Ciliopathy-Associated Protein Kinase ICK Requires Its Non-Catalytic Carboxyl-Terminal Domain for Regulation of Ciliogenesis

 ,
, {kind=link}
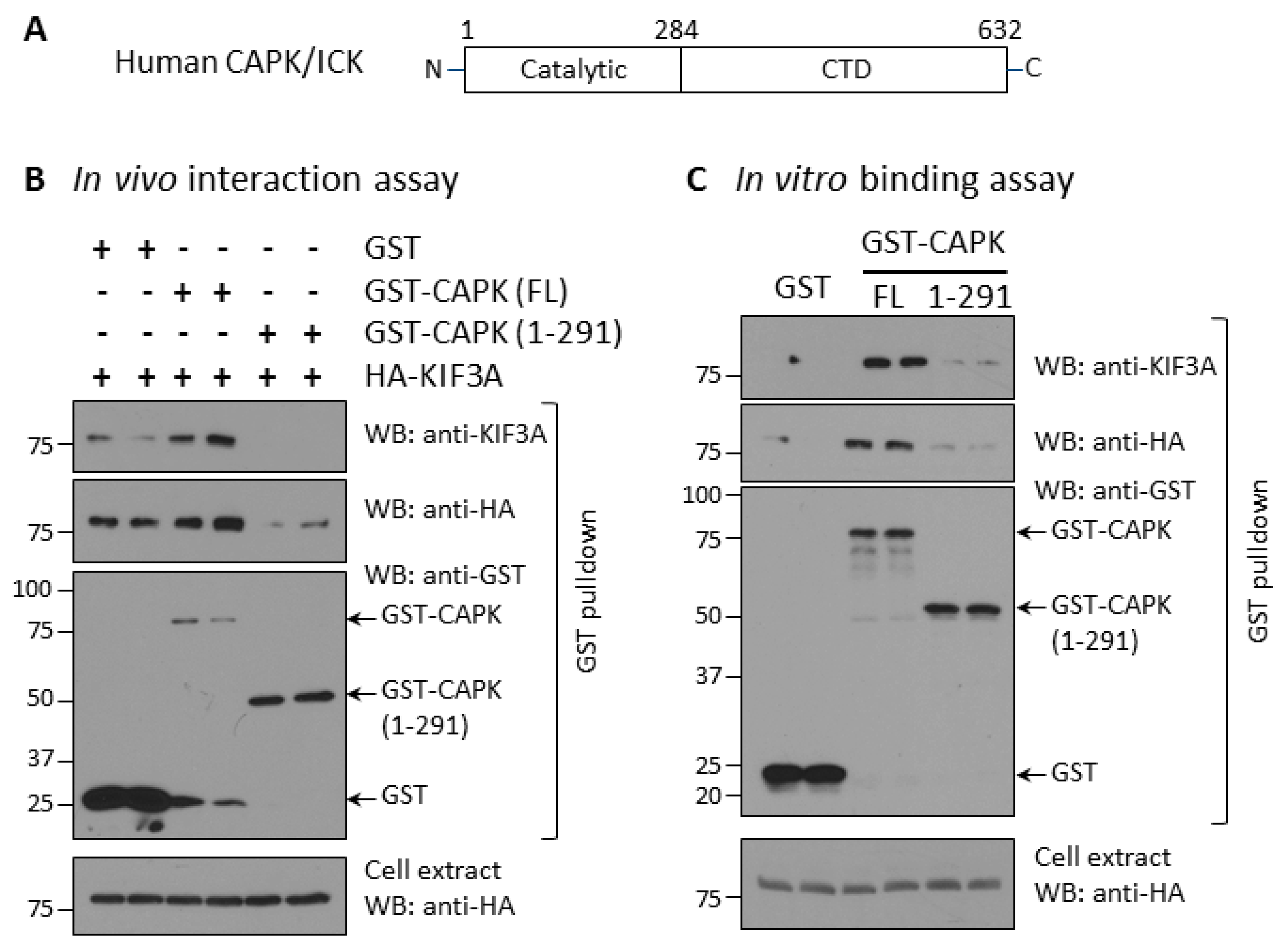{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids and Antibodies
2.2. Cell Culture and Transfection
2.3. GST Pull-Down, Immunoprecipitation, and Immunoblotting
2.4. In Vitro and In Vivo Phosphorylation Assay
2.5. In Vitro Binding Assay
2.6. Immunofluorescence Microscopy
2.7. Statistical Analysis
3. Results
3.1. CAPK Phosphorylates Human KIF3A-Thr672 In Vitro and In Vivo
3.2. Deletion of CAPK CTD Compromises Ability to Bind and Phosphorylate KIF3A
3.3. The CTD of CAPK Is Required for Ciliary Targeting
3.4. The CTD Truncation Abolishes the Suppressive Effect of CAPK on Ciliogenesis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abe, S.; Yagi, T.; Ishiyama, S.; Hiroe, M.; Marumo, F.; Ikawa, Y. Molecular cloning of a novel serine/threonine kinase, MRK, possibly involved in cardiac development. Oncogene 1995, 11, 2187–2195. [Google Scholar] [PubMed]
- Togawa, K.; Yan, Y.-X.; Inomoto, T.; Slaugenhaupt, S.; Rustgi, A.K.; Yan, Y. Intestinal cell kinase (ICK) localizes to the crypt region and requires a dual phosphorylation site found in map kinases. J. Cell. Physiol. 2000, 183, 129–139. [Google Scholar] [CrossRef]
- Fu, Z.; Schroeder, M.J.; Shabanowitz, J.; Kaldis, P.; Togawa, K.; Rustgi, A.K.; Hunt, D.F.; Sturgill, T.W. Activation of a Nuclear Cdc2-Related Kinase within a Mitogen-Activated Protein Kinase-Like TDY Motif by Autophosphorylation and Cyclin-Dependent Protein Kinase-Activating Kinase. Mol. Cell. Boil. 2005, 25, 6047–6064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Larson, K.A.; Chitta, R.K.; Parker, S.A.; Turk, B.E.; Lawrence, M.W.; Kaldis, P.; Galaktionov, K.; Cohn, S.M.; Shabanowitz, J.; et al. Identification of Yin-Yang Regulators and a Phosphorylation Consensus for Male Germ Cell-Associated Kinase (MAK)-Related Kinase. Mol. Cell. Boil. 2006, 26, 8639–8654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosakova, M.K.; Nita, A.; Gregor, T.; Varecha, M.; Gudernova, I.; Fafilek, B.; Barta, T.; Basheer, N.; Abraham, S.P.; Balek, L.; et al. Fibroblast growth factor receptor influences primary cilium length through an interaction with intestinal cell kinase. Proc. Natl. Acad. Sci. USA 2019, 116, 4316–4325. [Google Scholar] [CrossRef] [Green Version]
- Burghoorn, J.; Dekkers, M.P.J.; Rademakers, S.; De Jong, T.; Willemsen, R.; Jansen, G. Mutation of the MAP kinase DYF-5 affects docking and undocking of kinesin-2 motors and reduces their speed in the cilia of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2007, 104, 7157–7162. [Google Scholar] [CrossRef] [PubMed]
- Chaya, T.; Omori, Y.; Kuwahara, R.; Furukawa, T. ICK is essential for cell type-specific ciliogenesis and the regulation of ciliary transport. EMBO J. 2014, 33, 1227–1242. [Google Scholar] [CrossRef]
- Moon, H.; Song, J.; Shin, J.-O.; Lee, H.; Kim, H.-K.; Eggenschwiller, J.T.; Bok, J.; Ko, H.W. Intestinal cell kinase, a protein associated with endocrine-cerebro-osteodysplasia syndrome, is a key regulator of cilia length and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 8541–8546. [Google Scholar] [CrossRef] [Green Version]
- Ko, H.W.; Norman, R.X.; Tran, J.; Fuller, K.P.; Fukuda, M.; Eggenschwiler, J.T. Broad-minded links cell cycle-related kinase to cilia assembly and Hedgehog signal transduction. Dev. Cell 2010, 18, 237–247. [Google Scholar] [CrossRef]
- Snouffer, A.; Brown, D.; Lee, H.; Walsh, J.; Lupu, F.; Norman, R.; Lechtreck, K.; Ko, H.W.; Eggenschwiler, J. Cell Cycle-Related Kinase (CCRK) regulates ciliogenesis and Hedgehog signaling in mice. PLoS Genet. 2017, 13, e1006912. [Google Scholar] [CrossRef]
- Yang, Y.; Roine, N.; Mäkelä, T.P. CCRK depletion inhibits glioblastoma cell proliferation in a cilium-dependent manner. EMBO Rep. 2013, 14, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Malicki, J.J.; Johnson, C.A. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Boil. 2017, 27, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Boil. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Lahiry, P.; Wang, J.; Robinson, J.F.; Turowec, J.P.; Litchfield, D.W.; Lanktree, M.B.; Gloor, G.B.; Puffenberger, E.G.; Strauss, K.A.; Martens, M.B.; et al. A Multiplex Human Syndrome Implicates a Key Role for Intestinal Cell Kinase in Development of Central Nervous, Skeletal, and Endocrine Systems. Am. J. Hum. Genet. 2009, 84, 822. [Google Scholar] [CrossRef]
- Oud, M.M.; Bonnard, C.; Mans, D.A.; Altunoglu, U.; Tohari, S.; Ng, A.Y.J.; Eskin, A.; Lee, H.; Rupar, C.A.; De Wagenaar, N.P.; et al. A novel ICK mutation causes ciliary disruption and lethal endocrine-cerebro-osteodysplasia syndrome. Cilia 2016, 5, 134. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.P.; Bosakova, M.K.; Varecha, M.; Balek, L.; Barta, T.; Trantírek, L.; Jelinkova, I.; Duran, I.; Veselá, I.; Hampl, A.; et al. An inactivating mutation in intestinal cell kinase, ICK, impairs hedgehog signalling and causes short rib-polydactyly syndrome. Hum. Mol. Genet. 2016, 25, 3998–4011. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Park, S.H.; Wu, D.; Xu, W.; Guillot, S.J.; Jin, L.; Li, X.; Wang, Y.; Lin, C.-S.; Fu, Z. An essential role of intestinal cell kinase in lung development is linked to the perinatal lethality of human ECO syndrome. FEBS Lett. 2017, 591, 1247–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, M.; Jin, L.; Xie, L.; Park, S.H.; Tong, Y.; Wu, D.; Chhabra, A.B.; Fu, Z.; Li, X. A Murine Model for Human ECO Syndrome Reveals a Critical Role of Intestinal Cell Kinase in Skeletal Development. Calcif. Tissue Int. 2018, 102, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Engelke, M.F.; Waas, B.; Kearns, S.E.; Suber, A.; Boss, A.; Allen, B.L.; Verhey, K.J. Acute Inhibition of Heterotrimeric Kinesin-2 Function Reveals Mechanisms of Intraflagellar Transport in Mammalian Cilia. Curr. Boil. 2019, 29, 1137–1148.e4. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, B.S.; Barry, R.L.; Verhey, K.J.; Allen, B.L. The heterotrimeric kinesin-2 complex interacts with and regulates GLI protein function. J. Cell Sci. 2015, 128, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.W. Calcium-phosphate-mediated Transfection of Eukaryotic Cells with Plasmid DNAs. Cold Spring Harb. Protoc. 2006, 2006. [Google Scholar] [CrossRef]
- Fu, Z.; Kim, J.; Vidrich, A.; Sturgill, T.W.; Cohn, S.M. Intestinal cell kinase, a MAP kinase-related kinase, regulates proliferation and G1cell cycle progression of intestinal epithelial cells. Am. J. Physiol. Liver Physiol. 2009, 297, G632–G640. [Google Scholar] [CrossRef] [PubMed]
- Broekhuis, J.R.; Verhey, K.J.; Jansen, G. Regulation of Cilium Length and Intraflagellar Transport by the RCK-Kinases ICK and MOK in Renal Epithelial Cells. PLoS ONE 2014, 9, e108470. [Google Scholar] [CrossRef] [PubMed]
- Geer, L.Y.; Domrachev, M.; Lipman, D.J.; Bryant, S.H. CDART: Protein Homology by Domain Architecture. Genome Res. 2002, 12, 1619–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollastri, G.; McLysaght, A. Porter: A new, accurate server for protein secondary structure prediction. Bioinformatics 2005, 21, 1719–1720. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Ichinose, S.; Ogawa, T.; Hirokawa, N. Mechanism of Activity-Dependent Cargo Loading via the Phosphorylation of KIF3A by PKA and CaMKIIa. Neuron 2015, 87, 1022–1035. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, Y.S.; Wang, E.J.; Gailey, C.D.; Brautigan, D.L.; Allen, B.L.; Fu, Z. Ciliopathy-Associated Protein Kinase ICK Requires Its Non-Catalytic Carboxyl-Terminal Domain for Regulation of Ciliogenesis. Cells 2019, 8, 677. https://doi.org/10.3390/cells8070677
Oh YS, Wang EJ, Gailey CD, Brautigan DL, Allen BL, Fu Z. Ciliopathy-Associated Protein Kinase ICK Requires Its Non-Catalytic Carboxyl-Terminal Domain for Regulation of Ciliogenesis. Cells. 2019; 8(7):677. https://doi.org/10.3390/cells8070677
Chicago/Turabian StyleOh, Yoon Seon, Eric J. Wang, Casey D. Gailey, David L. Brautigan, Benjamin L. Allen, and Zheng Fu. 2019. "Ciliopathy-Associated Protein Kinase ICK Requires Its Non-Catalytic Carboxyl-Terminal Domain for Regulation of Ciliogenesis" Cells 8, no. 7: 677. https://doi.org/10.3390/cells8070677
APA StyleOh, Y. S., Wang, E. J., Gailey, C. D., Brautigan, D. L., Allen, B. L., & Fu, Z. (2019). Ciliopathy-Associated Protein Kinase ICK Requires Its Non-Catalytic Carboxyl-Terminal Domain for Regulation of Ciliogenesis. Cells, 8(7), 677. https://doi.org/10.3390/cells8070677