Fibronectin Regulation of Integrin B1 and SLUG in Circulating Tumor Cells



Abstract
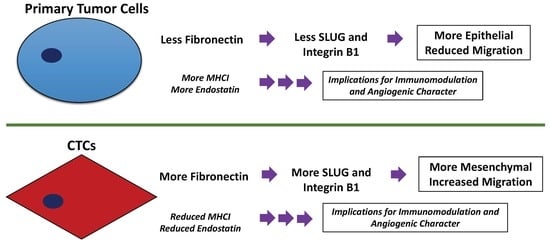
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
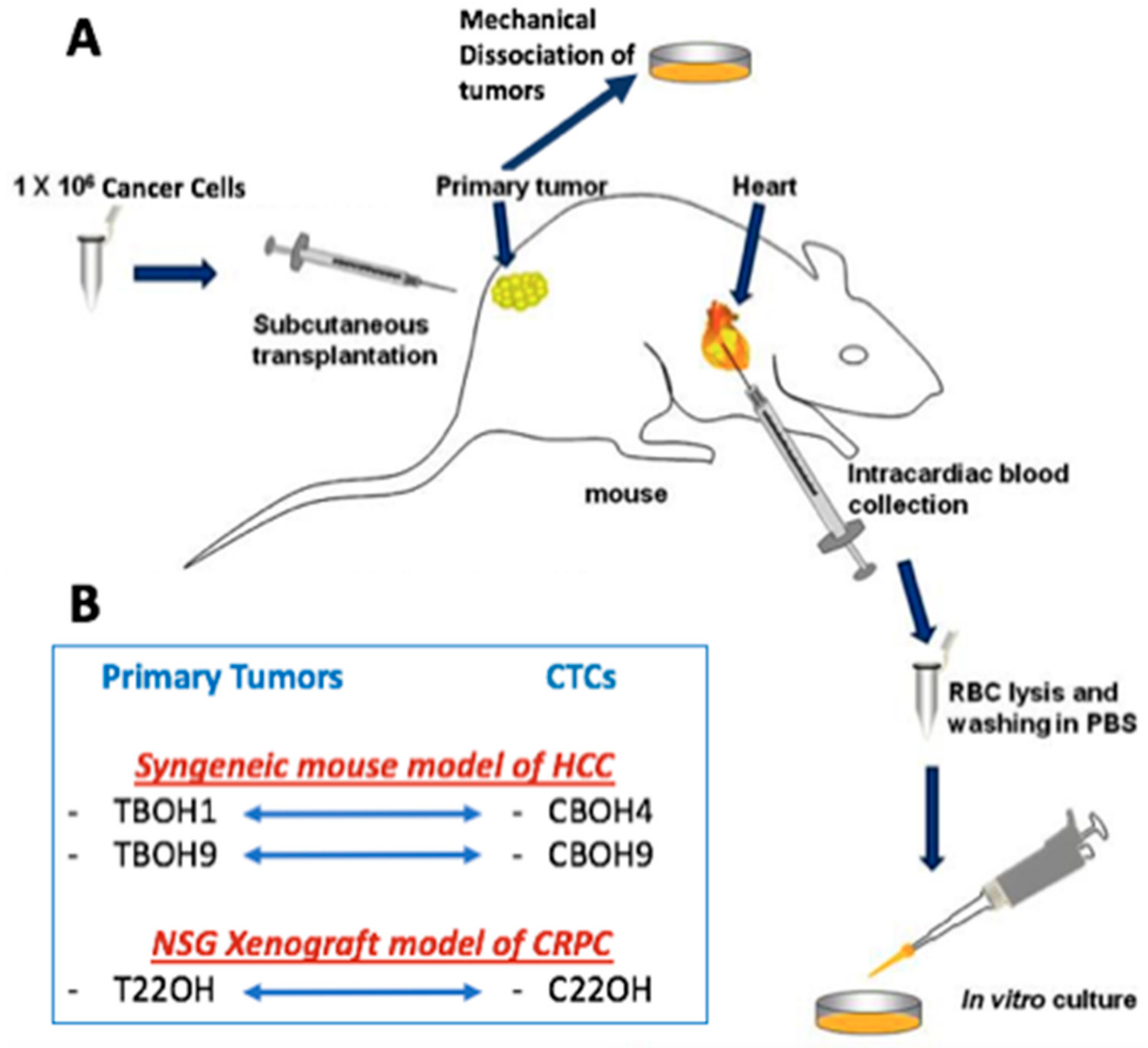
2.2. Mouse Tumor Studies
2.3. Immunofluorescence Staining
2.4. Migration Assays
2.5. Protein Extraction and Western Blotting
2.6. RNA Extraction and qPCR Analysis
2.7. Transfection of siRNAs
2.8. Flow Cytometry
2.9. Cytokine Array
2.10. Statistical Analyses
3. Results
3.1. CTCs Obtained from Blood Express Tissue-Specific Markers
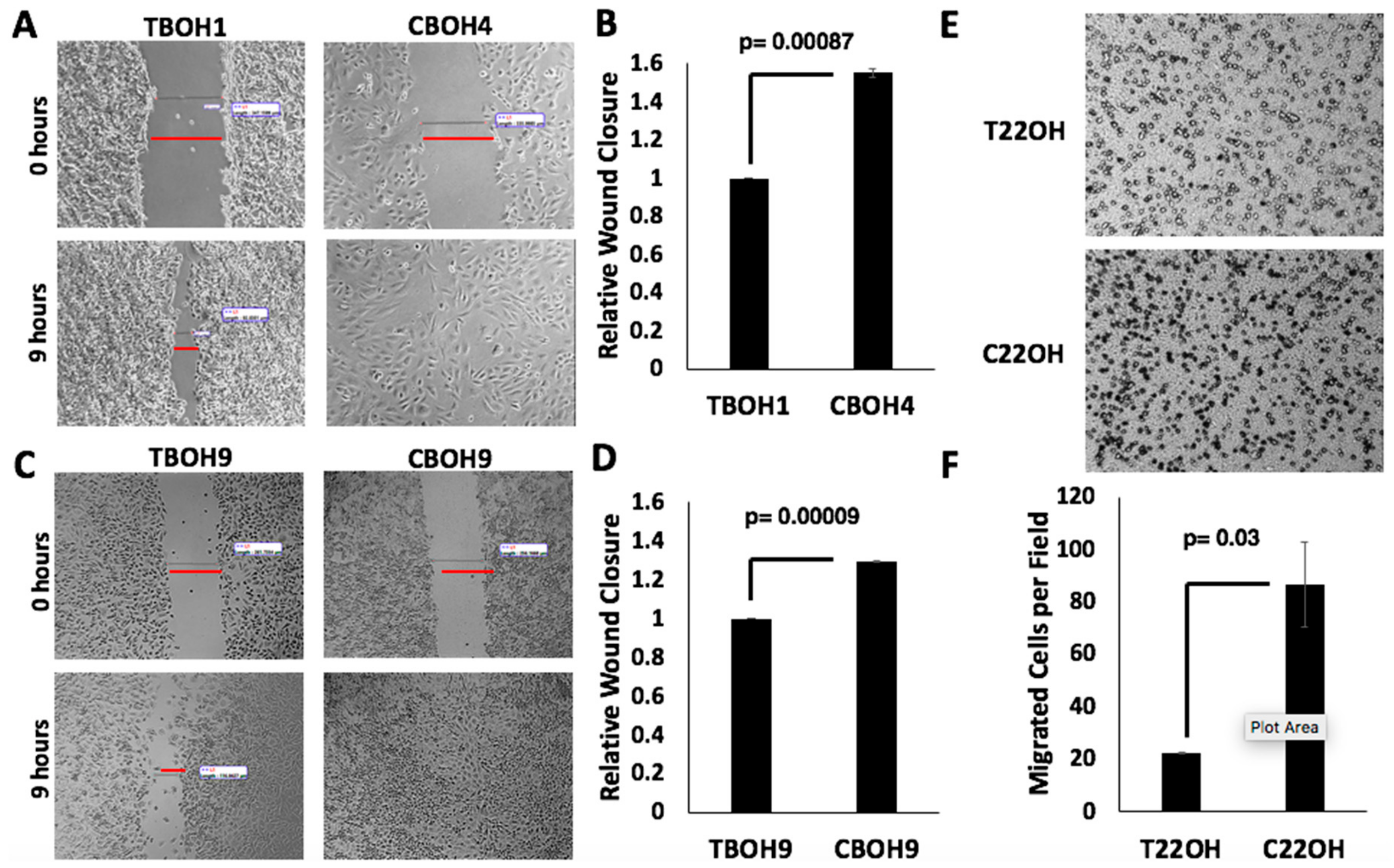
3.2. CTCs Have a Greater Migratory Capacity than Primary Tumor-Derived Cells
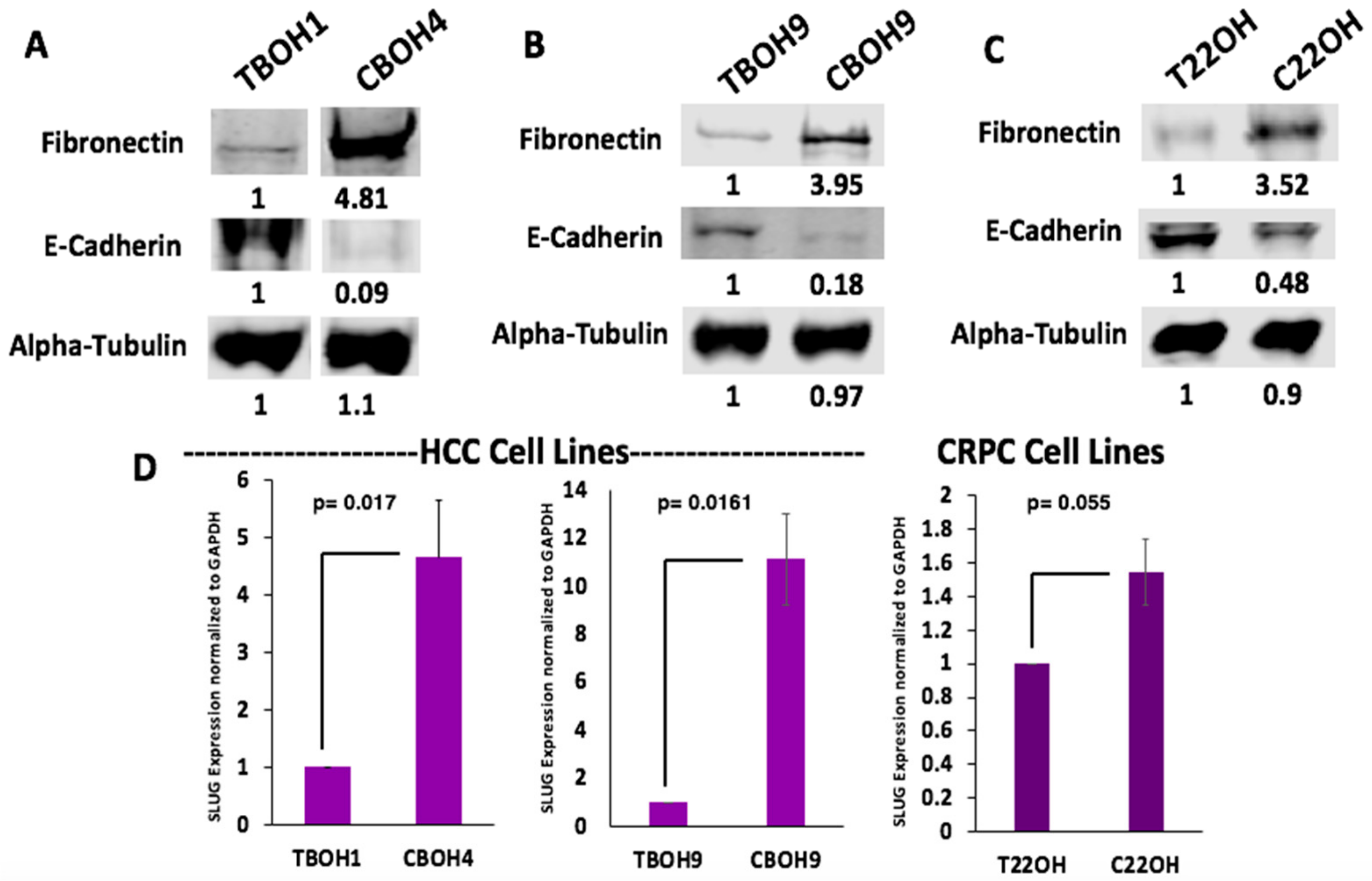
3.3. CTCs Exhibit Epithelial to Mesenchymal Transition (EMT)
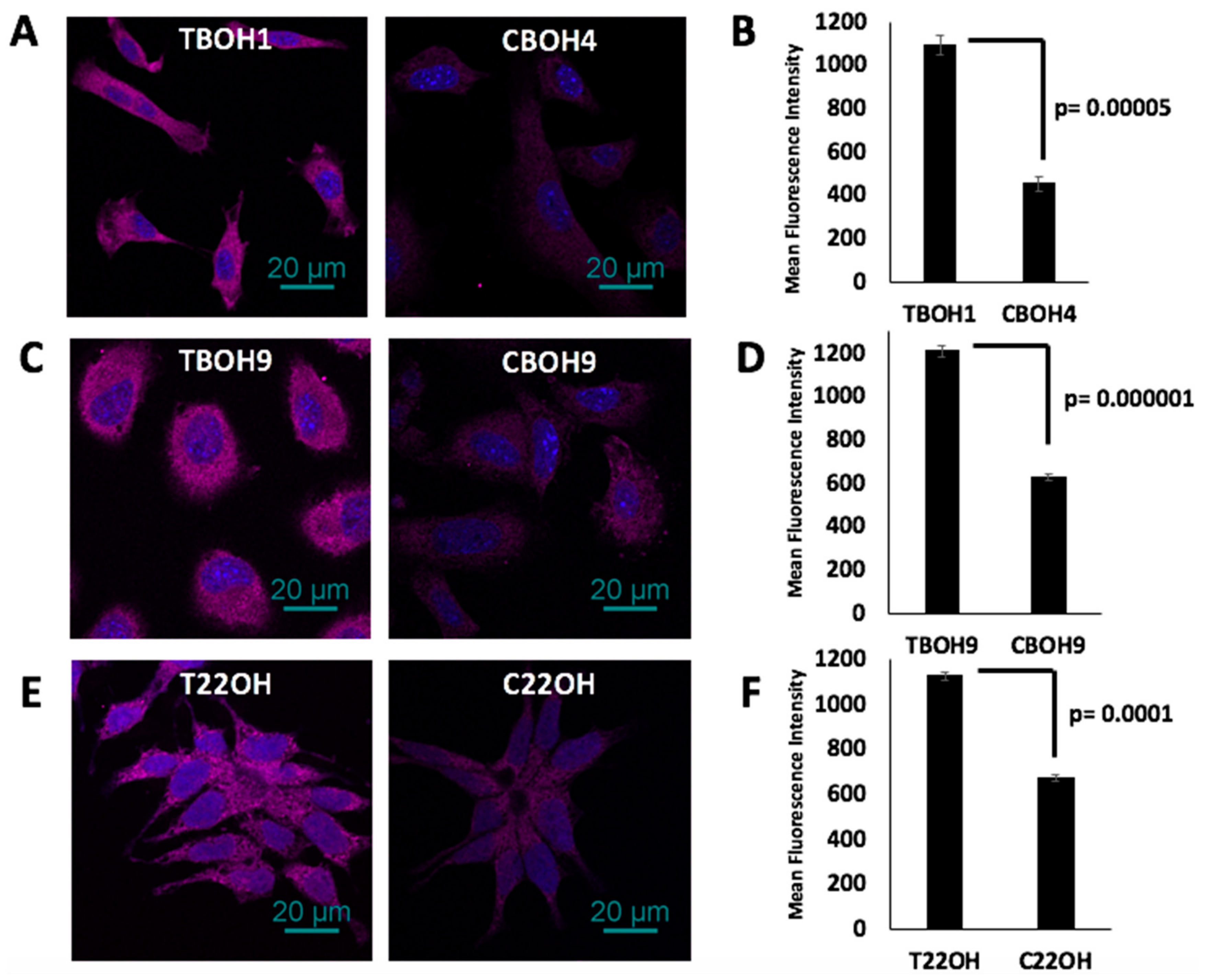
3.4. Fibronectin Expression Regulates Integrin B1 and SLUG Expression in CTCs
3.5. HCC CTCs have Decreased MHCI Cell Surface Expression
3.6. HCC CTCs have Significantly Decreased Secretion of Endostatin, CXCL5, and Proliferin
3.7. CTCs, in Comparison to Primary Tumor-Derived Cells, Have Decreased Endostatin Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, W.C.; Kopetz, S.; Wistuba, I.I.; Zhang, J. Metastasis of cancer: When and how? Ann. Oncol. 2017, 28, 2045–2047. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Van de Stolpe, A.; Pantel, K.; Sleijfer, S.; Terstappen, L.W.; Den Toonder, J.M. Circulating tumor cell isolation and diagnostics: Toward routine clinical use. Cancer Res. 2011, 71, 5955–5960. [Google Scholar] [CrossRef]
- Gallerani, G.; Fici, P.; Fabbri, F. Circulating Tumor Cells: Back to the Future. Front. Oncol. 2016, 6, 275. [Google Scholar] [CrossRef] [PubMed]
- Haber, D.A.; Velculescu, V.E. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Brakenhoff, R.H.; Brandt, B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat. Rev. Cancer 2008, 8, 329–340. [Google Scholar] [CrossRef]
- Bailey, P.M.; Martin, S.S. Insights on CTC Biology and Clinical Impact Emerging from Advances in Capture Technology. Cells 2019, 8, 553. [Google Scholar] [CrossRef]
- George, T.J., Jr.; Ogunwobi, O.O.; Sheng, W.; Fan, Z.H.; Liu, C. “Tissue is the issue”: Circulating tumor cells in pancreatic cancer. J. Gastrointest. Cancer 2014, 45, 222–225. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Alvarez Cubero, M.J.; Lorente, J.A.; Robles-Fernandez, I.; Rodriguez-Martinez, A.; Puche, J.L.; Serrano, M.J. Circulating Tumor Cells: Markers and Methodologies for Enrichment and Detection. Methods Mol. Biol. 2017, 1634, 283–303. [Google Scholar] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.C.; Doyle, G.V.; Terstappen, L.W. Significance of Circulating Tumor Cells Detected by the CellSearch System in Patients with Metastatic Breast Colorectal and Prostate Cancer. J. Oncol. 2010, 2010, 617421. [Google Scholar] [CrossRef] [PubMed]
- Zieglschmid, V.; Hollmann, C.; Bocher, O. Detection of disseminated tumor cells in peripheral blood. Crit. Rev. Clin. Lab. Sci. 2005, 42, 155–196. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts and Figures 2018. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2018/cancer-facts-and-figures-2018.pdf (accessed on 19 June 2019).
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Ogunwobi, O.H.; Harricharran, T.; Huaman, J.; Galuza, A.; Odumuwagun, O.; Tan, Y.; Ma, G.X.; Nguyen, M.T. Mechanisms of hepatocellular carcinoma progression. World J. Gastroenterol. 2019, 25, 2279–2293. [Google Scholar] [CrossRef]
- Keating, G.M. Sorafenib: A Review in Hepatocellular Carcinoma. Target Oncol. 2017, 12, 243–253. [Google Scholar] [CrossRef]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef]
- Suzman, D.L.; Antonarakis, E.S. Castration-resistant prostate cancer: Latest evidence and therapeutic implications. Ther. Adv. Med. Oncol. 2014, 6, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Pentyala, S.; Whyard, T.; Pentyala, S.; Muller, J.; Pfail, J.; Parmar, S.; Helguero, C.G.; Khan, S. Prostate cancer markers: An update. Biomed. Rep. 2016, 4, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litwin, M.S.; Tan, H.J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Kim, I.Y. Nonmetastatic castration-resistant prostate cancer. Korean J. Urol. 2014, 55, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Naidoo, M.K.; Ilboudo, A.; DuBois, P.; Durojaiye, V.; Liu, C.; Ogunwobi, O.O. Isolation and Propagation of Circulating Tumor Cells from a Mouse Cancer Model. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.O.; Puszyk, W.; Dong, H.J.; Liu, C. Epigenetic upregulation of HGF and c-Met drives metastasis in hepatocellular carcinoma. PLoS ONE 2013, 8, e63765. [Google Scholar] [CrossRef] [PubMed]
- Omori, Y.; Imai, J.; Watanabe, M.; Komatsu, T.; Suzuki, Y.; Kataoka, K.; Watanabe, S.; Tanigami, A.; Sugano, S. CREB-H: A novel mammalian transcription factor belonging to the CREB/ATF family and functioning via the box-B element with a liver-specific expression. Nucleic Acids Res. 2001, 29, 2154–2162. [Google Scholar] [CrossRef]
- Barbosa, S.; Fasanella, G.; Carreira, S.; Llarena, M.; Fox, R.; Barreca, C.; Andrew, D.; O’Hare, P. An orchestrated program regulating secretory pathway genes and cargos by the transmembrane transcription factor CREB-H. Traffic 2013, 14, 382–398. [Google Scholar] [CrossRef]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018, 174, 433–447. [Google Scholar] [CrossRef]
- Wadosky, K.M.; Koochekpour, S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 2017, 8, 18550–18576. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.; Christofori, G. Mechanisms of motility in metastasizing cells. Mol. Cancer Res. 2010, 8, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Tiwari, N.; Gheldof, A.; Tatari, M.; Christofori, G. EMT as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 2012, 22, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Mader, S.; Pantel, K. Epithelial-mesenchymal plasticity in circulating tumor cells. J. Mol. Med. 2017, 95, 133–142. [Google Scholar] [CrossRef]
- Meng, X.N.; Jin, Y.; Yu, Y.; Bai, J.; Liu, G.Y.; Zhu, J.; Zhao, Y.Z.; Wang, Z.; Chen, F.; Lee, K.Y.; et al. Characterisation of fibronectin-mediated FAK signalling pathways in lung cancer cell migration and invasion. Br. J. Cancer 2009, 101, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Schwarzbauer, J.E. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 2014, 33, 1649–1657. [Google Scholar] [CrossRef]
- Yousif, N.G. Fibronectin promotes migration and invasion of ovarian cancer cells through up-regulation of FAK-PI3K/Akt pathway. Cell. Biol. Int. 2014, 38, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Ramos Gde, O.; Bernardi, L.; Lauxen, I.; Sant’Ana Filho, M.; Horwitz, A.R.; Lamers, M.L. Fibronectin Modulates Cell Adhesion and Signaling to Promote Single Cell Migration of Highly Invasive Oral Squamous Cell Carcinoma. PLoS ONE 2016, 11, e0151338. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Hielscher, A. Fibronectin: How its Aberrant Expression in Tumors may Improve Therapeutic Targeting. J. Cancer 2017, 8, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Jeanes, A.; Gottardi, C.J.; Yap, A.S. Cadherins and cancer: How does cadherin dysfunction promote tumor progression? Oncogene 2008, 27, 6920–6929. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.M.; Fang, C.M.; Chuah, L.H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018, 121, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell. Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and cancer: Regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef]
- Blandin, A.F.; Renner, G.; Lehmann, M.; Lelong-Rebel, I.; Martin, S.; Dontenwill, M. beta1 Integrins as Therapeutic Targets to Disrupt Hallmarks of Cancer. Front. Pharmacol. 2015, 6, 279. [Google Scholar] [CrossRef]
- Howe, G.A.; Addison, C.L. beta1 integrin: An emerging player in the modulation of tumorigenesis and response to therapy. Cell Adh. Migr. 2012, 6, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Gu, A.; Jie, Y.; Yao, Q.; Zhang, Y.; Mingyan, E. Slug is associated with tumor metastasis and angiogenesis in ovarian cancer. Reprod. Sci. 2017, 24, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Uygur, B.; Wu, W.S. SLUG promotes prostate cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol. Cancer 2011, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Chen, Y.; Li, X.; Yang, R.; Zhang, L.; Huangfu, L.; Zheng, N.; Zhao, X.; Lv, L.; Hong, Y.; et al. YAP1 contributes to NSCLC invasion and migration by promoting Slug transcription via the transcription co-factor TEAD. Cell Death Dis. 2018, 9, 464. [Google Scholar] [CrossRef] [PubMed]
- Bubenik, J. MHC class I down-regulation: Tumour escape from immune surveillance? (review). Int. J. Oncol. 2004, 25, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Shastri, N.; Nagarajan, N.; Lind, K.C.; Kanaseki, T. Monitoring peptide processing for MHC class I molecules in the endoplasmic reticulum. Curr. Opin. Immunol. 2014, 26, 123–127. [Google Scholar] [CrossRef]
- Jang, M.H.; Seoh, J.Y.; Miyasaka, M. Cytokines, chemokines, and their receptors: Targets for immunomodulation. Conference report: International Cytokine Society Conference 2005. J. Leukoc. Biol. 2006, 80, 217–219. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Rhee, I. Cytokine Signaling in Tumor Progression. Immune Netw. 2017, 17, 214–227. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Langer, R. A review of Judah Folkman’s remarkable achievements in biomedicine. Proc. Natl. Acad. Sci. USA 2008, 105, 13203–13205. [Google Scholar] [CrossRef]
- Dhanabal, M.; Ramchandran, R.; Volk, R.; Stillman, I.E.; Lombardo, M.; Iruela-Arispe, M.L.; Simons, M.; Sukhatme, V.P. Endostatin: Yeast production, mutants, and antitumor effect in renal cell carcinoma. Cancer Res. 1999, 59, 189–197. [Google Scholar] [PubMed]
- Walz, A.; Burgener, R.; Car, B.; Baggiolini, M.; Kunkel, S.L.; Strieter, R.M. Structure and neutrophil-activating properties of a novel inflammatory peptide (ENA-78) with homology to interleukin 8. J. Exp. Med. 1991, 174, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Xu, X.; Huang, P.; He, M.; Wang, X. The potential of CXCL5 as a target for liver cancer - what do we know so far? Expert Opin. Ther. Targets 2015, 19, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Linzer, D.I.; Lee, S.J.; Ogren, L.; Talamantes, F.; Nathans, D. Identification of proliferin mRNA and protein in mouse placenta. Proc. Natl. Acad. Sci. USA 1985, 82, 4356–4359. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Volpert, O.V.; Bouck, N.; Linzer, D.I. Stimulation and inhibition of angiogenesis by placental proliferin and proliferin-related protein. Science 1994, 266, 1581–1584. [Google Scholar] [CrossRef] [PubMed]
- Toft, D.J.; Rosenberg, S.B.; Bergers, G.; Volpert, O.; Linzer, D.I. Reactivation of proliferin gene expression is associated with increased angiogenesis in a cell culture model of fibrosarcoma tumor progression. Proc. Natl. Acad. Sci. USA 2001, 98, 13055–13059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabassum, D.P.; Polyak, K. Tumorigenesis: It takes a village. Nat. Rev. Cancer 2015, 15, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Cayrefourcq, L.; Mazard, T.; Joosse, S.; Solassol, J.; Ramos, J.; Assenat, E.; Schumacher, U.; Costes, V.; Maudelonde, T.; Pantel, K.; et al. Establishment and characterization of a cell line from human circulating colon cancer cells. Cancer Res. 2015, 75, 892–901. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Hamilton, G.; Burghuber, O.; Zeillinger, R. Circulating tumor cells in small cell lung cancer: Ex vivo expansion. Lung 2015, 193, 451–452. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Bobek, V.; Kacprzak, G.; Rzechonek, A.; Kolostova, K. Detection and cultivation of circulating tumor cells in malignant pleural mesothelioma. Anticancer Res. 2014, 34, 2565–2569. [Google Scholar] [PubMed]
- Bobek, V.; Matkowski, R.; Gurlich, R.; Grabowski, K.; Szelachowska, J.; Lischke, R.; Schutzner, J.; Harustiak, T.; Pazdro, A.; Rzechonek, A.; et al. Cultivation of circulating tumor cells in esophageal cancer. Folia Histochem. Cytobiol. 2014, 52, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Cegan, M.; Kolostova, K.; Matkowski, R.; Broul, M.; Schraml, J.; Fiutowski, M.; Bobek, V. In vitro culturing of viable circulating tumor cells of urinary bladder cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 7164–7171. [Google Scholar] [PubMed]
- Kang, J.H.; Krause, S.; Tobin, H.; Mammoto, A.; Kanapathipillai, M.; Ingber, D.E. A combined micromagnetic-microfluidic device for rapid capture and culture of rare circulating tumor cells. Lab Chip 2012, 12, 2175–2181. [Google Scholar] [CrossRef] [PubMed]
- Kolostova, K.; Matkowski, R.; Gurlich, R.; Grabowski, K.; Soter, K.; Lischke, R.; Schutzner, J.; Bobek, V. Detection and cultivation of circulating tumor cells in gastric cancer. Cytotechnology 2016, 68, 1095–1102. [Google Scholar] [CrossRef]
- Kolostova, K.; Zhang, Y.; Hoffman, R.M.; Bobek, V. In vitro culture and characterization of human lung cancer circulating tumor cells isolated by size exclusion from an orthotopic nude-mouse model expressing fluorescent protein. J. Fluoresc. 2014, 24, 1531–1536. [Google Scholar] [CrossRef]
- Kulasinghe, A.; Perry, C.; Warkiani, M.E.; Blick, T.; Davies, A.; O’Byrne, K.; Thompson, E.W.; Nelson, C.C.; Vela, I.; Punyadeera, C. Short term ex-vivo expansion of circulating head and neck tumour cells. Oncotarget 2016, 7, 60101–60109. [Google Scholar] [CrossRef] [Green Version]
- Ting, D.T.; Wittner, B.S.; Ligorio, M.; Vincent Jordan, N.; Shah, A.M.; Miyamoto, D.T.; Aceto, N.; Bersani, F.; Brannigan, B.W.; Xega, K.; et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014, 8, 1905–1918. [Google Scholar] [CrossRef]
- Zhang, Z.; Shiratsuchi, H.; Lin, J.; Chen, G.; Reddy, R.M.; Azizi, E.; Fouladdel, S.; Chang, A.C.; Lin, L.; Jiang, H.; et al. Expansion of CTCs from early stage lung cancer patients using a microfluidic co-culture model. Oncotarget 2014, 5, 12383–12397. [Google Scholar] [CrossRef]
- Zhang, Z.; Shiratsuchi, H.; Palanisamy, N.; Nagrath, S.; Ramnath, N. Expanded Circulating Tumor Cells from a Patient with ALK-Positive Lung Cancer Present with EML4-ALK Rearrangement Along with Resistance Mutation and Enable Drug Sensitivity Testing: A Case Study. J. Thorac. Oncol. 2017, 12, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Nuhn, P.; De Bono, J.S.; Fizazi, K.; Freedland, S.J.; Grilli, M.; Kantoff, P.W.; Sonpavde, G.; Sternberg, C.N.; Yegnasubramanian, S.; Antonarakis, E.S. Update on Systemic Prostate Cancer Therapies: Management of Metastatic Castration-resistant Prostate Cancer in the Era of Precision Oncology. Eur. Urol. 2019, 75, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pan, D.; Yan, Q.; Song, Y. Activation mechanisms of alphaVbeta3 integrin by binding to fibronectin: A computational study. Protein Sci. 2017, 26, 1124–1137. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, F.; Ray, A.M.; Dontenwill, M. Integrin alpha5beta1, the Fibronectin Receptor, as a Pertinent Therapeutic Target in Solid Tumors. Cancers 2013, 5, 27–47. [Google Scholar] [CrossRef]
- Folkman, J. Antiangiogenesis in cancer therapy—Endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar] [CrossRef]
- Wang, S.; Lu, X.-A.; Liu, P.; Fu, Y.; Jia, L.; Zhan, S.; Luo, Y. Endostatin Has ATPase Activity, Which Mediates Its Antiangiogenic and Antitumor Activities. Mol. Cancer Ther. 2015, 14, 1192. [Google Scholar] [CrossRef]
- Sato, Y. Endostatin as a Biomarker of Basement Membrane Degradation. J. Atheroscler. Thromb. 2017, 24, 1014–1015. [Google Scholar] [CrossRef] [Green Version]
- Walia, A.; Yang, J.F.; Huang, Y.-H.; Rosenblatt, M.I.; Chang, J.-H.; Azar, D.T. Endostatin’s Emerging Roles in Angiogenesis, Lymphangiogenesis, Disease, and Clinical Applications. Biochim. Biophys. Acta 2015, 1850, 2422–2438. [Google Scholar] [CrossRef]
- Ocana, A.; Nieto-Jimenez, C.; Pandiella, A.; Templeton, A.J. Neutrophils in cancer: Prognostic role and therapeutic strategies. Mol. Cancer 2017, 16, 137. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Wellenstein, M.D.; De Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef]
- Granot, Z.; Henke, E.; Comen, E.A.; King, T.A.; Norton, L.; Benezra, R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 2011, 20, 300–314. [Google Scholar] [CrossRef] [PubMed]
- Speetjens, F.M.; Kuppen, P.J.; Sandel, M.H.; Menon, A.G.; Burg, D.; Van de Velde, C.J.; Tollenaar, R.A.; De Bont, H.J.; Nagelkerke, J.F. Disrupted expression of CXCL5 in colorectal cancer is associated with rapid tumor formation in rats and poor prognosis in patients. Clin. Cancer Res. 2008, 14, 2276–2284. [Google Scholar] [CrossRef] [PubMed]
- Blaisdell, A.; Crequer, A.; Columbus, D.; Daikoku, T.; Mittal, K.; Dey, S.K.; Erlebacher, A. Neutrophils Oppose Uterine Epithelial Carcinogenesis via Debridement of Hypoxic Tumor Cells. Cancer Cell 2015, 28, 785–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, D.; Johnson, R.S. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Faye, C.; Moreau, C.; Chautard, E.; Jetne, R.; Fukai, N.; Ruggiero, F.; Humphries, M.J.; Olsen, B.R.; Ricard-Blum, S. Molecular interplay between endostatin, integrins, and heparan sulfate. J. Biol. Chem. 2009, 284, 22029–22040. [Google Scholar] [CrossRef]
- Rehn, M.; Veikkola, T.; Kukk-Valdre, E.; Nakamura, H.; Ilmonen, M.; Lombardo, C.; Pihlajaniemi, T.; Alitalo, K.; Vuori, K. Interaction of endostatin with integrins implicated in angiogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 1024–1029. [Google Scholar] [CrossRef]
- Sudhakar, A.; Sugimoto, H.; Yang, C.; Lively, J.; Zeisberg, M.; Kalluri, R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc. Natl. Acad. Sci. USA 2003, 100, 4766–4771. [Google Scholar] [CrossRef]
- Wickstrom, S.A.; Alitalo, K.; Keski-Oja, J. Endostatin associates with integrin alpha5beta1 and caveolin-1, and activates Src via a tyrosyl phosphatase-dependent pathway in human endothelial cells. Cancer Res. 2002, 62, 5580–5589. [Google Scholar]
- Yokoyama, Y.; Ramakrishnan, S. Binding of endostatin to human ovarian cancer cells inhibits cell attachment. Int. J. Cancer 2007, 121, 2402–2409. [Google Scholar] [CrossRef]
- Gast, C.E.; Silk, A.D.; Zarour, L.; Riegler, L.; Burkhart, J.G.; Gustafson, K.T.; Parappilly, M.S.; Roh-Johnson, M.; Goodman, J.R.; Olson, B.; et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 2018, 4, eaat7828. [Google Scholar] [CrossRef] [Green Version]
- Laberge, G.S.; Duvall, E.; Haedicke, K.; Pawelek, J. Leukocyte-cancer cell fusion—Genesis of a deadly journey. Cells 2019, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Baggiolini, M. Chemokines and leukocyte traffic. Nat. Immunol. 2008, 9, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.K.; Funasaka, Y.; Ichihashi, M.; Pawelek, J.M. Upregulation of alpha and beta integrin subunits in metastatic macrophage-melanoma fusion hybrids. Melanoma Res. 2009, 19, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Knowles, L.M.; Gurski, L.A.; Engel, C.; Gnarra, J.R.; Maranchie, J.K.; Pilch, J. Integrin alphavbeta3 and fibronectin upregulate Slug in cancer cells to promote clot invasion and metastasis. Cancer Res. 2013, 73, 6175–6184. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huaman, J.; Naidoo, M.; Zang, X.; Ogunwobi, O.O. Fibronectin Regulation of Integrin B1 and SLUG in Circulating Tumor Cells. Cells 2019, 8, 618. https://doi.org/10.3390/cells8060618
Huaman J, Naidoo M, Zang X, Ogunwobi OO. Fibronectin Regulation of Integrin B1 and SLUG in Circulating Tumor Cells. Cells. 2019; 8(6):618. https://doi.org/10.3390/cells8060618
Chicago/Turabian StyleHuaman, Jeannette, Michelle Naidoo, Xingxing Zang, and Olorunseun O. Ogunwobi. 2019. "Fibronectin Regulation of Integrin B1 and SLUG in Circulating Tumor Cells" Cells 8, no. 6: 618. https://doi.org/10.3390/cells8060618
APA StyleHuaman, J., Naidoo, M., Zang, X., & Ogunwobi, O. O. (2019). Fibronectin Regulation of Integrin B1 and SLUG in Circulating Tumor Cells. Cells, 8(6), 618. https://doi.org/10.3390/cells8060618