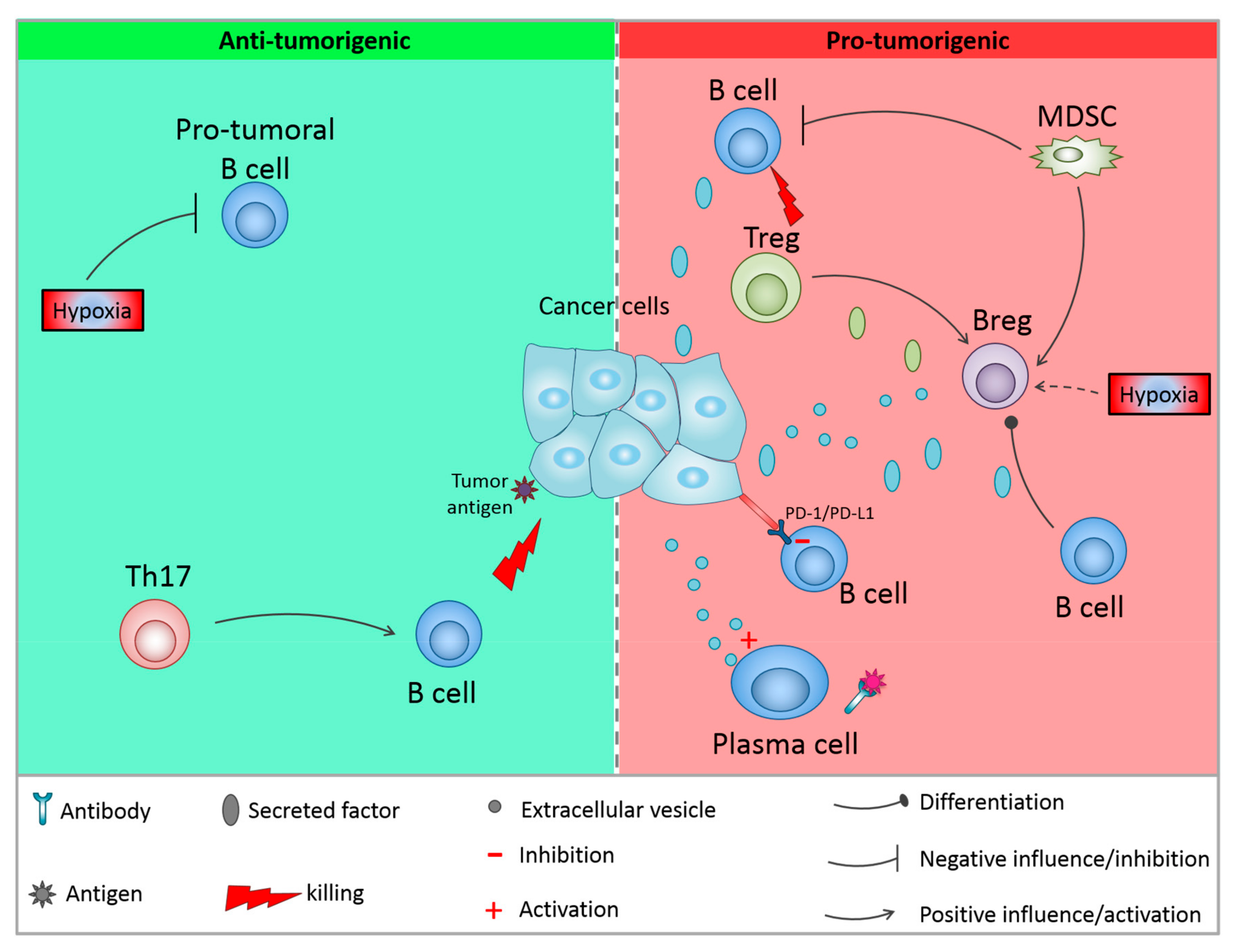{kind=link}
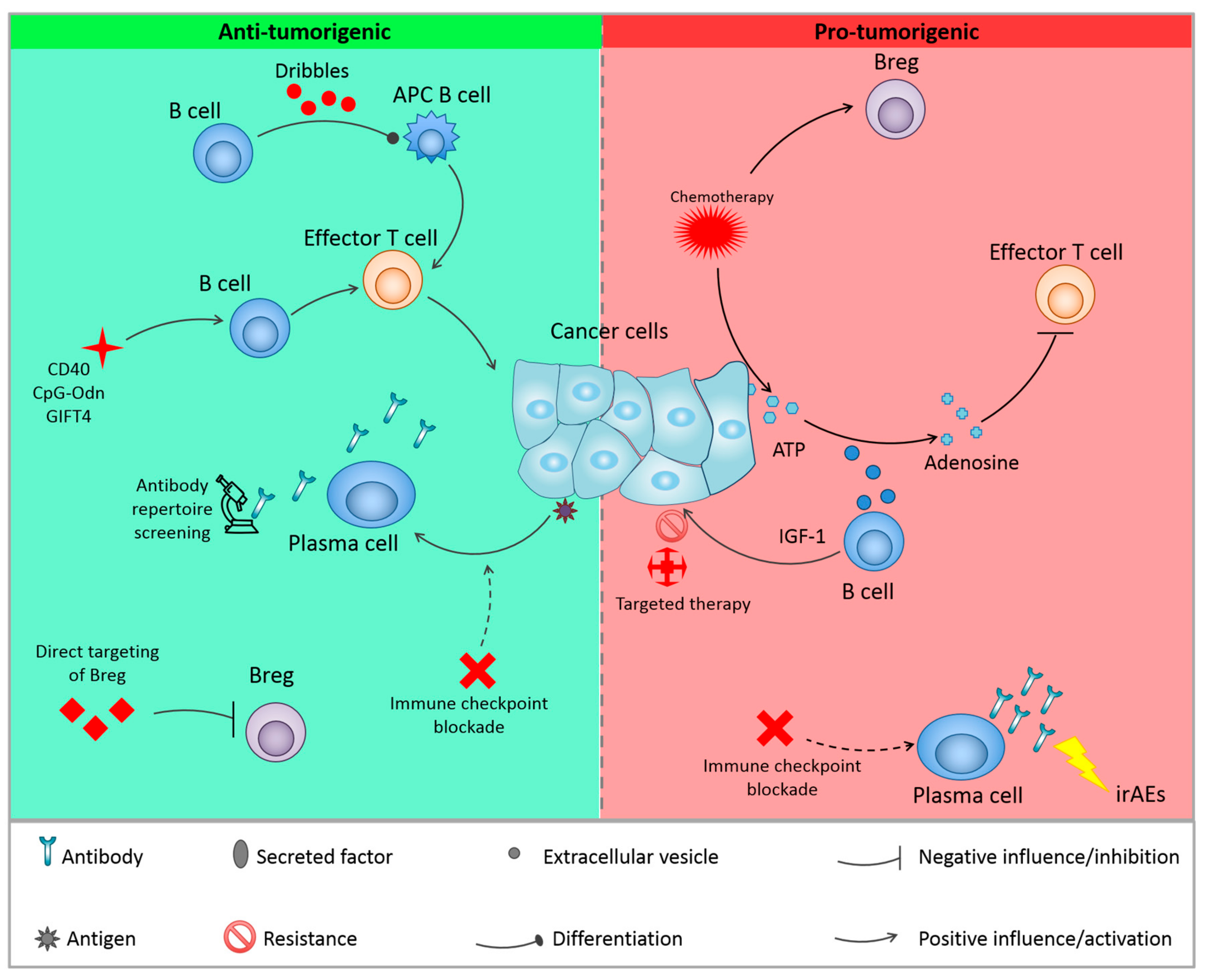{kind=link}
| Effector Molecules/Mechanism | Function | Reference | |
|---|---|---|---|
| Pro-Tumorigenic | Anti-Tumorigenic | ||
| Antibodies | Circulating immune complexes activate Fcγ receptors on immunosupressive myeloid cells, facilitate angiogenesis | Antibodies against tumor antigens, mediate complement -dependent lysis, ADCC, FcR-mediated phagocytosis, antigen presentation by DCs | [22,23,24,25] |
| Fas/FasL | Bregs inducing apoptosis in CD4+ T cells | Killing of tumor cells | [6] |
| TRAIL/Apo2L | Killing of tumor cells | [26] | |
| IL-10 | Produced by Bregs, exacerbate inflammation and support cancer growth, inhibit CD4+ T cells | [5,6,7,22,23,27] | |
| TGF-β | Convert naïve CD4+ T cells into Foxp3+ Tregs, upregulate reactive oxygen species (ROS) and nitric oxide (NO) in MDSCs | [28] | |
| Granzyme B | Transfer to T cells, degrading the T cell receptor ξ chain without inducing T cell apoptosis | Induce apoptosis in B-chronic lymphocytic leukemia cells | [29,30] |
| Lymphotoxin | Activates non-canonical and canonical NF-κB signaling and STAT3, inhibitory effect of B cells, survival signals to tumor cells | [27] | |
| IL-35 | Stimulates tumor growth | [31] | |
| IFN-y | Facilitate the killing of tumor cells by NK cells, polarize T cells towards Th1 or Th2 response | [5,6,7] | |
| Effect | Type of Therapy | B Cell Subtype/Function | Cancer Type/Mouse Model/Cell Line | References |
|---|---|---|---|---|
| Resistance | Oxaliplatin | IgA+ IL10+ PD-L1+ | Prostate | [74] |
| Platinum/Taxol-based chemotherapy | CD20+ | Carcinoma | [75] | |
| Chemotherapy (oxaliplatin, doxorubicin, phosphoramide mustard, cyclophosphamide | CD20+ EVs | B16F10, MC38 | [76] | |
| BRAF/MEK inhibitor | IGF-1 producing CD20+ | B16F10 | [56] | |
| Positive impact on response | Anti-CTLA4 | Presence of tumor specific antibodies | Melanoma | [79] |
| Anti-CTLA4, anti-PD-1, chemotherapy | Somatic hypermutation, IgG class switch, clonal expansion of B cells | Melanoma, lung and renal cell carcinomas | [80] | |
| immunotherapy | CD20+ enrichment | Melanoma | [81] | |
| Prediction of irAEs | Anti-CTLA4 and anti-PD-1 | CD21lo PD-1hi enrichment | Melanoma | [87] |
| BCG + anti-CTLA4 | Increase in anti–tumor antibodies repertoire | Melanoma | [88,89] |
| Type of Therapy | Target Cells/Effect on B Cells | Consequence | Cancer Type/Animal Model/Cell Line | References |
|---|---|---|---|---|
| Activation of B cells | ||||
| B cell–based vaccine–CD40 dependent activation | CD19+ | Activation of T cells, migration to secondary lymphoid organs | HPV16, B16-F10, E.G7, 4T1 metastasis, sarcoma, spontaneous NHL. | [25,92,94,95,96,97] |
| CpG-ODN | CD19+ | Metastasis regression, decrease immunosuppressive TME | B16-F10 | [98] |
| GIFT4 | Up-regulation of CD25, CD27, CD40, CD69, MHC class I/II, CD80, CD83 and CD86 expression | Activation of CTL | B16-F10 | [99] |
| DRibbles | MHC class I and II molecules, CD86 and CD40 | Activation of tumor specific T cells | Lymphoma, HCC | [100,101] |
| Inhibition/Depletion of B cells | ||||
| Anti-CD20 antibody | Depletion of CD20+ cells | Health benefit | colon cancer, melanoma, cutaneous T cell lymphoma | [110,111,112,113] |
| No effect or deleterious effect | B16-F10 Renal cell carcinoma, melanoma | [98,114,115,116] | ||
| BTK inhibitor | Depletion of IgMlo CD23+ CD5− and IgMlo CD23− B cells | Reduction in tumor growth | Pancreatic ductal adenocarcinoma | [117] |
| CXCL13-CpG-ODN | Depletion of CXCR5+ Bregs | Activation of CTL | 4T1 | [116] |
| Anti-IL10 antibody | Inhibition of Bregs | Increase efficiency of CD40-activated B cells | 4T1 | [6] |
| Resveratrol | Decrease in Breg number | Block metastasis formation | 4T1 | [120] |
| Total glucosides of paeony (TGP) | Decrease in Breg number | Improved survival | HCC | [121] |
| Lipoxin A4 | Inhibition of Breg conversion from naïve B cells | Decrease tumor growth | B16-F10, H22 | [122] |
| MK886 | Inhibition of Breg conversion from naïve B cells | Decrease tumor growth | B16-F10 | [53] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Largeot, A.; Pagano, G.; Gonder, S.; Moussay, E.; Paggetti, J. The B-Side of Cancer Immunity: The Underrated Tune. Cells 2019, 8, 449. https://doi.org/10.3390/cells8050449
Largeot A, Pagano G, Gonder S, Moussay E, Paggetti J. The B-Side of Cancer Immunity: The Underrated Tune. Cells. 2019; 8(5):449. https://doi.org/10.3390/cells8050449
Chicago/Turabian StyleLargeot, Anne, Giulia Pagano, Susanne Gonder, Etienne Moussay, and Jerome Paggetti. 2019. "The B-Side of Cancer Immunity: The Underrated Tune" Cells 8, no. 5: 449. https://doi.org/10.3390/cells8050449
APA StyleLargeot, A., Pagano, G., Gonder, S., Moussay, E., & Paggetti, J. (2019). The B-Side of Cancer Immunity: The Underrated Tune. Cells, 8(5), 449. https://doi.org/10.3390/cells8050449