Insights on CTC Biology and Clinical Impact Emerging from Advances in Capture Technology

Abstract
:1. Introduction
2. Diagnostic Importance of CTCs
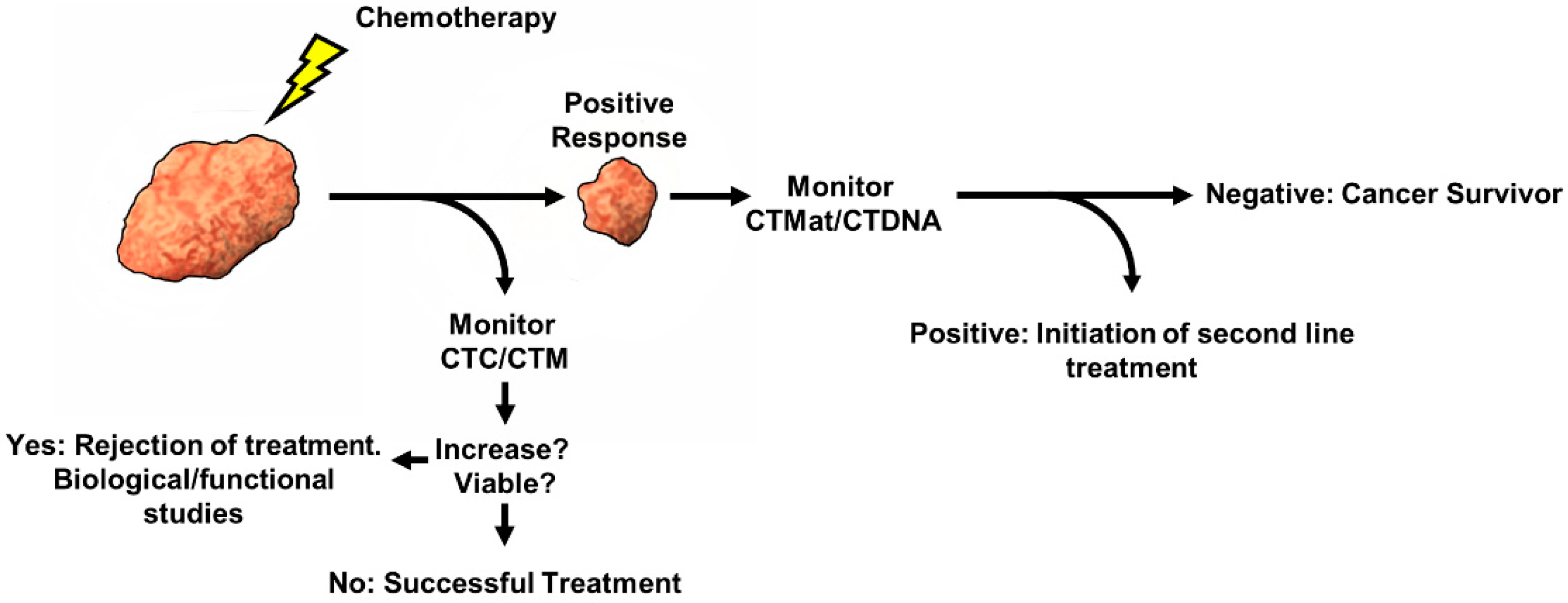
3. Clinical Relevance
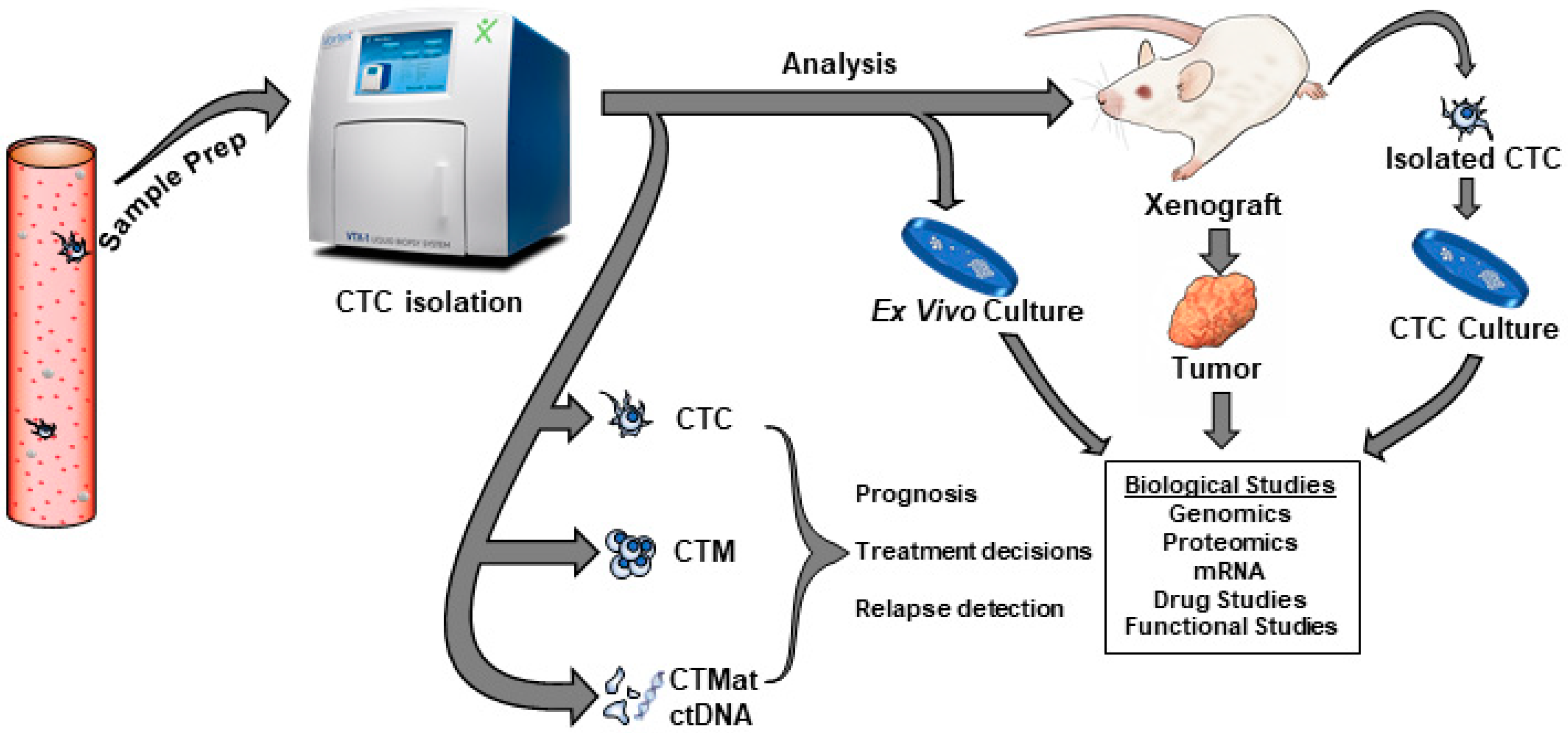
4. Isolation of Cells
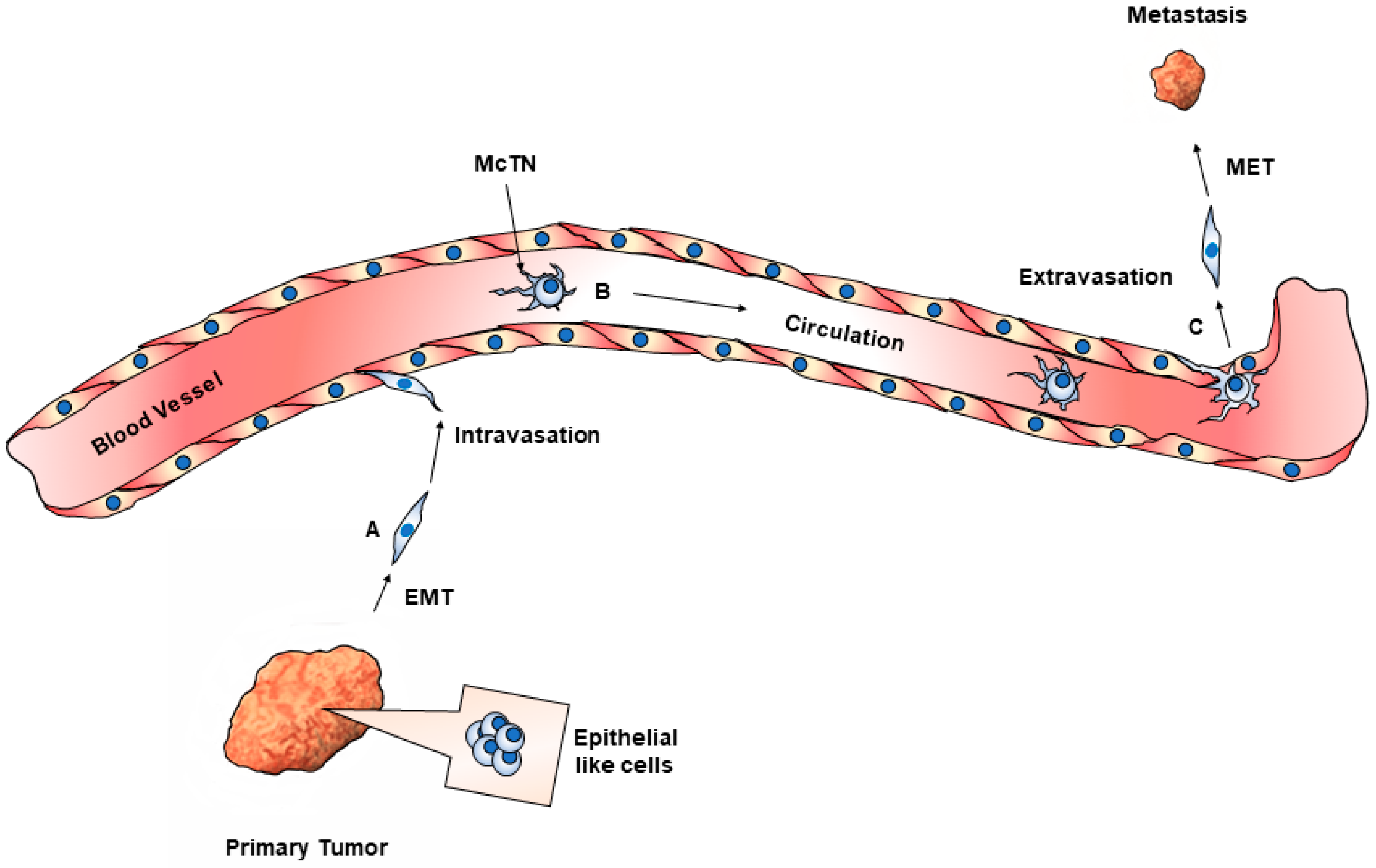
5. CTC Biology
6. Functional CTC Studies
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Nie, D.; Chakrabarty, S. Growth factors in tumor microenvironment. Front. Biosci. Landmark Ed. 2010, 15, 151–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, T.P.; Gullino, P.M. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975, 35, 512–516. [Google Scholar] [PubMed]
- Ross, A.A.; Cooper, B.W.; Lazarus, H.M.; Mackay, W.; Moss, T.J.; Ciobanu, N.; Tallman, M.S.; Kennedy, M.J.; Davidson, N.E.; Sweet, D.; et al. Detection and viability of tumor cells in peripheral blood stem cell collections from breast cancer patients using immunocytochemical and clonogenic assay techniques. Blood 1993, 82, 2605–2610. [Google Scholar] [PubMed]
- Hong, B.; Zu, Y. Detecting circulating tumor cells: Current challenges and new trends. Theranostics 2013, 3, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.M.; Krebs, M.G.; Lancashire, L.; Sloane, R.; Backen, A.; Swain, R.K.; Priest, L.J.; Greystoke, A.; Zhou, C.; Morris, K.; et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J. Clin. Oncol. 2012, 30, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Edge, S.; Greene, F.; Byrd, D.R.; Brookland, R.K.; Washington, M.K.; Gershenwald, J.E.; Compton, C.C.; Hess, K.R.; Sullivan, D.C.; et al. American Joint Committee on Cancer; American Cancer Society. AJCC Cancer Staging Manual, 8th ed.; American Joint Committee on Cancer; Springer: Chicago, IL, USA, 2017; p. xvii. 1024p. [Google Scholar]
- Mittendorf, E.A.; Bartlett, J.M.S.; Lichtensztajn, D.L.; Chandarlapaty, S. Incorporating Biology Into Breast Cancer Staging: American Joint Committee on Cancer, Eighth Edition, Revisions and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2018, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Sturgeon, C.M.; Duffy, M.J.; Stenman, U.H.; Lilja, H.; Brunner, N.; Chan, D.W.; Babaian, R.; Bast, R.C., Jr.; Dowell, B.; Esteva, F.J.; et al. National Academy of Clinical Biochemistry laboratory medicine practice guidelines for use of tumor markers in testicular, prostate, colorectal, breast, and ovarian cancers. Clin. Chem. 2008, 54, e11–e79. [Google Scholar] [CrossRef]
- Van der Toom, E.E.; Verdone, J.E.; Gorin, M.A.; Pienta, K.J. Technical challenges in the isolation and analysis of circulating tumor cells. Oncotarget 2016, 7, 62754–62766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, P.K.; Cummins, A.G.; Price, T.J.; Roberts-Thomson, I.C.; Hardingham, J.E. Circulating tumour cells: the evolving concept and the inadequacy of their enrichment by EpCAM-based methodology for basic and clinical cancer research. Ann. Oncol. 2014, 25, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Balasubramanian, P.; Chen, A.P.; Kummar, S.; Evrard, Y.A.; Kinders, R.J. Promise and limits of the CellSearch platform for evaluating pharmacodynamics in circulating tumor cells. Semin. Oncol. 2016, 43, 464–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, L.M.; Kindelberger, D.W.; Ligon, A.H.; Capelletti, M.; Fiorentino, M.; Loda, M.; Cibas, E.S.; Janne, P.A.; Krop, I.E. Improving the yield of circulating tumour cells facilitates molecular characterisation and recognition of discordant HER2 amplification in breast cancer. Br. J. Cancer 2010, 102, 1495–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, G.C.; Larson, C.; Repollet, M.; Rutner, H.; Terstappen, L.W.; O’hara, S.M.; Gross, S. Analysis of Circulating Tumor Cells, Fragments, and Debris. U.S. Patent 8,329,422, 11 December 2012. [Google Scholar]
- Coumans, F.A.; Doggen, C.J.; Attard, G.; de Bono, J.S.; Terstappen, L.W. All circulating EpCAM+CK+CD45- objects predict overall survival in castration-resistant prostate cancer. Ann. Oncol. 2010, 21, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Alix-Panabieres, C.; Muller, I.; Letang, N.; Vendrell, J.P.; Rebillard, X.; Pantel, K. Cell-free tumor DNA in blood plasma as a marker for circulating tumor cells in prostate cancer. Clin. Cancer Res. 2009, 15, 1032–1038. [Google Scholar] [CrossRef]
- Leary, R.J.; Kinde, I.; Diehl, F.; Schmidt, K.; Clouser, C.; Duncan, C.; Antipova, A.; Lee, C.; McKernan, K.; De La Vega, F.M.; et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci. Transl. Med. 2010, 2, 20ra14. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J. Medicine. Keeping tabs on tumor DNA. Science 2010, 327, 1074. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra392. [Google Scholar] [CrossRef]
- Schmiegel, W.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: Concordance of results from circulating tumor DNA and tissue-based RAS testing. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Bardelli, A. Genotyping cell-free tumor DNA in the blood to detect residual disease and drug resistance. Genome Biol. 2014, 15, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulz, P.; Heitzer, E.; Geigl, J.B.; Speicher, M.R. Patient monitoring through liquid biopsies using circulating tumor DNA. Int. J. Cancer 2017, 141, 887–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chera, B.S.; Kumar, S.; Beaty, B.T.; Marron, D.; Jefferys, S.R.; Green, R.L.; Goldman, E.C.; Amdur, R.; Sheets, N.; Dagan, R.; et al. Rapid Clearance Profile of Plasma Circulating Tumor HPV Type 16 DNA during Chemoradiotherapy Correlates with Disease Control in HPV-Associated Oropharyngeal Cancer. Clin Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.M.; Krebs, M.; Ward, T.; Morris, K.; Sloane, R.; Blackhall, F.; Dive, C. Circulating tumor cells, enumeration and beyond. Cancers 2010, 2, 1236–1250. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Morat, L.; Chauveinc, L.; Prapotnich, D.; De Crevoisier, R.; Escudier, B.; Cathelineau, X.; Rozet, F.; Vallancien, G.; Sabatier, L.; et al. High detection rate of circulating tumor cells in blood of patients with prostate cancer using telomerase activity. Ann. Oncol. 2007, 18, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Sapi, E.; Okpokwasili, N.I.; Rutherford, T. Detection of telomerase-positive circulating epithelial cells in ovarian cancer patients. Cancer Detect. Prev. 2002, 26, 158–167. [Google Scholar] [CrossRef]
- Soria, J.C.; Gauthier, L.R.; Raymond, E.; Granotier, C.; Morat, L.; Armand, J.P.; Boussin, F.D.; Sabatier, L. Molecular detection of telomerase-positive circulating epithelial cells in metastatic breast cancer patients. Clin. Cancer Res. 1999, 5, 971–975. [Google Scholar]
- Gauthier, L.R.; Granotier, C.; Soria, J.C.; Faivre, S.; Boige, V.; Raymond, E.; Boussin, F.D. Detection of circulating carcinoma cells by telomerase activity. Br. J. Cancer 2001, 84, 631–635. [Google Scholar] [CrossRef] [Green Version]
- Maurelli, R.; Zambruno, G.; Guerra, L.; Abbruzzese, C.; Dimri, G.; Gellini, M.; Bondanza, S.; Dellambra, E. Inactivation of p16INK4a (inhibitor of cyclin-dependent kinase 4A) immortalizes primary human keratinocytes by maintaining cells in the stem cell compartment. FASEB J. 2006, 20, 1516–1518. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Deneve, E.; Nocca, D.; Coffy, A.; Vendrell, J.P.; Maudelonde, T.; Riethdorf, S.; Alix-Panabieres, C. Circulating epithelial cells in patients with benign colon diseases. Clin. Chem. 2012, 58, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.A.; Anderson, R.L.; Narayan, K.; MacManus, M.P. Does the mobilization of circulating tumour cells during cancer therapy cause metastasis? Nat. Rev. Clin. Oncol. 2017, 14, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Mego, M.; Gao, H.; Lee, B.N.; Cohen, E.N.; Tin, S.; Giordano, A.; Wu, Q.; Liu, P.; Nieto, Y.; Champlin, R.E.; et al. Prognostic Value of EMT-Circulating Tumor Cells in Metastatic Breast Cancer Patients Undergoing High-Dose Chemotherapy with Autologous Hematopoietic Stem Cell Transplantation. J. Cancer 2012, 3, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Inhestern, J.; Oertel, K.; Stemmann, V.; Schmalenberg, H.; Dietz, A.; Rotter, N.; Veit, J.; Gorner, M.; Sudhoff, H.; Junghanss, C.; et al. Prognostic Role of Circulating Tumor Cells during Induction Chemotherapy Followed by Curative Surgery Combined with Postoperative Radiotherapy in Patients with Locally Advanced Oral and Oropharyngeal Squamous Cell Cancer. PLoS ONE 2015, 10, e0132901. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Pastoriza, J.M.; Wang, Y.; Harney, A.S.; Entenberg, D.; Pignatelli, J.; Sharma, V.P.; Xue, E.A.; Cheng, E.; D’Alfonso, T.M.; et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, W.; Patel, S.S.; Cong, J.; Zhang, N.; Sabbatino, F.; Liu, X.; Qi, Y.; Huang, P.; Lee, H.; et al. Blocking the formation of radiation-induced breast cancer stem cells. Oncotarget 2014, 5, 3743–3755. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Ghisolfi, L.; Keates, A.C.; Zhang, J.; Xiang, S.; Lee, D.K.; Li, C.J. Induction of cancer cell stemness by chemotherapy. Cell Cycle 2012, 11, 2691–2698. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.Y.; Tang, J.N.; Xie, H.X.; Du, Y.A.; Huang, L.; Yu, P.F.; Cheng, X.D. 5-Fluorouracil chemotherapy of gastric cancer generates residual cells with properties of cancer stem cells. Int. J. Biol. Sci. 2015, 11, 284–294. [Google Scholar] [CrossRef]
- Kaplan, H.S.; Murphy, E.D. The effect of local roentgen irradiation on the biological behavior of a transplantable mouse carcinoma; increased frequency of pulmonary metastasis. J. Natl. Cancer Inst. 1949, 9, 407–413. [Google Scholar]
- Sheldon, P.W.; Fowler, J.F. The effect of low-dose pre-operative X-irradiation of implanted mouse mammary carcinomas on local recurrence and metastasis. Br. J. Cancer 1976, 34, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camphausen, K.; Moses, M.A.; Beecken, W.D.; Khan, M.K.; Folkman, J.; O’Reilly, M.S. Radiation therapy to a primary tumor accelerates metastatic growth in mice. Cancer Res. 2001, 61, 2207–2211. [Google Scholar] [PubMed]
- Eriksson, D.; Stigbrand, T. Radiation-induced cell death mechanisms. Tumour Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Butof, R.; Dubrovska, A.; Baumann, M. Clinical perspectives of cancer stem cell research in radiation oncology. Radiother. Oncol. 2013, 108, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Galappathi, K.; Mathiesen, B.S. Tumor interstitial fluid pressure-a link between tumor hypoxia, microvascular density, and lymph node metastasis. Neoplasia 2014, 16, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Gorski, D.H.; Beckett, M.A.; Jaskowiak, N.T.; Calvin, D.P.; Mauceri, H.J.; Salloum, R.M.; Seetharam, S.; Koons, A.; Hari, D.M.; Kufe, D.W.; et al. Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 1999, 59, 3374–3378. [Google Scholar] [PubMed]
- Martin, O.A.; Anderson, R.L.; Russell, P.A.; Cox, R.A.; Ivashkevich, A.; Swierczak, A.; Doherty, J.P.; Jacobs, D.H.; Smith, J.; Siva, S.; et al. Mobilization of viable tumor cells into the circulation during radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Polascik, T.J.; Wang, Z.P.; Shue, M.; Di, S.; Gurganus, R.T.; Hortopan, S.C.; Ts’o, P.O.; Partin, A.W. Influence of sextant prostate needle biopsy or surgery on the detection and harvest of intact circulating prostate cancer cells. J. Urol. 1999, 162, 749–752. [Google Scholar] [CrossRef]
- Kusukawa, J.; Suefuji, Y.; Ryu, F.; Noguchi, R.; Iwamoto, O.; Kameyama, T. Dissemination of cancer cells into circulation occurs by incisional biopsy of oral squamous cell carcinoma. J. Oral Pathol. Med. 2000, 29, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Jones, O.M.; Rees, M.; John, T.G.; Bygrave, S.; Plant, G. Biopsy of resectable colorectal liver metastases causes tumour dissemination and adversely affects survival after liver resection. Br. J. Surg. 2005, 92, 1165–1168. [Google Scholar] [CrossRef]
- Weitz, J.; Kienle, P.; Lacroix, J.; Willeke, F.; Benner, A.; Lehnert, T.; Herfarth, C.; von Knebel Doeberitz, M. Dissemination of tumor cells in patients undergoing surgery for colorectal cancer. Clin. Cancer Res. 1998, 4, 343–348. [Google Scholar] [PubMed]
- Ferreira, M.M.; Ramani, V.C.; Jeffrey, S.S. Circulating tumor cell technologies. Mol. Oncol. 2016, 10, 374–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galletti, G.; Sung, M.S.; Vahdat, L.T.; Shah, M.A.; Santana, S.M.; Altavilla, G.; Kirby, B.J.; Giannakakou, P. Isolation of breast cancer and gastric cancer circulating tumor cells by use of an anti HER2-based microfluidic device. Lab Chip 2014, 14, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Mikolajczyk, S.D.; Millar, L.S.; Tsinberg, P.; Coutts, S.M.; Zomorrodi, M.; Pham, T.; Bischoff, F.Z.; Pircher, T.J. Detection of EpCAM-Negative and Cytokeratin-Negative Circulating Tumor Cells in Peripheral Blood. J. Oncol. 2011, 2011, 252361. [Google Scholar] [CrossRef] [PubMed]
- Kamande, J.W.; Hupert, M.L.; Witek, M.A.; Wang, H.; Torphy, R.J.; Dharmasiri, U.; Njoroge, S.K.; Jackson, J.M.; Aufforth, R.D.; Snavely, A.; et al. Modular microsystem for the isolation, enumeration, and phenotyping of circulating tumor cells in patients with pancreatic cancer. Anal. Chem. 2013, 85, 9092–9100. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, K.; Liu, J.; Yu, Z.T.; Xu, X.; Zhao, L.; Lee, T.; Lee, E.K.; Reiss, J.; Lee, Y.K.; et al. Highly efficient capture of circulating tumor cells by using nanostructured silicon substrates with integrated chaotic micromixers. Angew. Chem. Int. Ed. Engl. 2011, 50, 3084–3088. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Ogunwobi, O.O.; Chen, T.; Zhang, J.; George, T.J.; Liu, C.; Fan, Z.H. Capture, release and culture of circulating tumor cells from pancreatic cancer patients using an enhanced mixing chip. Lab Chip 2014, 14, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.A.; Okagbare, P.I.; Feng, J.; Hupert, M.L.; Patterson, D.; Gottert, J.; McCarley, R.L.; Nikitopoulos, D.; Murphy, M.C.; Soper, S.A. Highly efficient circulating tumor cell isolation from whole blood and label-free enumeration using polymer-based microfluidics with an integrated conductivity sensor. J. Am. Chem. Soc. 2008, 130, 8633–8641. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.J.; Kim, T.H.; Zhang, Z.; Azizi, E.; Pham, T.M.; Paoletti, C.; Lin, J.; Ramnath, N.; Wicha, M.S.; Hayes, D.F.; et al. Sensitive capture of circulating tumour cells by functionalized graphene oxide nanosheets. Nat. Nanotechnol. 2013, 8, 735–741. [Google Scholar] [CrossRef]
- Stott, S.L.; Hsu, C.H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saliba, A.E.; Saias, L.; Psychari, E.; Minc, N.; Simon, D.; Bidard, F.C.; Mathiot, C.; Pierga, J.Y.; Fraisier, V.; Salamero, J.; et al. Microfluidic sorting and multimodal typing of cancer cells in self-assembled magnetic arrays. Proc. Natl. Acad. Sci. USA 2010, 107, 14524–14529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer-Jones, J.P.; Vahidi, B.; Arquilevich, N.; Fang, C.; Ferguson, S.; Harkins, D.; Hill, C.; Klem, E.; Pagano, P.C.; Peasley, C.; et al. Circulating tumor cells: Clinically relevant molecular access based on a novel CTC flow cell. PLoS ONE 2014, 9, e86717. [Google Scholar] [CrossRef] [PubMed]
- Harb, W.; Fan, A.; Tran, T.; Danila, D.C.; Keys, D.; Schwartz, M.; Ionescu-Zanetti, C. Mutational Analysis of Circulating Tumor Cells Using a Novel Microfluidic Collection Device and qPCR Assay. Transl. Oncol. 2013, 6, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Musella, V.; Pietrantonio, F.; Di Buduo, E.; Iacovelli, R.; Martinetti, A.; Sottotetti, E.; Bossi, I.; Maggi, C.; Di Bartolomeo, M.; de Braud, F.; et al. Circulating tumor cells as a longitudinal biomarker in patients with advanced chemorefractory, RAS-BRAF wild-type colorectal cancer receiving cetuximab or panitumumab. Int. J. Cancer 2015, 137, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Pluim, D.; Devriese, L.A.; Beijnen, J.H.; Schellens, J.H. Validation of a multiparameter flow cytometry method for the determination of phosphorylated extracellular-signal-regulated kinase and DNA in circulating tumor cells. Cytometry A 2012, 81, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Zhang, Y.; Sun, S.; Wang, Z.; Wang, M.; Yu, B.; Czajkowsky, D.M.; Liu, B.; Li, Y.; Wei, W.; et al. An integrated microfluidic chip system for single-cell secretion profiling of rare circulating tumor cells. Sci. Rep. 2014, 4, 7499. [Google Scholar] [CrossRef]
- Saucedo-Zeni, N.; Mewes, S.; Niestroj, R.; Gasiorowski, L.; Murawa, D.; Nowaczyk, P.; Tomasi, T.; Weber, E.; Dworacki, G.; Morgenthaler, N.G.; et al. A novel method for the in vivo isolation of circulating tumor cells from peripheral blood of cancer patients using a functionalized and structured medical wire. Int. J. Oncol. 2012, 41, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Fusi, A.; Klopocki, E.; Schmittel, A.; Tinhofer, I.; Nonnenmacher, A.; Keilholz, U. Negative enrichment by immunomagnetic nanobeads for unbiased characterization of circulating tumor cells from peripheral blood of cancer patients. J. Transl. Med. 2011, 9, 70. [Google Scholar] [CrossRef]
- Lara, O.; Tong, X.; Zborowski, M.; Chalmers, J.J. Enrichment of rare cancer cells through depletion of normal cells using density and flow-through, immunomagnetic cell separation. Exp. Hematol. 2004, 32, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Lara, O.; Tong, X.; Zborowski, M.; Farag, S.S.; Chalmers, J.J. Comparison of two immunomagnetic separation technologies to deplete T cells from human blood samples. Biotechnol. Bioeng. 2006, 94, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Ozkumur, E.; Shah, A.M.; Ciciliano, J.C.; Emmink, B.L.; Miyamoto, D.T.; Brachtel, E.; Yu, M.; Chen, P.I.; Morgan, B.; Trautwein, J.; et al. Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci. Transl. Med. 2013, 5, 179ra147. [Google Scholar] [CrossRef] [PubMed]
- Harouaka, R.A.; Nisic, M.; Zheng, S.Y. Circulating tumor cell enrichment based on physical properties. J. Lab. Autom. 2013, 18, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.; Gertler, R.; Friederichs, J.; Fuehrer, K.; Dahm, M.; Phelps, R.; Thorban, S.; Nekarda, H.; Siewert, J.R. Comparison of two density gradient centrifugation systems for the enrichment of disseminated tumor cells in blood. Cytometry 2002, 49, 150–158. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Kularatne, S.A.; Kalli, K.R.; Prendergast, F.G.; Amato, R.J.; Klee, G.G.; Hartmann, L.C.; Low, P.S. Quantitation of circulating tumor cells in blood samples from ovarian and prostate cancer patients using tumor-specific fluorescent ligands. Int. J. Cancer 2008, 123, 1968–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campton, D.E.; Ramirez, A.B.; Nordberg, J.J.; Drovetto, N.; Clein, A.C.; Varshavskaya, P.; Friemel, B.H.; Quarre, S.; Breman, A.; Dorschner, M.; et al. High-recovery visual identification and single-cell retrieval of circulating tumor cells for genomic analysis using a dual-technology platform integrated with automated immunofluorescence staining. BMC Cancer 2015, 15, 360. [Google Scholar] [CrossRef]
- Xu, L.; Mao, X.; Imrali, A.; Syed, F.; Mutsvangwa, K.; Berney, D.; Cathcart, P.; Hines, J.; Shamash, J.; Lu, Y.J. Optimization and Evaluation of a Novel Size Based Circulating Tumor Cell Isolation System. PLoS ONE 2015, 10, e0138032. [Google Scholar] [CrossRef]
- Mohamed, H.; Murray, M.; Turner, J.N.; Caggana, M. Isolation of tumor cells using size and deformation. J. Chromatogr. A 2009, 1216, 8289–8295. [Google Scholar] [CrossRef]
- Yanagita, M.; Luke, J.J.; Hodi, F.S.; Janne, P.A.; Paweletz, C.P. Isolation and characterization of circulating melanoma cells by size filtration and fluorescent in-situ hybridization. Melanoma Res. 2018, 28, 89–95. [Google Scholar] [CrossRef]
- Qin, X.; Park, S.; Duffy, S.P.; Matthews, K.; Ang, R.R.; Todenhofer, T.; Abdi, H.; Azad, A.; Bazov, J.; Chi, K.N.; et al. Size and deformability based separation of circulating tumor cells from castrate resistant prostate cancer patients using resettable cell traps. Lab Chip 2015, 15, 2278–2286. [Google Scholar] [CrossRef] [PubMed]
- Harouaka, R.A.; Zhou, M.D.; Yeh, Y.T.; Khan, W.J.; Das, A.; Liu, X.; Christ, C.C.; Dicker, D.T.; Baney, T.S.; Kaifi, J.T.; et al. Flexible micro spring array device for high-throughput enrichment of viable circulating tumor cells. Clin. Chem. 2014, 60, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.D.; Hao, S.; Williams, A.J.; Harouaka, R.A.; Schrand, B.; Rawal, S.; Ao, Z.; Brenneman, R.; Gilboa, E.; Lu, B.; et al. Separable bilayer microfiltration device for viable label-free enrichment of circulating tumour cells. Sci. Rep. 2014, 4, 7392. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.J.; Lakshmi, R.L.; Chen, P.; Lim, W.T.; Yobas, L.; Lim, C.T. Versatile label free biochip for the detection of circulating tumor cells from peripheral blood in cancer patients. Biosens. Bioelectron. 2010, 26, 1701–1705. [Google Scholar] [CrossRef]
- Vona, G.; Sabile, A.; Louha, M.; Sitruk, V.; Romana, S.; Schutze, K.; Capron, F.; Franco, D.; Pazzagli, M.; Vekemans, M.; et al. Isolation by size of epithelial tumor cells: A new method for the immunomorphological and molecular characterization of circulatingtumor cells. Am. J. Pathol. 2000, 156, 57–63. [Google Scholar] [CrossRef]
- Adams, D.L.; Zhu, P.; Makarova, O.V.; Martin, S.S.; Charpentier, M.; Chumsri, S.; Li, S.; Amstutz, P.; Tang, C.M. The systematic study of circulating tumor cell isolation using lithographic microfilters. RSC Adv. 2014, 9, 4334–4342. [Google Scholar] [CrossRef]
- Sarioglu, A.F.; Aceto, N.; Kojic, N.; Donaldson, M.C.; Zeinali, M.; Hamza, B.; Engstrom, A.; Zhu, H.; Sundaresan, T.K.; Miyamoto, D.T.; et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods 2015, 12, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Sollier, E.; Go, D.E.; Che, J.; Gossett, D.R.; O’Byrne, S.; Weaver, W.M.; Kummer, N.; Rettig, M.; Goldman, J.; Nickols, N.; et al. Size-selective collection of circulating tumor cells using Vortex technology. Lab Chip 2014, 14, 63–77. [Google Scholar] [CrossRef]
- Sun, J.; Li, M.; Liu, C.; Zhang, Y.; Liu, D.; Liu, W.; Hu, G.; Jiang, X. Double spiral microchannel for label-free tumor cell separation and enrichment. Lab Chip 2012, 12, 3952–3960. [Google Scholar] [CrossRef] [Green Version]
- Bhagat, A.A.; Hou, H.W.; Li, L.D.; Lim, C.T.; Han, J. Pinched flow coupled shear-modulated inertial microfluidics for high-throughput rare blood cell separation. Lab Chip 2011, 11, 1870–1878. [Google Scholar] [CrossRef]
- Warkiani, M.E.; Guan, G.; Luan, K.B.; Lee, W.C.; Bhagat, A.A.; Chaudhuri, P.K.; Tan, D.S.; Lim, W.T.; Lee, S.C.; Chen, P.C.; et al. Slanted spiral microfluidics for the ultra-fast, label-free isolation of circulating tumor cells. Lab Chip 2014, 14, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Jafferji, I.; Garza, M.; Melnikova, V.O.; Hasegawa, D.K.; Pethig, R.; Davis, D.W. ApoStream(), a new dielectrophoretic device for antibody independent isolation and recovery of viable cancer cells from blood. Biomicrofluidics 2012, 6, 24133. [Google Scholar] [CrossRef] [PubMed]
- Polzer, B.; Medoro, G.; Pasch, S.; Fontana, F.; Zorzino, L.; Pestka, A.; Andergassen, U.; Meier-Stiegen, F.; Czyz, Z.T.; Alberter, B.; et al. Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol. Med. 2014, 6, 1371–1386. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.F.; Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Miller, M.C.; Matera, J.; Allard, W.J.; Doyle, G.V.; Terstappen, L.W. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin. Cancer Res. 2006, 12, 4218–4224. [Google Scholar] [CrossRef] [PubMed]
- Riethdorf, S.; Fritsche, H.; Muller, V.; Rau, T.; Schindlbeck, C.; Rack, B.; Janni, W.; Coith, C.; Beck, K.; Janicke, F.; et al. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: A validation study of the CellSearch system. Clin. Cancer Res. 2007, 13, 920–928. [Google Scholar] [CrossRef]
- Hofman, P.; Popper, H.H. Pathologists and liquid biopsies: To be or not to be? Virchows Arch. 2016, 469, 601–609. [Google Scholar] [CrossRef]
- De Wit, S.; van Dalum, G.; Lenferink, A.T.; Tibbe, A.G.; Hiltermann, T.J.; Groen, H.J.; van Rijn, C.J.; Terstappen, L.W. The detection of EpCAM(+) and EpCAM(-) circulating tumor cells. Sci. Rep. 2015, 5, 12270. [Google Scholar] [CrossRef]
- Connelly, M.; Wang, Y.; Doyle, G.V.; Terstappen, L.; McCormack, R. Re: Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J. Natl. Cancer Inst. 2009, 101, 895. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Kirby, B.J.; Jodari, M.; Loftus, M.S.; Gakhar, G.; Pratt, E.D.; Chanel-Vos, C.; Gleghorn, J.P.; Santana, S.M.; Liu, H.; Smith, J.P.; et al. Functional characterization of circulating tumor cells with a prostate-cancer-specific microfluidic device. PLoS ONE 2012, 7, e35976. [Google Scholar] [CrossRef]
- Sequist, L.V.; Nagrath, S.; Toner, M.; Haber, D.A.; Lynch, T.J. The CTC-chip: An exciting new tool to detect circulating tumor cells in lung cancer patients. J. Thorac. Oncol. 2009, 4, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bauerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lang, J.C.; Balasubramanian, P.; Jatana, K.R.; Schuller, D.; Agrawal, A.; Zborowski, M.; Chalmers, J.J. Optimization of an enrichment process for circulating tumor cells from the blood of head and neck cancer patients through depletion of normal cells. Biotechnol. Bioeng. 2009, 102, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Lustberg, M.; Jatana, K.R.; Zborowski, M.; Chalmers, J.J. Emerging technologies for CTC detection based on depletion of normal cells. Recent Results Cancer Res. 2012, 195, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.; Mathur, S.R.; Mukhopadhyay, A. Fusion-derived epithelial cancer cells express hematopoietic markers and contribute to stem cell and migratory phenotype in ovarian carcinoma. Cancer Res. 2013, 73, 5360–5370. [Google Scholar] [CrossRef]
- Dolfi, S.C.; Chan, L.L.; Qiu, J.; Tedeschi, P.M.; Bertino, J.R.; Hirshfield, K.M.; Oltvai, Z.N.; Vazquez, A. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 2013, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.L.; Martin, S.S.; Alpaugh, R.K.; Charpentier, M.; Tsai, S.; Bergan, R.C.; Ogden, I.M.; Catalona, W.; Chumsri, S.; Tang, C.M.; et al. Circulating giant macrophages as a potential biomarker of solid tumors. Proc. Natl. Acad. Sci. USA 2014, 111, 3514–3519. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.L.; Adams, D.K.; Alpaugh, R.K.; Cristofanilli, M.; Martin, S.S.; Chumsri, S.; Tang, C.M.; Marks, J.R. Circulating Cancer-Associated Macrophage-Like Cells Differentiate Malignant Breast Cancer and Benign Breast Conditions. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- Kaifi, J.T.; Kunkel, M.; Das, A.; Harouaka, R.A.; Dicker, D.T.; Li, G.; Zhu, J.; Clawson, G.A.; Yang, Z.; Reed, M.F.; et al. Circulating tumor cell isolation during resection of colorectal cancer lung and liver metastases: A prospective trial with different detection techniques. Cancer Biol. Ther. 2015, 16, 699–708. [Google Scholar] [CrossRef]
- Hao, S.; Nisic, M.; He, H.; Tai, Y.C.; Zheng, S.Y. Separable Bilayer Microfiltration Device for Label-Free Enrichment of Viable Circulating Tumor Cells. Methods Mol. Biol. 2017, 1634, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, K.R.; Andorko, J.I.; Whipple, R.A.; Zhang, P.; Sooklal, E.L.; Martin, S.S.; Jewell, C.M. Lipid tethering of breast tumor cells enables real-time imaging of free-floating cell dynamics and drug response. Oncotarget 2016, 7, 10486–10497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, H.W.; Warkiani, M.E.; Khoo, B.L.; Li, Z.R.; Soo, R.A.; Tan, D.S.; Lim, W.T.; Han, J.; Bhagat, A.A.; Lim, C.T. Isolation and retrieval of circulating tumor cells using centrifugal forces. Sci. Rep. 2013, 3, 1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoo, B.L.; Lee, S.C.; Kumar, P.; Tan, T.Z.; Warkiani, M.E.; Ow, S.G.; Nandi, S.; Lim, C.T.; Thiery, J.P. Short-term expansion of breast circulating cancer cells predicts response to anti-cancer therapy. Oncotarget 2015, 6, 15578–15593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, S.; Stemke-Hale, K.; Noshari, J.; Becker, F.F.; Gascoyne, P.R. Dielectrophoresis has broad applicability to marker-free isolation of tumor cells from blood by microfluidic systems. Biomicrofluidics 2013, 7, 11808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manaresi, N.; Romani, A.; Medoro, G.; Altomare, L.; Leonardi, A.; Tartagni, M.; Guerrieri, R. A CMOS chip for individual cell manipulation and detection. IEEE J. Solid State Circuits 2003, 38, 2297–2305. [Google Scholar] [CrossRef]
- Carpenter, E.L.; Rader, J.; Ruden, J.; Rappaport, E.F.; Hunter, K.N.; Hallberg, P.L.; Krytska, K.; O’Dwyer, P.J.; Mosse, Y.P. Dielectrophoretic capture and genetic analysis of single neuroblastoma tumor cells. Front. Oncol. 2014, 4, 201. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, S.V.; Bingham, C.; Fittipaldi, P.; Austin, L.; Palazzo, J.; Palmer, G.; Alpaugh, K.; Cristofanilli, M. TP53 mutations detected in circulating tumor cells present in the blood of metastatic triple negative breast cancer patients. Breast Cancer Res. 2014, 16, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, D.J.; De Laere, B.; Van den Eynden, G.G.; Van Laere, S.J.; Rothe, F.; Ignatiadis, M.; Sieuwerts, A.M.; Lambrechts, D.; Rutten, A.; van Dam, P.A.; et al. Semiautomated isolation and molecular characterisation of single or highly purified tumour cells from CellSearch enriched blood samples using dielectrophoretic cell sorting. Br. J. Cancer 2013, 108, 1358–1367. [Google Scholar] [CrossRef] [Green Version]
- Zill, A.; Mortimer, S.; Banks, K.; Nagy, R.; Chudova, D.; Jackson, C.; Baca, A.; Ye, J.Z.; Lanman, B.; Talasaz, A.; et al. Somatic genomic landscape of over 15,000 patients with advanced-stage cancer from clinical next-generation sequencing analysis of circulating tumor DNA. Proceedings of ASCO Annual Meeting, Chicago, IL, USA, 31 May–4 June 2019. [Google Scholar]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Kallergi, G.; Aggouraki, D.; Zacharopoulou, N.; Stournaras, C.; Georgoulias, V.; Martin, S.S. Evaluation of alpha-tubulin, detyrosinated alpha-tubulin, and vimentin in CTCs: Identification of the interaction between CTCs and blood cells through cytoskeletal elements. Breast Cancer Res. 2018, 20, 67. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurzu, S.; Turdean, S.; Kovecsi, A.; Contac, A.O.; Jung, I. Epithelial-mesenchymal, mesenchymal-epithelial, and endothelial-mesenchymal transitions in malignant tumors: An update. World J. Clin. Cases 2015, 3, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Paterlini-Brechot, P.; Benali, N.L. Circulating tumor cells (CTC) detection: Clinical impact and future directions. Cancer Lett. 2007, 253, 180–204. [Google Scholar] [CrossRef] [PubMed]
- Au, S.H.; Storey, B.D.; Moore, J.C.; Tang, Q.; Chen, Y.L.; Javaid, S.; Sarioglu, A.F.; Sullivan, R.; Madden, M.W.; O’Keefe, R.; et al. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc. Natl. Acad. Sci. USA 2016, 113, 4947–4952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112 e114. [Google Scholar] [CrossRef]
- Christiansen, J.J.; Rajasekaran, A.K. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006, 66, 8319–8326. [Google Scholar] [CrossRef]
- Nagy, J.A.; Chang, S.H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, D.; Zhao, N.; Zhao, X. Epithelial-to-endothelial transition and cancer stem cells: Two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget 2017, 8, 30502–30510. [Google Scholar] [CrossRef]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5, 180ra148. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shiratsuchi, H.; Lin, J.; Chen, G.; Reddy, R.M.; Azizi, E.; Fouladdel, S.; Chang, A.C.; Lin, L.; Jiang, H.; et al. Expansion of CTCs from early stage lung cancer patients using a microfluidic co-culture model. Oncotarget 2014, 5, 12383–12397. [Google Scholar] [CrossRef]
- Rossi, E.; Rugge, M.; Facchinetti, A.; Pizzi, M.; Nardo, G.; Barbieri, V.; Manicone, M.; De Faveri, S.; Chiara Scaini, M.; Basso, U.; et al. Retaining the long-survive capacity of Circulating Tumor Cells (CTCs) followed by xeno-transplantation: not only from metastatic cancer of the breast but also of prostate cancer patients. Oncoscience 2014, 1, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, C.L.; Morrow, C.J.; Li, Y.; Metcalf, R.L.; Rothwell, D.G.; Trapani, F.; Polanski, R.; Burt, D.J.; Simpson, K.L.; Morris, K.; et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat. Med. 2014, 20, 897–903. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subcategory | Platform | Enrichment Principle | Live Cell Analysis Reported * | Company |
|---|---|---|---|---|
| Label-Based | ||||
| Positive Enrichment Immunoaffinity | ||||
| Micropost Arrays | CTC-Chip [55] | EpCAM | Yes | |
| GEDI Chip [56] | PSMA/HER2, Size | No | ||
| OncoCEE [57] | Antibody Cocktail | No | Biocept Inc. San Diego, CA, USA | |
| Microfluidic Surface Capture | Biofluidica CTC system [58] | EpCAM | Yes | Biofluidica Inc.San Diego, CA, USA |
| CytoTrapNano [59] | EpCAM | No | Cytolumina. Los Angeles, CA, USA | |
| GEM Chip [60] | EpCAM | Yes | ||
| HTMSU [61] | EpCAM | No | ||
| Graphene Oxide Chip [62] | EpCAM | No | ||
| Herringbone Chip [63] | EpCAM | No | ||
| Microfluidic Magnetic | Ephesia [64] | EpCAM | Yes | |
| Magnetic Sifter [60] | EpCAM | No | ||
| LiquidBiopsy [65] | Antibody Cocktail | No | Thermo Fisher, Waltham, MA, USA | |
| Isoflux [66] | EpCAM | No | Fluxion Biosciences, Alameda, CA, USA | |
| Magnetic | CellSearch [67] | EpCAM | No | Silicon Biosystems, Huntington Valley, PA, USA |
| AdnaTest [68] | Antibody Cocktail | No | Qiagen, Hilden, Germany | |
| MACS [69] | EpCAM | No | Miltenyi Biotec, Bergisch Gladbach, North Rhine-Westphalia, Germany | |
| MagSweeper [70] | EpCAM | No | ||
| Magnetic in vivo | CellCollector [71] | EpCAM | Yes | GILUPI, Potsdam, Germany |
| Negative Enrichment Immunoaffinity | ||||
| Magnetic | EasySep [72] | CD45 | No | STEMCELL, Vancouver, BC, Canada |
| QMS [73] | Yes | |||
| MACS [74] | Yes | Miltenyi Biotec, Bergisch Gladbach, North Rhine-Westphalia, Germany | ||
| Microfluidic/Magnetic | CTC-iChip [75] | CD45, CD66b, Size | Yes | |
| Label-Free | ||||
| Density | ||||
| Ficoll-Paque [76] | Density | Yes | GE Healthcare Bio-Sciences, Pittsburg, PA, USA | |
| OncoQuick [77] | Density, Size | Yes | Greiner Bio-One, Kremsmünster, Austria | |
| RosetteSep [78] | Density, Antibody Cocktail | Yes | STEMCELL, Vancouver, BC, Canada | |
| Accucyte and CyteSealer [79] | Density | Yes | Rarecyte, Seattle, WA, USA | |
| Size | ||||
| Filtration | Parsortix [80] | Size, Deformability | Yes | Angle, King of Prussia, PA, USA |
| Microwall Chip [81] | Yes | |||
| ScreenCell [82] | Yes | ScreenCell, Westford, MA, USA | ||
| Resettable Cell Trap [83] | Yes | |||
| Flexible Micro Spring Array (FMSA) [84] | Yes | |||
| FaCTchecker [85] | Yes | Circulogix, Hallandale Beach, FL, USA | ||
| Crescent Chip [86] | Yes | |||
| ISET [87] | Yes | RareCells Diagnostics, Paris Cedes, France | ||
| CellSieve [88] | Yes | Creatv Microtech, Potomac, MD, USA | ||
| Cluster Chip [89] | Yes | |||
| Fluid Dynamics | Vortex [90] | Size | Yes | Vortex Biosciences, Pleasanton, CA, USA |
| Double Spiral Chip [91] | Yes | |||
| Micropinching Chip [92] | Yes | |||
| ClearCell FX [93] | Yes | Genomax Technologies, Singapore | ||
| Electric | ||||
| ApoStream [94] | Electrical Signature | Yes | Apocell, Houston, TX, USA | |
| DEPArray [95] | Yes | Silicon Biosystems, Huntington Valley, PA, USA | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailey, P.C.; Martin, S.S. Insights on CTC Biology and Clinical Impact Emerging from Advances in Capture Technology. Cells 2019, 8, 553. https://doi.org/10.3390/cells8060553
Bailey PC, Martin SS. Insights on CTC Biology and Clinical Impact Emerging from Advances in Capture Technology. Cells. 2019; 8(6):553. https://doi.org/10.3390/cells8060553
Chicago/Turabian StyleBailey, Patrick C., and Stuart S. Martin. 2019. "Insights on CTC Biology and Clinical Impact Emerging from Advances in Capture Technology" Cells 8, no. 6: 553. https://doi.org/10.3390/cells8060553
APA StyleBailey, P. C., & Martin, S. S. (2019). Insights on CTC Biology and Clinical Impact Emerging from Advances in Capture Technology. Cells, 8(6), 553. https://doi.org/10.3390/cells8060553