Inflammatory Bowel Disease Types Differ in Markers of Inflammation, Gut Barrier and in Specific Anti-Bacterial Response

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Antibody Array Assay for Serum Biomarkers
2.3. ELISA for Serum Biomarkers
2.4. Bacterial Antigen Preparation
2.5. Peripheral Blood Mononuclear Cells (PBMC)
2.6. Indirect Enzyme-Linked Immunosorbent Assays (ELISA)
2.7. Flow Cytometry Analysis (FACS)
2.8. Ethics Statement
2.9. Statistical Analysis
3. Results
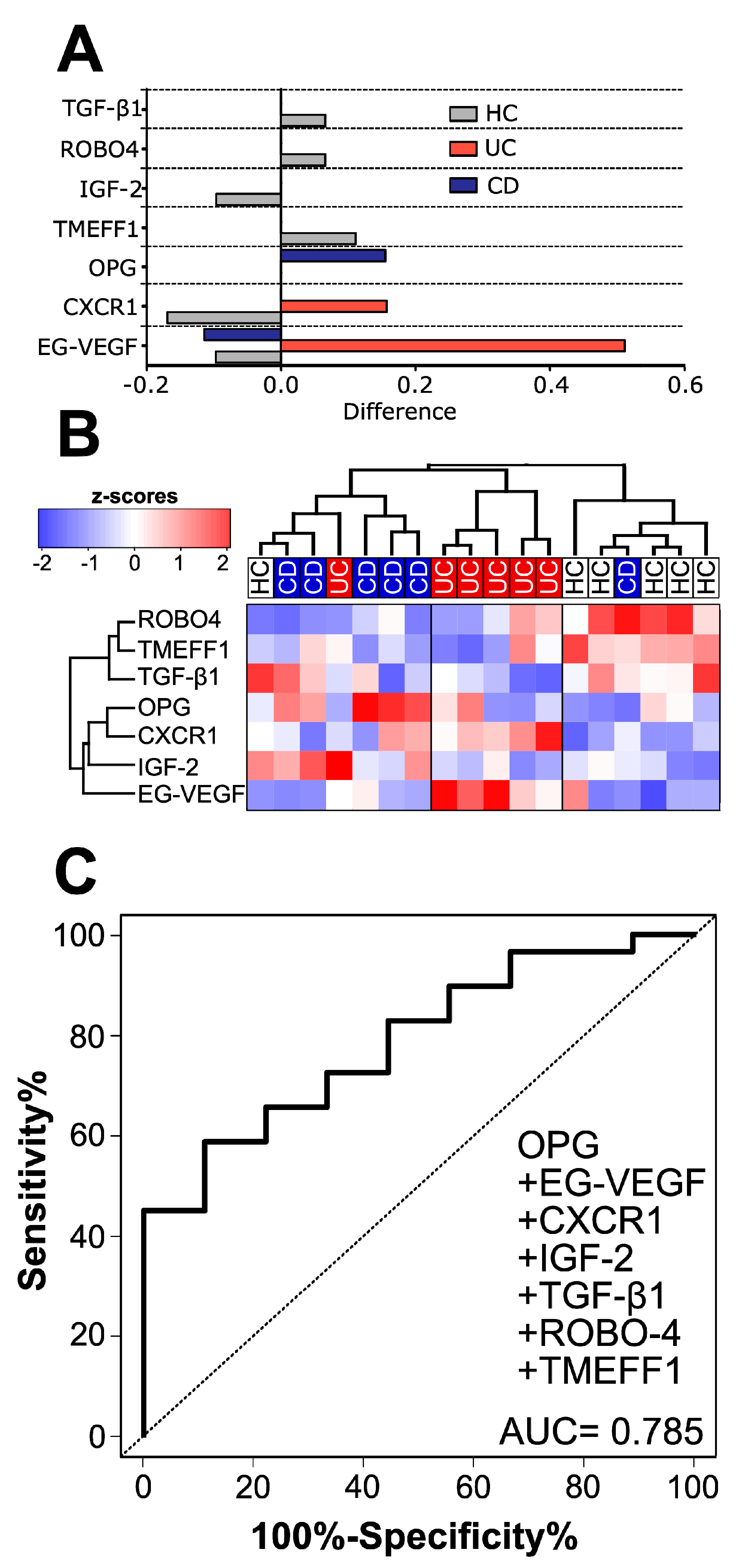
3.1. Healthy Subjects, CD and UC Patients Each Have a Distinct Cytokine Signature in Human Serum
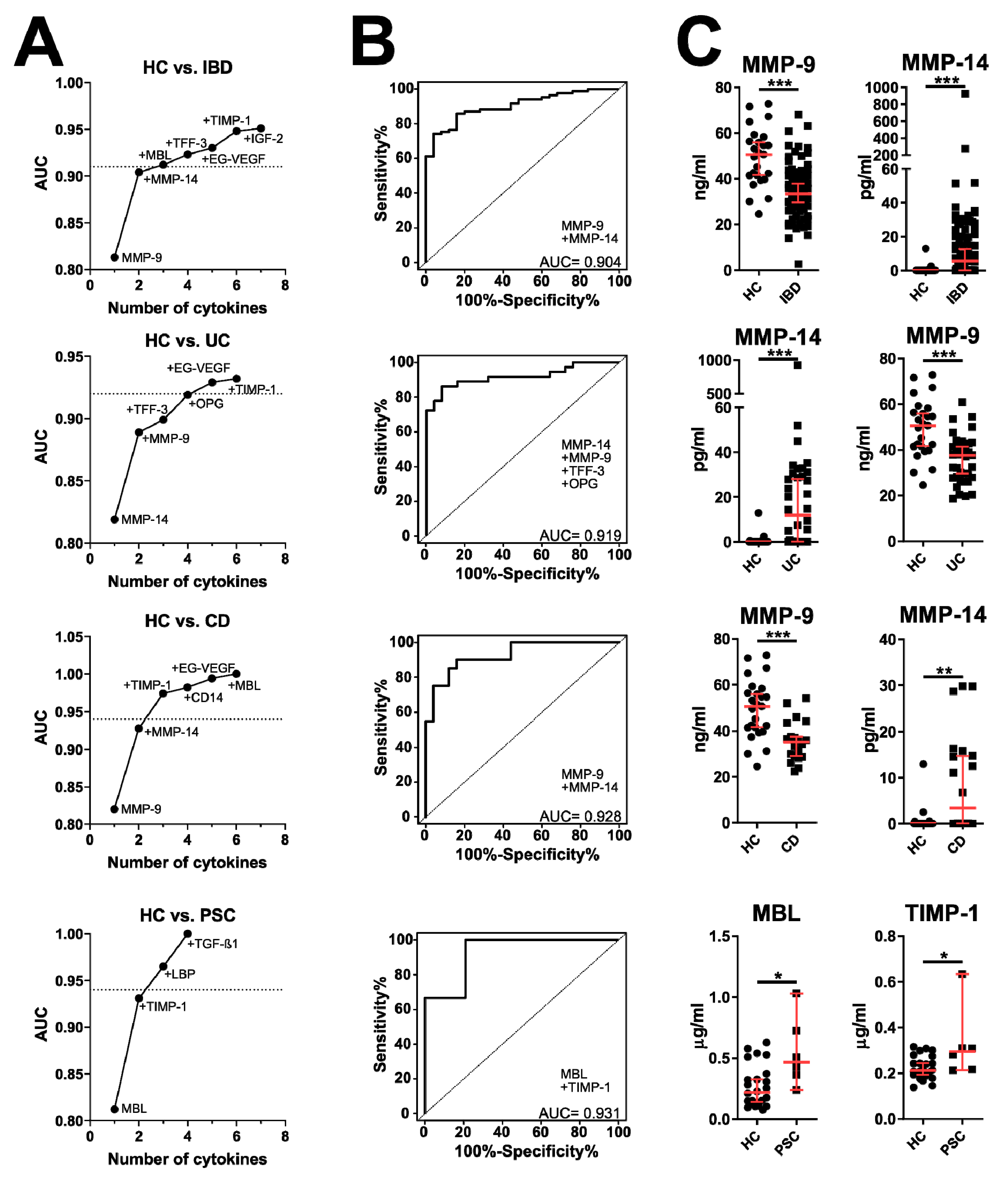
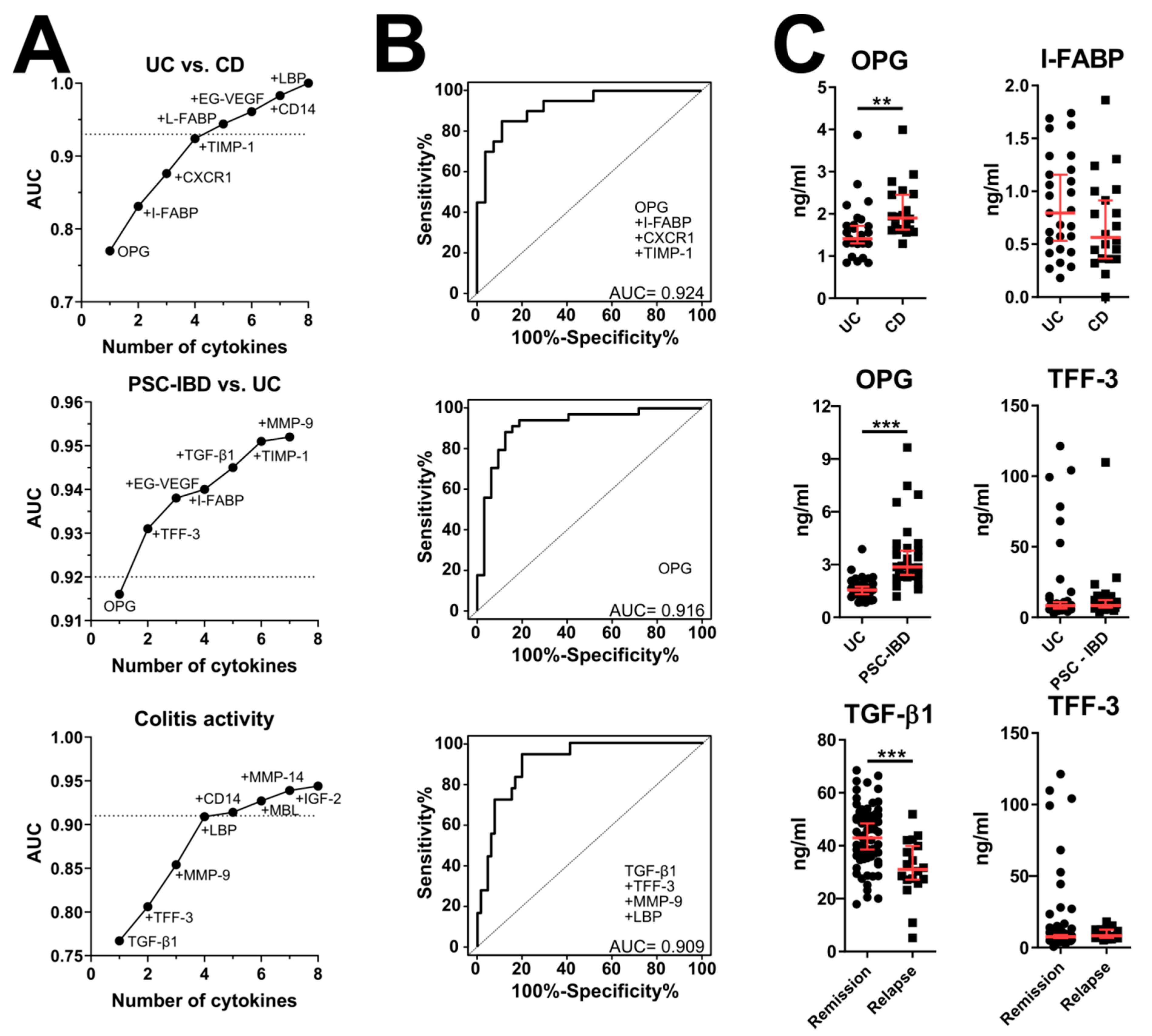
3.2. Validation of Microarray Data by ELISA
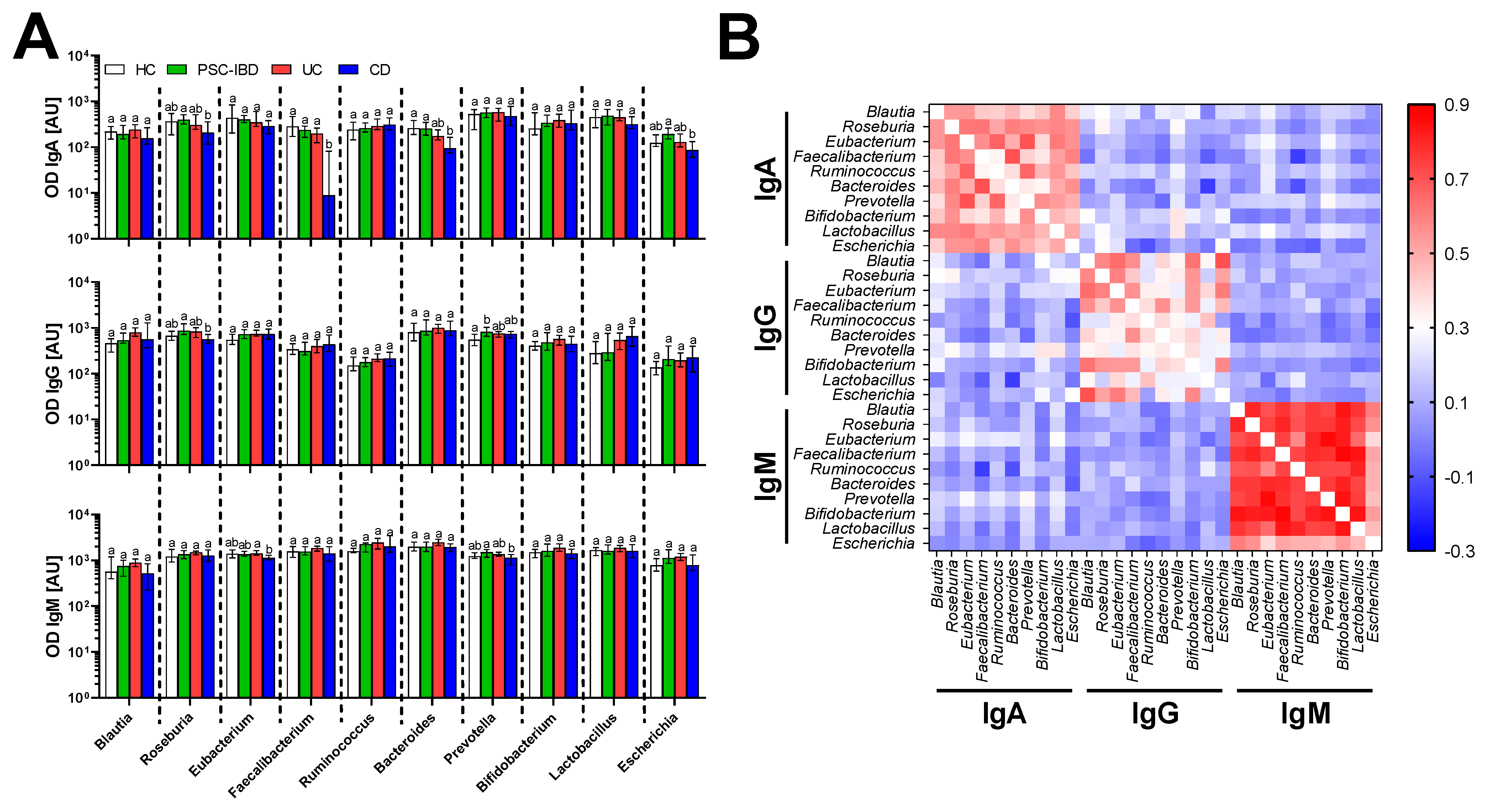
3.3. Serum Antibodies Against Bacteria
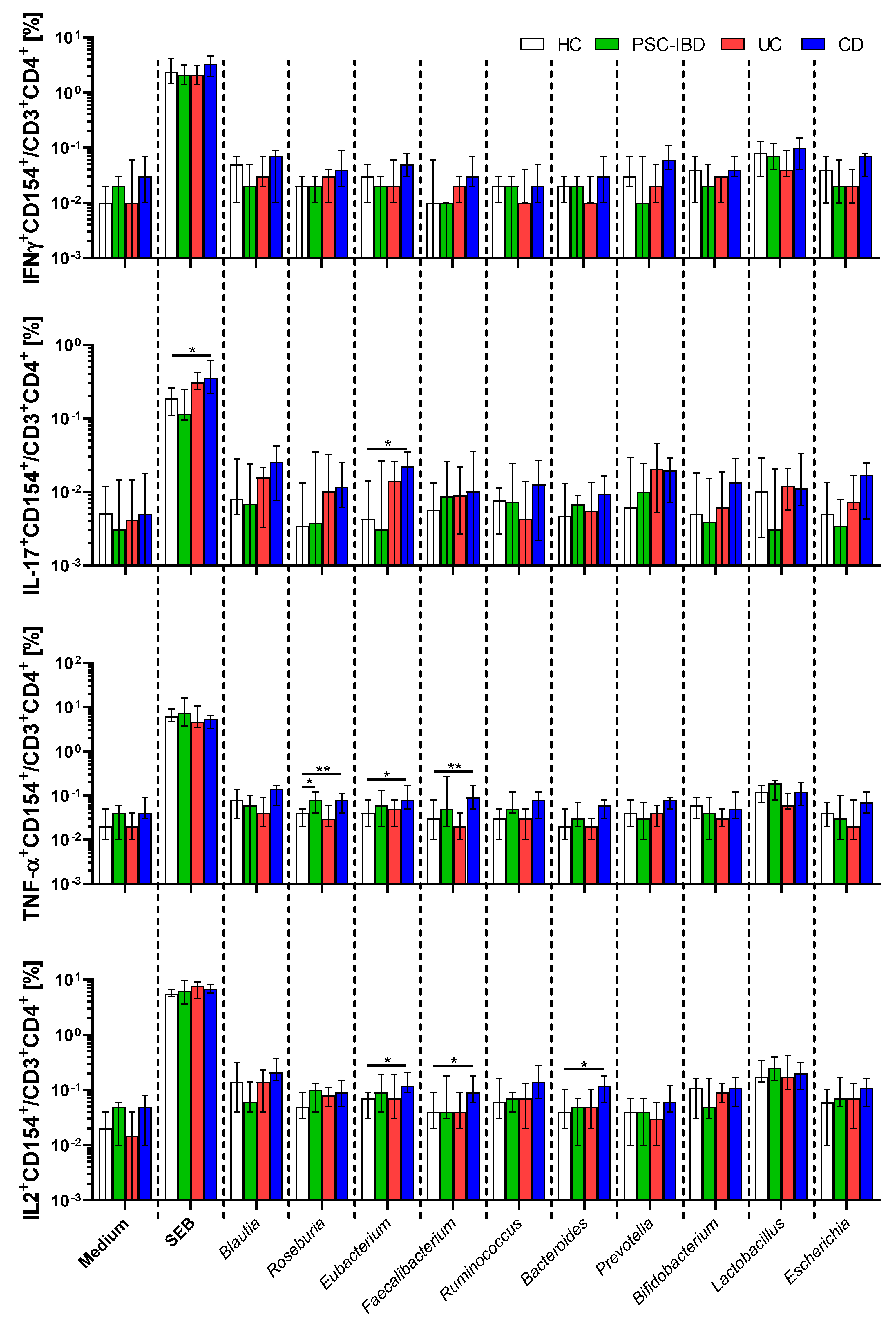
3.4. Circulating Gut Microbiota Reactive T-cells
3.5. Effect of IBD Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- Sartor, R.B. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008, 134, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Loftus, E.V., Jr.; Harewood, G.C.; Loftus, C.G.; Tremaine, W.J.; Harmsen, W.S.; Zinsmeister, A.R.; Jewell, D.A.; Sandborn, W.J. PSC-IBD: A unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut 2005, 54, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Sandborn, W.J. Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef]
- Stein, R.B.; Hanauer, S.B. Comparative tolerability of treatments for inflammatory bowel disease. Drug Saf. 2000, 23, 429–448. [Google Scholar] [CrossRef] [PubMed]
- Swoger, J.M.; Binion, D.G. Supportive therapy in IBD: What additional diagnoses and conditions must be treated? Dig. Dis. 2010, 28, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and fungi of the human gut microbiome: Correlations with diet and bacterial residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef]
- Columpsi, P.; Sacchi, P.; Zuccaro, V.; Cima, S.; Sarda, C.; Mariani, M.; Gori, A.; Bruno, R. Beyond the gut bacterial microbiota: The gut virome. J. Med. Virol. 2016, 88, 1467–1472. [Google Scholar] [CrossRef]
- Tlaskalova-Hogenova, H.; Sterzl, J.; Stepankova, R.; Dlabac, V.; Veticka, V.; Rossmann, P.; Mandel, L.; Rejnek, J. Development of immunological capacity under germfree and conventional conditions. Ann. N. Y. Acad. Sci. 1983, 409, 96–113. [Google Scholar] [CrossRef]
- Du, Z.; Hudcovic, T.; Mrazek, J.; Kozakova, H.; Srutkova, D.; Schwarzer, M.; Tlaskalova-Hogenova, H.; Kostovcik, M.; Kverka, M. Development of gut inflammation in mice colonized with mucosa-associated bacteria from patients with ulcerative colitis. Gut Pathog. 2015, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; Di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J.F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef]
- Mirsepasi-Lauridsen, H.C.; Du, Z.; Struve, C.; Charbon, G.; Karczewski, J.; Krogfelt, K.A.; Petersen, A.M.; Wells, J.M. Secretion of Alpha-Hemolysin by Escherichia coli Disrupts Tight Junctions in Ulcerative Colitis Patients. Clin. Transl. Gastroenterol. 2016, 7, e149. [Google Scholar] [CrossRef] [PubMed]
- Tannock, G.W. Molecular analysis of the intestinal microflora in IBD. Mucosal Immunol. 2008, 1, S15–S18. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kverka, M.; Tlaskalova-Hogenova, H. Intestinal Microbiota: Facts and Fiction. Dig. Dis. 2017, 35, 139–147. [Google Scholar] [CrossRef]
- McGuckin, M.A.; Eri, R.; Simms, L.A.; Florin, T.H.; Radford-Smith, G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm. Bowel Dis. 2009, 15, 100–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tlaskalova-Hogenova, H.; Stepankova, R.; Kozakova, H.; Hudcovic, T.; Vannucci, L.; Tuckova, L.; Rossmann, P.; Hrncir, T.; Kverka, M.; Zakostelska, Z.; et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: Contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol. Immunol. 2011, 8, 110–120. [Google Scholar] [CrossRef]
- Chang, J.; Leong, R.W.; Wasinger, V.C.; Ip, M.; Yang, M.; Phan, T.G. Impaired Intestinal Permeability Contributes to Ongoing Bowel Symptoms in Patients With Inflammatory Bowel Disease and Mucosal Healing. Gastroenterology 2017, 153, 723–731 e721. [Google Scholar] [CrossRef]
- Johansson, M.E.; Gustafsson, J.K.; Holmen-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjovall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Guthmann, F.; Borchers, T.; Wolfrum, C.; Wustrack, T.; Bartholomaus, S.; Spener, F. Plasma concentration of intestinal-and liver-FABP in neonates suffering from necrotizing enterocolitis and in healthy preterm neonates. Mol. Cell Biochem. 2002, 239, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Coufal, S.; Kokesova, A.; Tlaskalova-Hogenova, H.; Snajdauf, J.; Rygl, M.; Kverka, M. Urinary Intestinal Fatty Acid-Binding Protein Can Distinguish Necrotizing Enterocolitis from Sepsis in Early Stage of the Disease. J. Immunol. Res. 2016, 2016, 5727312. [Google Scholar] [CrossRef] [PubMed]
- Kokesova, A.; Coufal, S.; Frybova, B.; Kverka, M.; Rygl, M. The intestinal fatty acid-binding protein as a marker for intestinal damage in gastroschisis. PLoS ONE 2019, 14, e0210797. [Google Scholar] [CrossRef] [PubMed]
- Faubion, W.A., Jr.; Fletcher, J.G.; O’Byrne, S.; Feagan, B.G.; de Villiers, W.J.; Salzberg, B.; Plevy, S.; Proctor, D.D.; Valentine, J.F.; Higgins, P.D.; et al. EMerging BiomARKers in Inflammatory Bowel Disease (EMBARK) study identifies fecal calprotectin, serum MMP9, and serum IL-22 as a novel combination of biomarkers for Crohn’s disease activity: Role of cross-sectional imaging. Am. J. Gastroenterol. 2013, 108, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Farkas, K.; Sarodi, Z.; Balint, A.; Foldesi, I.; Tiszlavicz, L.; Szucs, M.; Nyari, T.; Tajti, J.; Nagy, F.; Szepes, Z.; et al. The diagnostic value of a new fecal marker, matrix metalloprotease-9, in different types of inflammatory bowel diseases. J. Crohn’s Colitis 2015, 9, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Mathew, C.G. New links to the pathogenesis of Crohn disease provided by genome-wide association scans. Nat. Rev. Genet. 2008, 9, 9–14. [Google Scholar] [CrossRef] [PubMed]
- McGovern, D.P.; Gardet, A.; Torkvist, L.; Goyette, P.; Essers, J.; Taylor, K.D.; Neale, B.M.; Ong, R.T.; Lagace, C.; Li, C.; et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010, 42, 332–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumberg, R.S. Inflammation in the intestinal tract: Pathogenesis and treatment. Dig. Dis. 2009, 27, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.J.; Heazlewood, S.P.; Gilshenan, K.S.; O’Brien, M.; McGuckin, M.A.; Florin, T.H. IgG antibodies against common gut bacteria are more diagnostic for Crohn’s disease than IgG against mannan or flagellin. Am. J. Gastroenterol. 2008, 103, 386–396. [Google Scholar] [CrossRef]
- Ruemmele, F.M.; Targan, S.R.; Levy, G.; Dubinsky, M.; Braun, J.; Seidman, E.G. Diagnostic accuracy of serological assays in pediatric inflammatory bowel disease. Gastroenterology 1998, 115, 822–829. [Google Scholar] [CrossRef]
- Zholudev, A.; Zurakowski, D.; Young, W.; Leichtner, A.; Bousvaros, A. Serologic testing with ANCA, ASCA, and anti-OmpC in children and young adults with Crohn’s disease and ulcerative colitis: Diagnostic value and correlation with disease phenotype. Am. J. Gastroenterol. 2004, 99, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Silva, M.; Afonso, P.C.; de Souza, P.R.; Peghini, B.C.; Rodrigues-Junior, V.; de Barros Cardoso, C.R. Reappraisal of antibodies against Saccharomyces cerevisiae (ASCA) as persistent biomarkers in quiescent Crohn’s disease. Autoimmunity 2019, 52, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Sitaraman, S.V.; Klapproth, J.M.; Moore, D.A., 3rd; Landers, C.; Targan, S.; Williams, I.R.; Gewirtz, A.T. Elevated flagellin-specific immunoglobulins in Crohn’s disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G403–G406. [Google Scholar] [CrossRef] [PubMed]
- Iltanen, S.; Tervo, L.; Halttunen, T.; Wei, B.; Braun, J.; Rantala, I.; Honkanen, T.; Kronenberg, M.; Cheroutre, H.; Turovskaya, O.; et al. Elevated serum anti-I2 and anti-OmpW antibody levels in children with IBD. Inflamm. Bowel Dis. 2006, 12, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Papp, M.; Norman, G.L.; Altorjay, I.; Lakatos, P.L. Utility of serological markers in inflammatory bowel diseases: Gadget or magic? World J. Gastroenterol. 2007, 13, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Standaert-Vitse, A.; Branche, J.; Chamaillard, M. IBD serological panels: Facts and perspectives. Inflamm. Bowel Dis. 2007, 13, 1561–1566. [Google Scholar] [CrossRef]
- Lewis, J.D. The utility of biomarkers in the diagnosis and therapy of inflammatory bowel disease. Gastroenterology 2011, 140, 1817–1826.e1812. [Google Scholar] [CrossRef]
- Cathelin, R.; Lopez, F.; Klopp, C. AGScan: A pluggable microarray image quantification software based on the ImageJ library. Bioinformatics 2007, 23, 247–248. [Google Scholar] [CrossRef]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Chu, G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6567–6572. [Google Scholar] [CrossRef] [Green Version]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Kreher, C.R.; Dittrich, M.T.; Guerkov, R.; Boehm, B.O.; Tary-Lehmann, M. CD4+ and CD8+ cells in cryopreserved human PBMC maintain full functionality in cytokine ELISPOT assays. J. Immunol. Methods 2003, 278, 79–93. [Google Scholar] [CrossRef]
- Hegazy, A.N.; West, N.R.; Stubbington, M.J.T.; Wendt, E.; Suijker, K.I.M.; Datsi, A.; This, S.; Danne, C.; Campion, S.; Duncan, S.H.; et al. Circulating and Tissue-Resident CD4(+) T Cells With Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered During Inflammation. Gastroenterology 2017, 153, 1320–1337.e1316. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Hoerning, A.; Datta, D.; Edelbauer, M.; Stack, M.P.; Calzadilla, K.; Pal, S.; Briscoe, D.M. Cutting Edge: Vascular Endothelial Growth Factor-Mediated Signaling in Human CD45RO+ CD4+ T Cells Promotes Akt and ERK Activation and Costimulates IFN- Production. J. Immunol. 2009, 184, 545–549. [Google Scholar] [CrossRef]
- Danese, S. VEGF in inflammatory bowel disease: A master regulator of mucosal immune-driven angiogenesis. Dig. Liver Dis. 2008, 40, 680–683. [Google Scholar] [CrossRef]
- Zdravkovic, N.D.; Jovanovic, I.P.; Radosavljevic, G.D.; Arsenijevic, A.N.; Zdravkovic, N.D.; Mitrovic, S.L.; Arsenijevic, N.N. Potential Dual Immunomodulatory Role of VEGF in Ulcerative Colitis and Colorectal Carcinoma. Int. J. Med Sci. 2014, 11, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Dorsch, M.; Qiu, Y.; Soler, D.; Frank, N.; Duong, T.; Goodearl, A.; O’Neil, S.; Lora, J.; Fraser, C.C. PK1/EG-VEGF induces monocyte differentiation and activation. J. Leukoc. Biol. 2005, 78, 426–434. [Google Scholar] [CrossRef]
- Wu, L.; Ruffing, N.; Shi, X.; Newman, W.; Soler, D.; Mackay, C.R.; Qin, S. Discrete steps in binding and signaling of interleukin-8 with its receptor. J. Biol. Chem. 1996, 271, 31202–31209. [Google Scholar] [CrossRef]
- Sabroe, I.; Jones, E.C.; Whyte, M.K.; Dower, S.K. Regulation of human neutrophil chemokine receptor expression and function by activation of Toll-like receptors 2 and 4. Immunology 2005, 115, 90–98. [Google Scholar] [CrossRef]
- Muthas, D.; Reznichenko, A.; Balendran, C.A.; Bottcher, G.; Clausen, I.G.; Karrman Mardh, C.; Ottosson, T.; Uddin, M.; MacDonald, T.T.; Danese, S.; et al. Neutrophils in ulcerative colitis: A review of selected biomarkers and their potential therapeutic implications. Scand. J. Gastroenterol. 2017, 52, 125–135. [Google Scholar] [CrossRef]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Liu, W.; Xu, C.; Zhao, H.; Xia, P.; Song, R.; Gu, J.; Liu, X.; Bian, J.; Yuan, Y.; Liu, Z. Osteoprotegerin Induces Apoptosis of Osteoclasts and Osteoclast Precursor Cells via the Fas/Fas Ligand Pathway. PLoS ONE 2015, 10, e0142519. [Google Scholar] [CrossRef] [PubMed]
- Franchimont, N.; Reenaers, C.; Lambert, C.; Belaiche, J.; Bours, V.; Malaise, M.; Delvenne, P.; Louis, E. Increased expression of receptor activator of NF-kappaB ligand (RANKL), its receptor RANK and its decoy receptor osteoprotegerin in the colon of Crohn’s disease patients. Clin. Exp. Immunol. 2004, 138, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Ludwiczek, O.; Gabriel, M.; Obrist, P.; Wolf, A.M.; Tilg, H. The RANKL/OPG system is activated in inflammatory bowel disease and relates to the state of bone loss. Gut 2005, 54, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentine, J.F.; Sninsky, C.A. Prevention and treatment of osteoporosis in patients with inflammatory bowel disease. Am. J. Gastroenterol. 1999, 94, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Kapsoritakis, A.N.; Kapsoritaki, A.I.; Davidi, I.P.; Lotis, V.D.; Manolakis, A.C.; Mylonis, P.I.; Theodoridou, A.T.; Germenis, A.E.; Potamianos, S.P. Imbalance of tissue inhibitors of metalloproteinases (TIMP)—1 and—4 serum levels, in patients with inflammatory bowel disease. BMC Gastroenterol. 2008, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Somerville, R.P.; Oblander, S.A.; Apte, S.S. Matrix metalloproteinases: Old dogs with new tricks. Genome Biol. 2003, 4, 216. [Google Scholar] [CrossRef] [PubMed]
- Baugh, M.D.; Perry, M.J.; Hollander, A.P.; Davies, D.R.; Cross, S.S.; Lobo, A.J.; Taylor, C.J.; Evans, G.S. Matrix metalloproteinase levels are elevated in inflammatory bowel disease. Gastroenterology 1999, 117, 814–822. [Google Scholar] [CrossRef]
- Lakatos, G.; Hritz, I.; Varga, M.Z.; Juhász, M.; Miheller, P.; Cierny, G.; Tulassay, Z.; Herszényi, L. The impact of matrix metalloproteinases and their tissue inhibitors in inflammatory bowel diseases. Dig. Dis. 2012, 30, 289–295. [Google Scholar] [CrossRef]
- Kofla-Dlubacz, A.; Matusiewicz, M.; Krzystek-Korpacka, M.; Iwanczak, B. Correlation of MMP-3 and MMP-9 with Crohn’s Disease Activity in Children. Dig. Dis. Sci. 2011, 57, 706–712. [Google Scholar] [CrossRef]
- Makitalo, L.; Rintamaki, H.; Tervahartiala, T.; Sorsa, T.; Kolho, K.L. Serum MMPs 7-9 and their inhibitors during glucocorticoid and anti-TNF-alpha therapy in pediatric inflammatory bowel disease. Scand. J. Gastroenterol. 2012, 47, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Crowe, M.J.; Doetschman, T.; Greenhalgh, D.G. Delayed Wound Healing in Immunodeficient TGF-β1 Knockout Mice. J. Investig. Dermatol. 2000, 115, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Hahm, K.B. Loss of transforming growth factor beta signalling in the intestine contributes to tissue injury in inflammatory bowel disease. Gut 2001, 49, 190–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howe, K.L.; Reardon, C.; Wang, A.; Nazli, A.; McKay, D.M. Transforming Growth Factor-β Regulation of Epithelial Tight Junction Proteins Enhances Barrier Function and Blocks Enterohemorrhagic Escherichia coli O157:H7-Induced Increased Permeability. Am. J. Pathol. 2005, 167, 1587–1597. [Google Scholar] [CrossRef]
- Stallmach, A.; Schuppan, D.; Riese, H.H.; Matthes, H.; Riecken, E.O. Increased collagen type III synthesis by fibroblasts isolated from strictures of patients with Crohn’s disease. Gastroenterology 1992, 102, 1920–1929. [Google Scholar] [CrossRef]
- Kader, H.A.; Tchernev, V.T.; Satyaraj, E.; Lejnine, S.; Kotler, G.; Kingsmore, S.F.; Patel, D.D. Protein microarray analysis of disease activity in pediatric inflammatory bowel disease demonstrates elevated serum PLGF, IL-7, TGF-beta1, and IL-12p40 levels in Crohn’s disease and ulcerative colitis patients in remission versus active disease. Am. J. Gastroenterol. 2005, 100, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Miquel, S.; Martin, R.; Rossi, O.; Bermudez-Humaran, L.G.; Chatel, J.M.; Sokol, H.; Thomas, M.; Wells, J.M.; Langella, P. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 2013, 16, 255–261. [Google Scholar] [CrossRef]
- Fujimoto, T.; Imaeda, H.; Takahashi, K.; Kasumi, E.; Bamba, S.; Fujiyama, Y.; Andoh, A. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn’s disease. J. Gastroenterol. Hepatol. 2013, 28, 613–619. [Google Scholar] [CrossRef]
- Rajca, S.; Grondin, V.; Louis, E.; Vernier-Massouille, G.; Grimaud, J.C.; Bouhnik, Y.; Laharie, D.; Dupas, J.-L.; Pillant, H.; Picon, L.; et al. Alterations in the Intestinal Microbiome (Dysbiosis) as a Predictor of Relapse After Infliximab Withdrawal in Crohn’s Disease. Inflamm. Bowel Dis. 2014, 20, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermudez-Humaran, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Quevrain, E.; Maubert, M.A.; Michon, C.; Chain, F.; Marquant, R.; Tailhades, J.; Miquel, S.; Carlier, L.; Bermudez-Humaran, L.G.; Pigneur, B.; et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut 2016, 65, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Andoh, A. Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion 2016, 93, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.R.; Mirsepasi-Lauridsen, H.C.; Thysen, A.H.; Brynskov, J.; Krogfelt, K.A.; Petersen, A.M.; Pedersen, A.E.; Brix, S. Distinct inflammatory and cytopathic characteristics of Escherichia coli isolates from inflammatory bowel disease patients. Int. J. Med. Microbiol. 2015, 305, 925–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, N.; Oliver, J.R.; Wagner, F.H.; Lazenby, A.S.; Elson, C.O.; Weaver, C.T. T helper 1 and T helper 2 cells are pathogenic in an antigen-specific model of colitis. J. Exp. Med. 2002, 195, 71–84. [Google Scholar] [CrossRef]
- Feng, T.; Wang, L.; Schoeb, T.R.; Elson, C.O.; Cong, Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J. Exp. Med. 2010, 207, 1321–1332. [Google Scholar] [CrossRef]
- Calderon-Gomez, E.; Bassolas-Molina, H.; Mora-Buch, R.; Dotti, I.; Planell, N.; Esteller, M.; Gallego, M.; Marti, M.; Garcia-Martin, C.; Martinez-Torro, C.; et al. Commensal-specific CD4(+) cells from patients with Crohn’s disease have a t-helper 17 inflammatory profile. Gastroenterology 2016, 151, 489–500. [Google Scholar] [CrossRef]
- Casanova, M.J.; Chaparro, M.; Garcia-Sanchez, V.; Nantes, O.; Leo, E.; Rojas-Feria, M.; Jauregui-Amezaga, A.; Garcia-Lopez, S.; Huguet, J.M.; Arguelles-Arias, F.; et al. Evolution after anti-TNF discontinuation in patients with inflammatory bowel disease: A multicenter long-term follow-up study. Am. J. Gastroenterol. 2017, 112, 120–131. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HC n = 28 | PSC n = 47 | UC n = 52 | CD n = 20 | |
|---|---|---|---|---|
| Age (mean ±SD; years) | 42.5 ± 10.5 | 38.0 ± 11.6 | 39.7 ± 9.8 | 33.5 ± 7.8 |
| Sex (% of males) | 53.6; 15/13 | 74.5; 35/12 | 53.9; 28/24 | 45.0; 9/11 |
| Activity (% of active) | 0.0 | 14.0 | 26.9 | 20.0 |
| Extent of intestinal inflammation | ||||
| none (%; n) | 100.0; 28 | 12.8; 6 | 0.0; 0 | 0.0; 0 |
| partial (%; n) | 0.0; 0 | 10.6; 5 | 38.4; 20 | 45.0; 9 |
| pancolitis (%; n) | 0.0; 0 | 72.3; 34 | 61.5; 32 | 50.0; 10 |
| Therapy | ||||
| Mesalazine (5-ASA) (%; n) | 0.0; 0 | 70.2; 33 | 92.3; 48 | 85.0; 17 |
| Glucocorticoids (%; n) | 0.0; 0 | 38.3; 18 | 21.2; 11 | 15.0; 3 |
| Azathioprine (AZA) (%; n) | 0.0; 0 | 31.9; 15 | 40.4; 21 | 35.0; 7 |
| Anti-TNF-α (%; n) | 0.0; 0 | 0.0; 0 | 38.5; 20 | 45.0; 9 |
| E. coli Nissle 1917 (%; n) | 0.0; 0 | 8.5; 4 | 23.1; 12 | 20.0; 4 |
| Biomarker | Abbreviation | Manufacturer | Cat. No |
|---|---|---|---|
| Endocrine-Gland-derived Vascular Endothelial Growth Factor * | EG-VEGF | R&D systems | DY1209 |
| Interleukin-8 receptor, alpha * | CXCR1/IL8RA | LifeSpan BioScience | LS-F11255 |
| Osteoprotegerin | OPG | R&D systems | DY805 |
| Tomoregulin 1 | TMEFF1 | LifeSpan BioScience | LS-F52730 |
| Insulin-like Growth Factor 2 * | IGF2 | R&D systems | DY292 |
| Transforming Growth Factor-β1 * | TGF-β1 | R&D systems | DY240 |
| TROY protein | TNFRSF19 | LifeSpan BioScience | LS-F39966 |
| Roundabout Guidance Receptor 4 * | ROBO4 | RayBiotech | ELH-ROBO4 |
| Matrix Metalloproteinase 9 | MMP-9 | R&D systems | DY911 |
| Matrix Metalloproteinase 14 | MMP-14 | R&D systems | DY918 |
| Tissue Inhibitor of Metalloproteinases 1 | TIMP-1 | R&D systems | DY970 |
| Mannan-Binding Lectin | MBL | R&D systems | DY2307 |
| Soluble CD14 | CD14 | R&D systems | DY383 |
| Lipopolysaccharide-Binding Protein | LBP | R&D systems | DY870 |
| Trefoil Factor 3 | TFF-3 | R&D systems | DY4407 |
| Endotoxin-Core Antibody IgM | EndoCab | MyBiosource | MBS9352896 |
| Serum Amyloid A | SAA | HyCult Biotech | HK333 |
| Pre-haptoglobin 2 | Zonulin | MyBiosource | MBS2880564 |
| D-amino-acid oxidase | DAAO | MyBiosource | MBS2886321 |
| Intestinal Fatty Acid-Binding Protein | I-FABP | HyCult Biotech | HK406 |
| Liver Fatty Acid-Binding Protein | L-FABP | HyCult Biotech | HK404 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coufal, S.; Galanova, N.; Bajer, L.; Gajdarova, Z.; Schierova, D.; Jiraskova Zakostelska, Z.; Kostovcikova, K.; Jackova, Z.; Stehlikova, Z.; Drastich, P.; et al. Inflammatory Bowel Disease Types Differ in Markers of Inflammation, Gut Barrier and in Specific Anti-Bacterial Response. Cells 2019, 8, 719. https://doi.org/10.3390/cells8070719
Coufal S, Galanova N, Bajer L, Gajdarova Z, Schierova D, Jiraskova Zakostelska Z, Kostovcikova K, Jackova Z, Stehlikova Z, Drastich P, et al. Inflammatory Bowel Disease Types Differ in Markers of Inflammation, Gut Barrier and in Specific Anti-Bacterial Response. Cells. 2019; 8(7):719. https://doi.org/10.3390/cells8070719
Chicago/Turabian StyleCoufal, Stepan, Natalie Galanova, Lukas Bajer, Zuzana Gajdarova, Dagmar Schierova, Zuzana Jiraskova Zakostelska, Klara Kostovcikova, Zuzana Jackova, Zuzana Stehlikova, Pavel Drastich, and et al. 2019. "Inflammatory Bowel Disease Types Differ in Markers of Inflammation, Gut Barrier and in Specific Anti-Bacterial Response" Cells 8, no. 7: 719. https://doi.org/10.3390/cells8070719
APA StyleCoufal, S., Galanova, N., Bajer, L., Gajdarova, Z., Schierova, D., Jiraskova Zakostelska, Z., Kostovcikova, K., Jackova, Z., Stehlikova, Z., Drastich, P., Tlaskalova-Hogenova, H., & Kverka, M. (2019). Inflammatory Bowel Disease Types Differ in Markers of Inflammation, Gut Barrier and in Specific Anti-Bacterial Response. Cells, 8(7), 719. https://doi.org/10.3390/cells8070719