Fisetin Suppresses the Proliferation and Metastasis of Renal Cell Carcinoma through Upregulation of MEK/ERK-Targeting CTSS and ADAM9



 ,
, 
Abstract
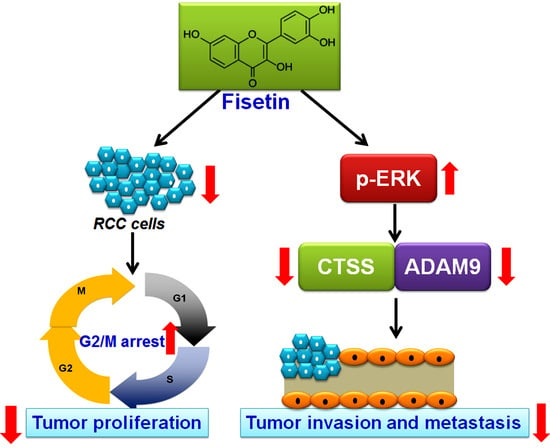
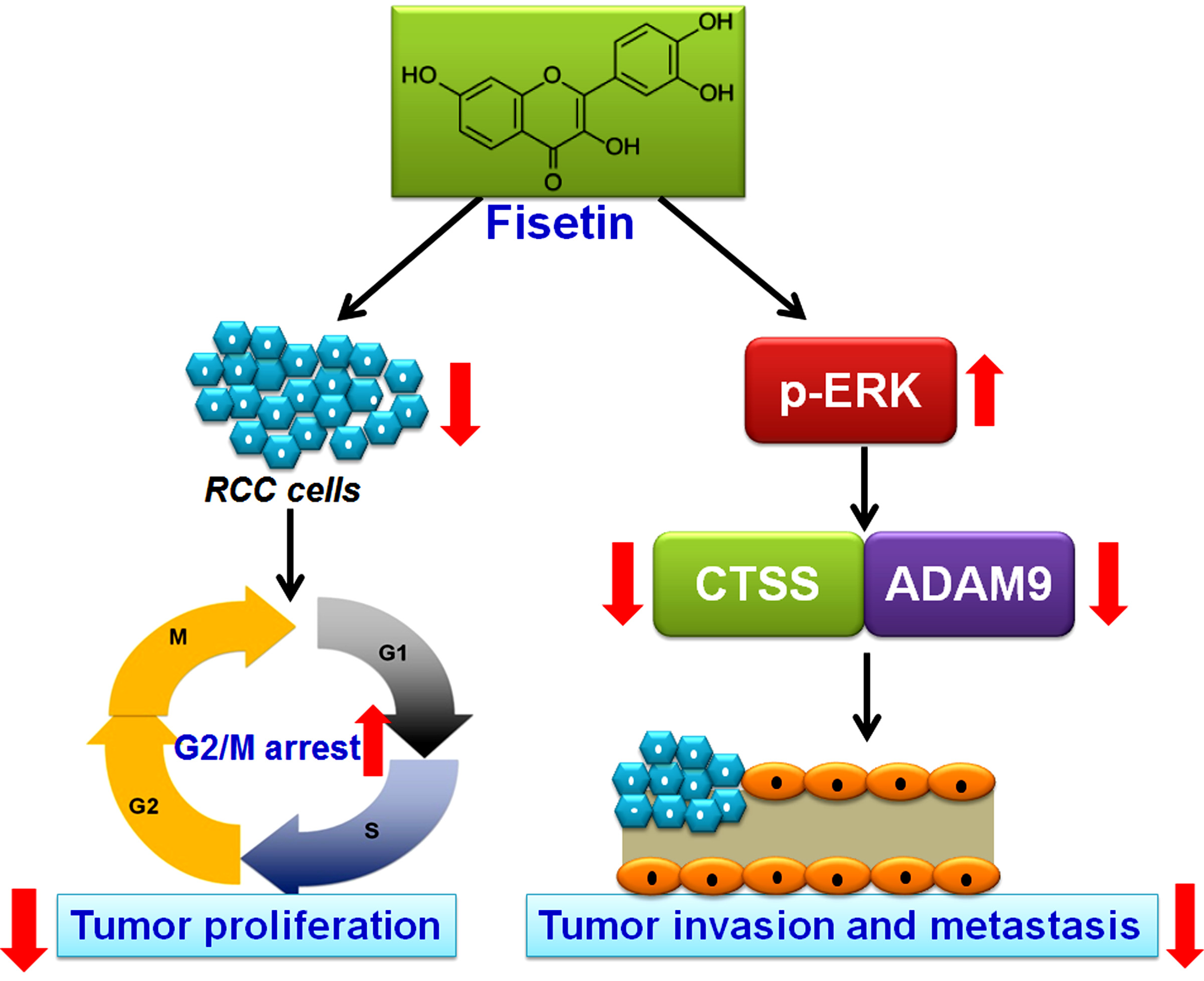
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemical, Reagents, and Antibody
2.2. Cell Culture
2.3. MTT Assay
2.4. Cell Toxicity Assay
2.5. Colony Formation Assay
2.6. Evaluation of Cell Cycle
2.7. Annen V/PI Staining by Flow Cytomrtey
2.8. Migration and Invasion Assay
2.9. Proteome Profiler Human Protease Array
2.10. Western Blot Analysis
2.11. Quantitative Reverse Transcription Polymerase Chain Reaction
2.12. Statistical Analysis
3. Results
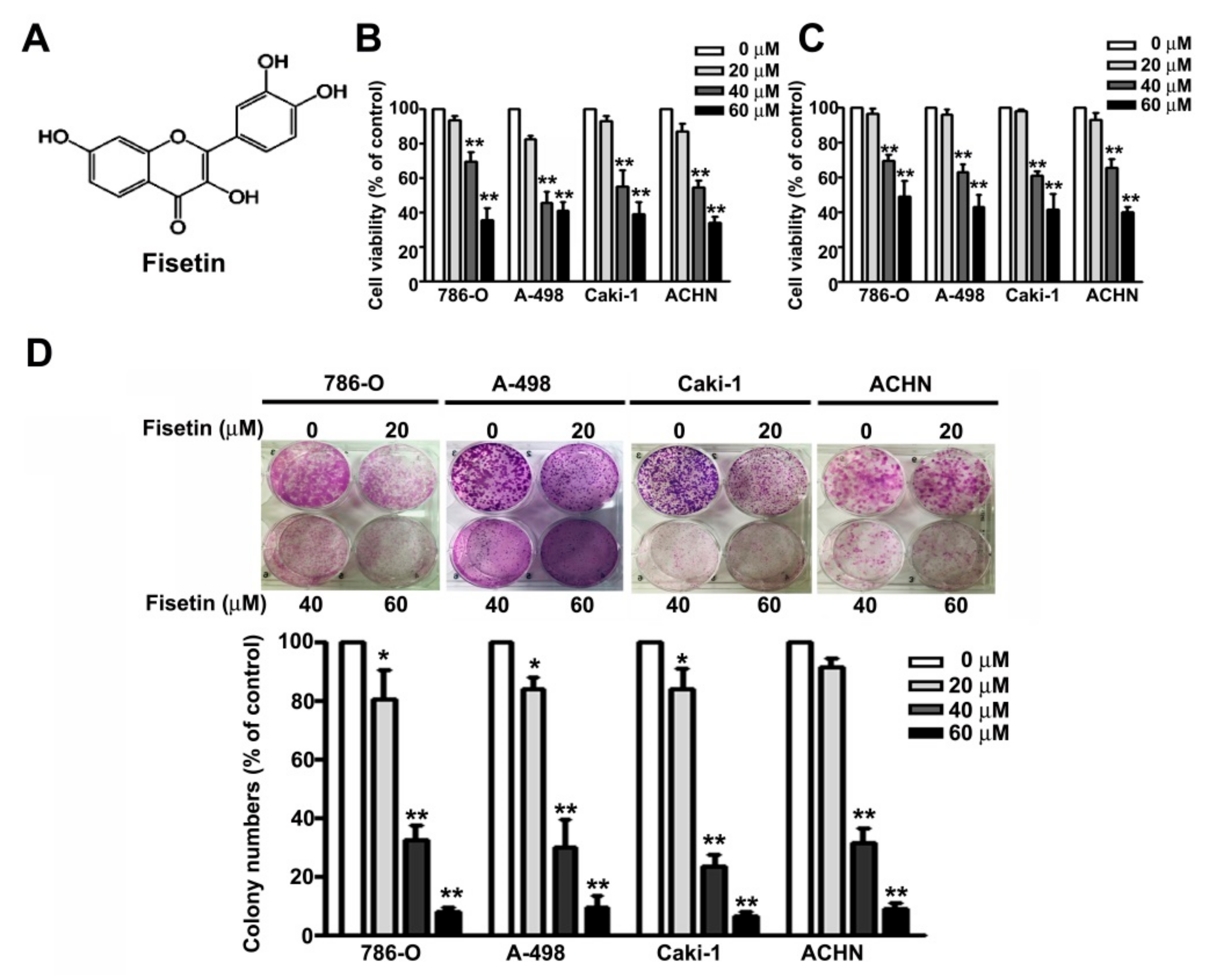
3.1. Fisetin Decreased RCC Cell Viability
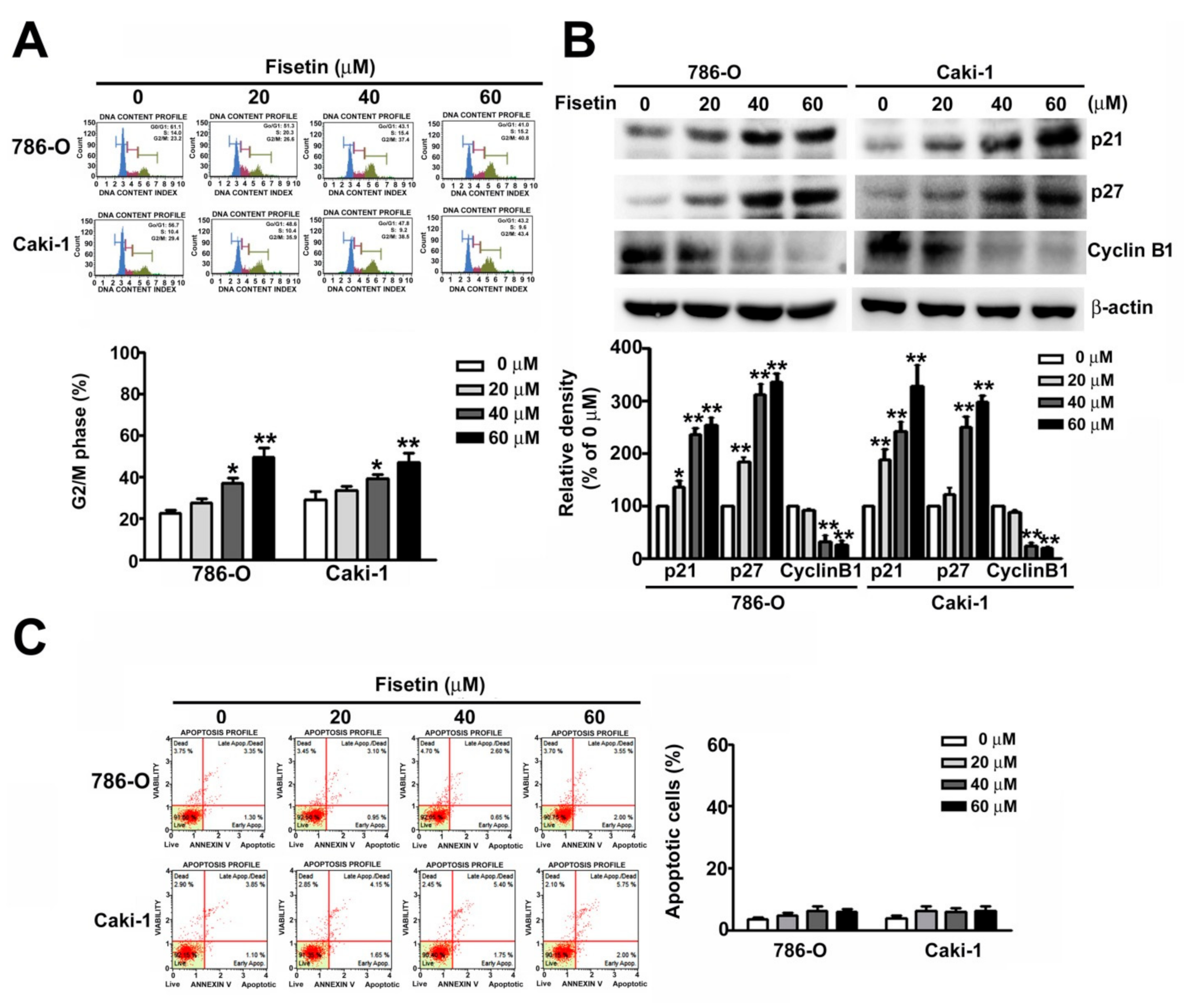
3.2. Fisetin Induced Cell Cycle Arrest in the G2/M Phase and Assessment of Related G2/M Proteins of RCC Cells
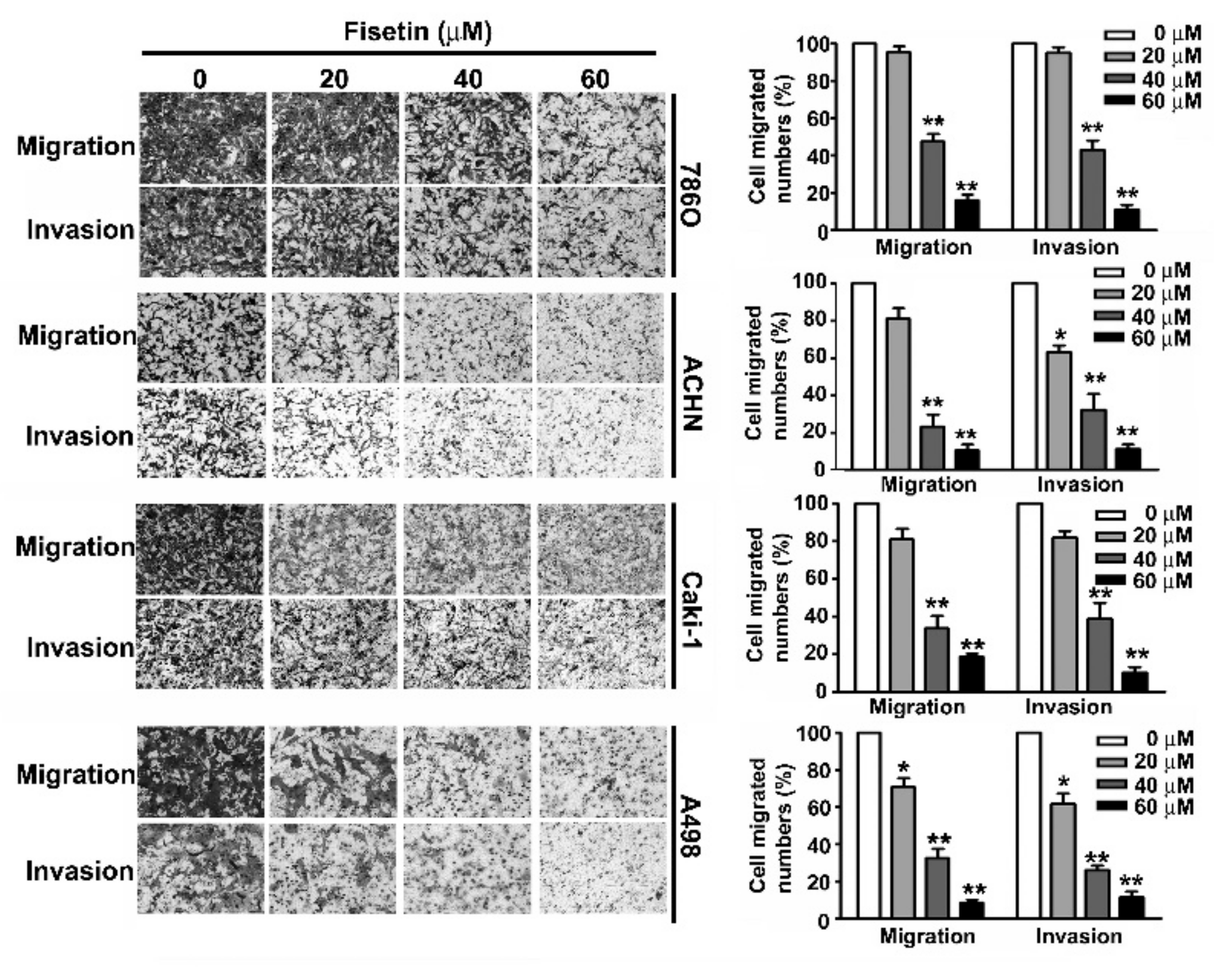
3.3. Fisetin Inhibited Migration and Invasion of RCC Cells
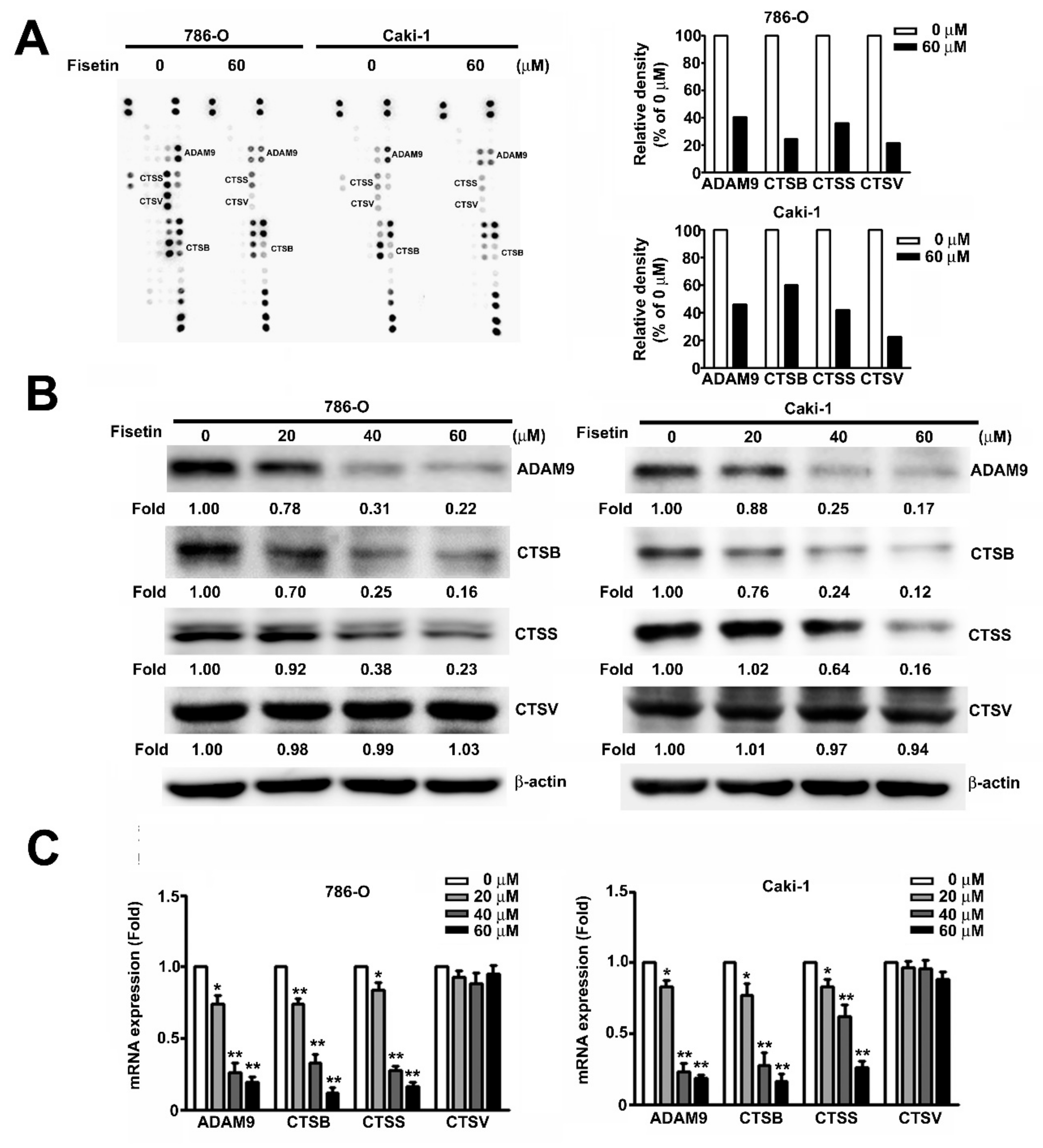
3.4. Fisetin Inhibited CTSB, CTSS, and ADAM9 in RCC Cells
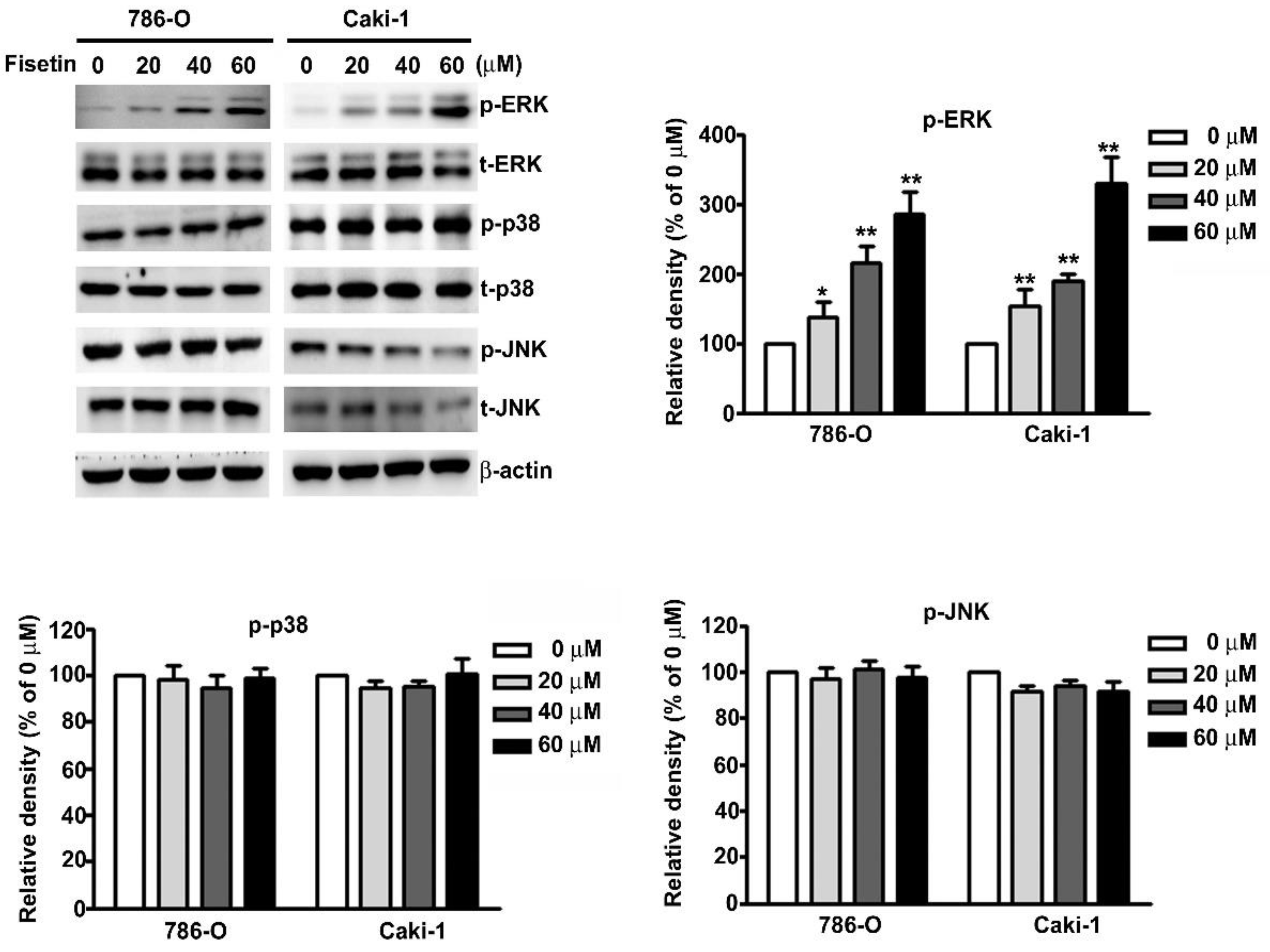
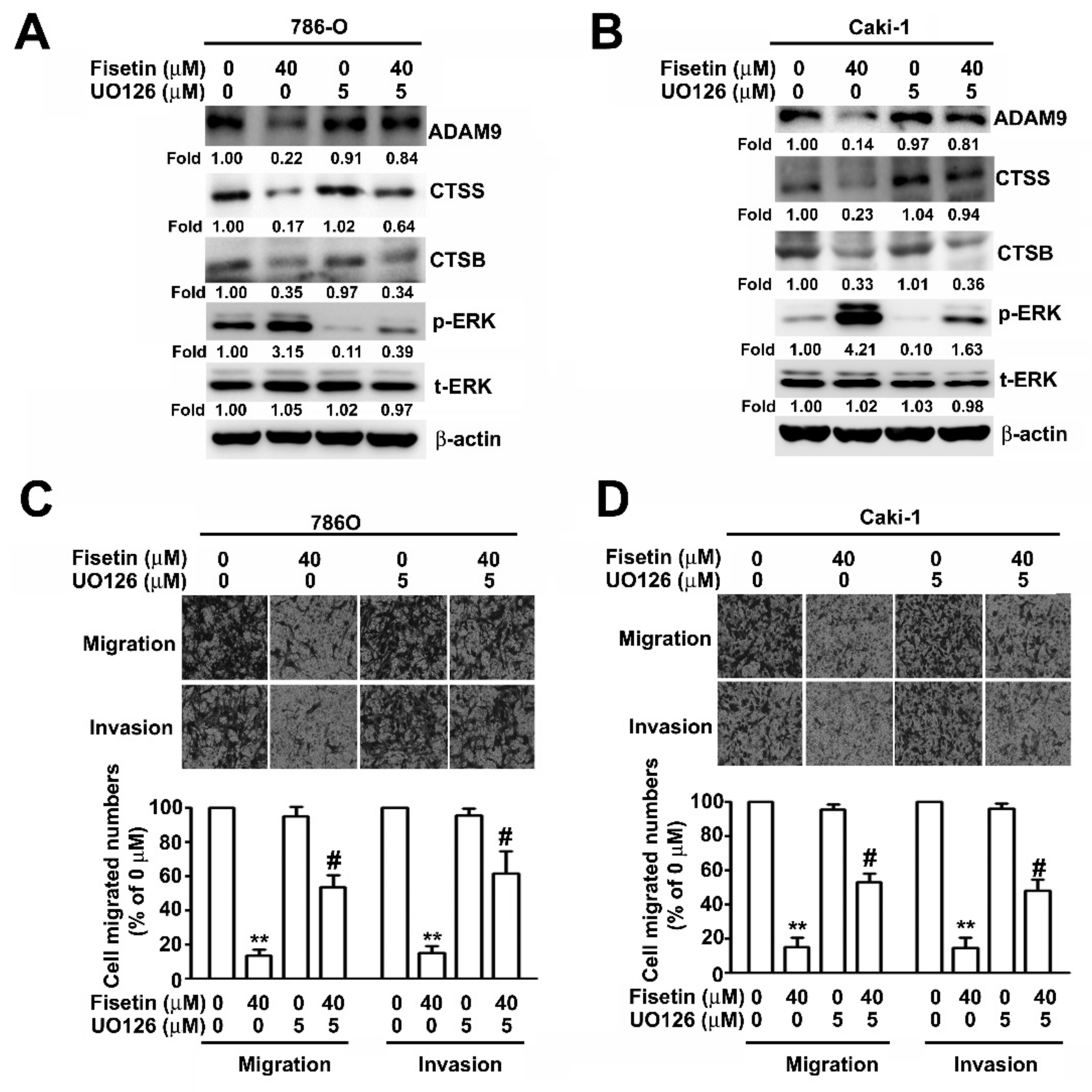
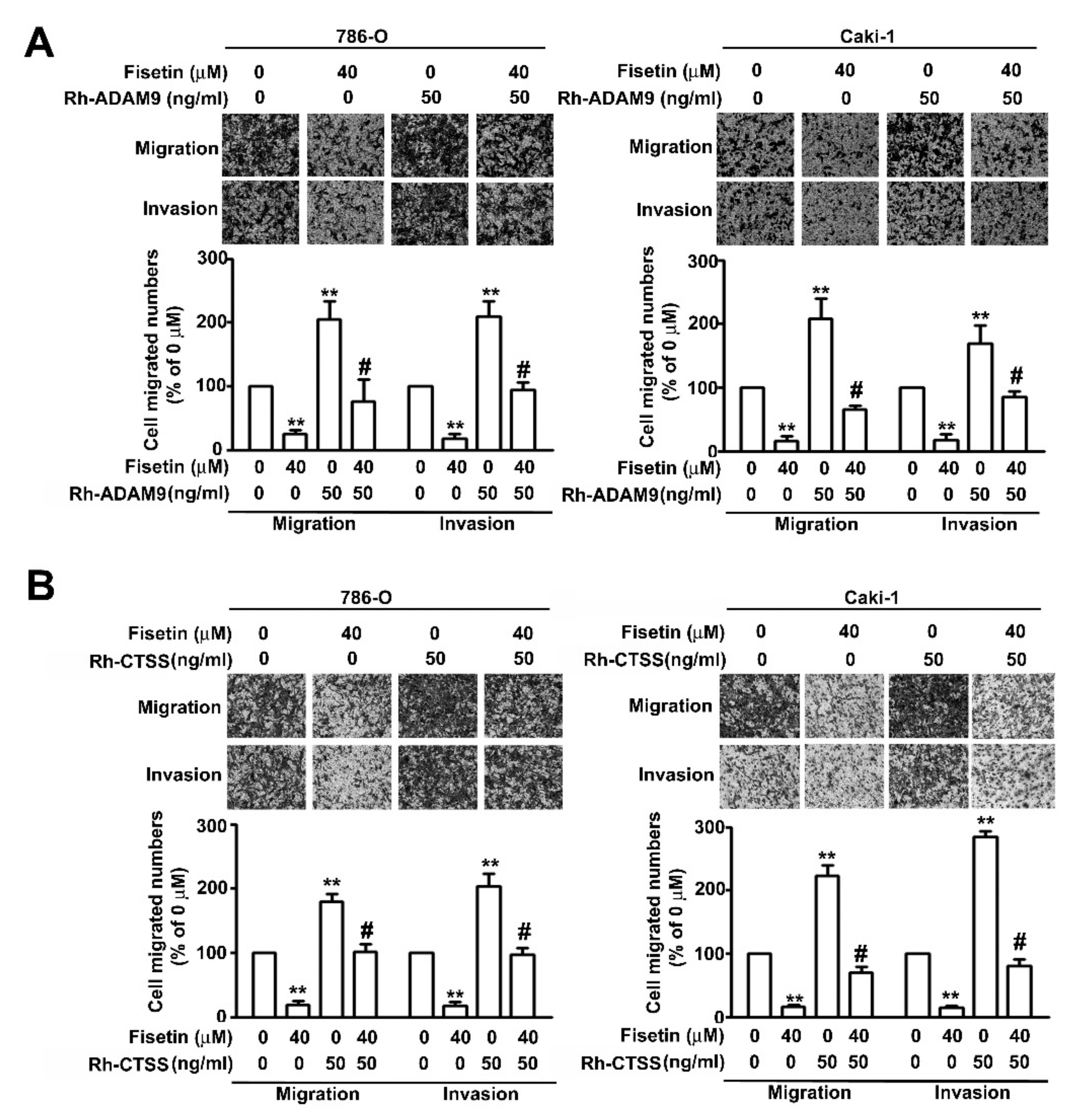
3.5. Upregulation of ERK Activation in Fisetin-Treated RCC Cell Migration and Invasion
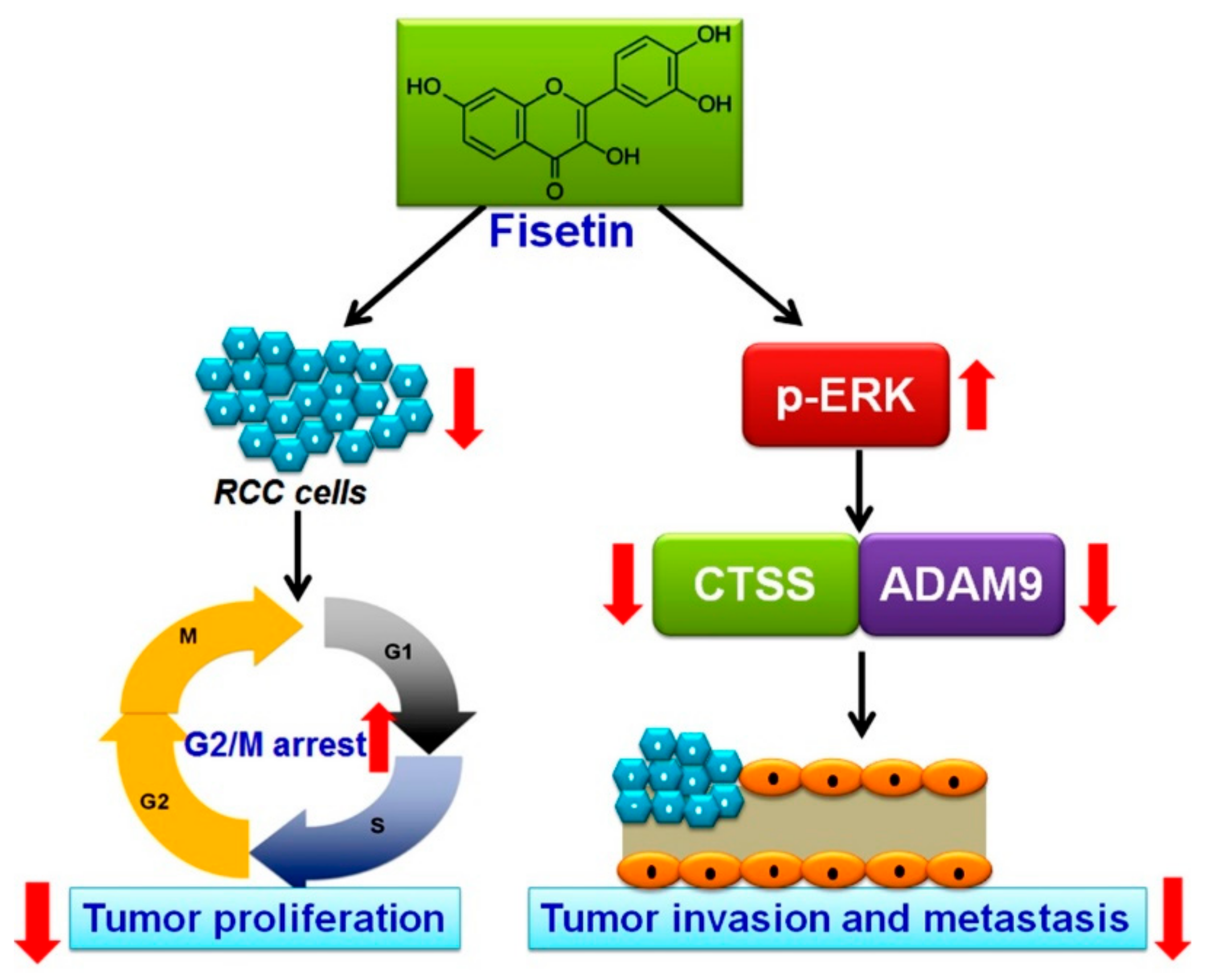
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Znaor, A.; Lortet-Tieulent, J.; Laversanne, M.; Jemal, A.; Bray, F. International Variations and Trends in Renal Cell Carcinoma Incidence and Mortality. Eur. Urol. 2015, 67, 519–530. [Google Scholar] [CrossRef]
- Conti, S.L.; Thomas, I.C.; Hagedorn, J.C.; Chung, B.I.; Chertow, G.M.; Wagner, T.H.; Brooks, J.D.; Srinivas, S.; Leppert, J.T. Utilization of cytoreductive nephrectomy and patient survival in the targeted therapy era. Int. J. Cancer 2014, 134, 2245–2252. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [Green Version]
- Chiong, E.; Tay, M.H.; Tan, M.H.; Kumar, S.; Sim, H.G.; Teh, B.T.; Umbas, R.; Chau, N.M. Management of kidney cancer in Asia: resource-stratified guidelines from the Asian Oncology Summit 2012. Lancet Oncol. 2012, 13, e482–e491. [Google Scholar] [CrossRef]
- Chen, C.-M.; Hsieh, S.-C.; Lin, C.-L.; Lin, Y.-S.; Tsai, J.-P.; Hsieh, Y.-H. Alpha-Mangostin Suppresses the Metastasis of Human Renal Carcinoma Cells by Targeting MEK/ERK Expression and MMP-9 Transcription Activity. Cell. Physiol. Biochem. 2017, 44, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lee, W.-C.; Huang, B.-M.; Chia, Y.-C.; Chen, Y.-C.; Chen, Y.-C. 16-Hydroxycleroda-3, 13-dien-15, 16-olide inhibits the proliferation and induces mitochondrial-dependent apoptosis through Akt, mTOR, and MEK-ERK pathways in human renal carcinoma cells. Phytomedicine 2017, 36, 95–107. [Google Scholar]
- Ren, W.; Qiao, Z.; Wang, H.; Zhu, L.; Zhang, L. Flavonoids: Promising anticancer agents. Med. Res. Rev. 2003, 23, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Hyun, J.W. Fisetin induces apoptosis in human nonsmall lung cancer cells via a mitochondria-mediated pathway. In vitro cellular & developmental biology. Animal 2015, 51, 300–309. [Google Scholar]
- Liao, Y.-C.; Shih, Y.-W.; Chao, C.-H.; Lee, X.-Y.; Chiang, T.-A. Involvement of the ERK Signaling Pathway in Fisetin Reduces Invasion and Migration in the Human Lung Cancer Cell Line A549. J. Agric. Food Chem. 2009, 57, 8933–8941. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Shen, S.-C.; Lee, W.-R.; Lin, H.-Y.; Ko, C.-H.; Shih, C.-M.; Yang, L.-L. Wogonin and fisetin induction of apoptosis through activation of caspase 3 cascade and alternative expression of p21 protein in hepatocellular carcinoma cells SK-HEP-1. Arch. Toxicol. 2002, 76, 351–359. [Google Scholar] [CrossRef]
- Khan, N.; Adhami, V.M.; Mukhtar, H. Apoptosis by dietary agents for prevention and treatment of prostate cancer. Endocr. Relat. Cancer 2010, 17, R39–R52. [Google Scholar] [CrossRef] [Green Version]
- Chou, R.-H.; Hsieh, S.-C.; Yu, Y.-L.; Huang, M.-H.; Huang, Y.-C.; Hsieh, Y.-H. Fisetin Inhibits Migration and Invasion of Human Cervical Cancer Cells by Down-Regulating Urokinase Plasminogen Activator Expression through Suppressing the p38 MAPK-Dependent NF-κB Signaling Pathway. PLoS ONE 2013, 8, e71983. [Google Scholar] [CrossRef]
- Ying, T.H.; Yang, S.F.; Tsai, S.J.; Hsieh, S.C.; Huang, Y.C.; Bau, D.T.; Hsieh, Y.H. Fisetin induces apoptosis in human cervical cancer HeLa cells through ERK1/2-mediated activation of caspase-8-/caspase-3-dependent pathway. Arch. Toxicol. 2012, 86, 263–273. [Google Scholar] [CrossRef]
- Sloane, B.; Dunn, J.R.; Honn, K. Lysosomal cathepsin B: Correlation with metastatic potential. Science 1981, 212, 1151–1153. [Google Scholar] [CrossRef]
- A Joyce, J.; Baruch, A.; Chehade, K.; Meyer-Morse, N.; Giraudo, E.; Tsai, F.-Y.; Greenbaum, D.C.; Hager, J.H.; Bogyo, M.; Hanahan, D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell 2004, 5, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Harbeck, N.; Alt, U.; Berger, U.; Krüger, A.; Thomssen, C.; Jänicke, F.; Höfler, H.; E Kates, R.; Schmitt, M. Prognostic impact of proteolytic factors (urokinase-type plasminogen activator, plasminogen activator inhibitor 1, and cathepsins B, D, and L) in primary breast cancer reflects effects of adjuvant systemic therapy. Clin. Cancer Res. 2001, 7, 2757–2764. [Google Scholar]
- A Gormley, J.; Hegarty, S.M.; O’Grady, A.; Stevenson, M.R.; E Burden, R.; Barrett, H.L.; Scott, C.J.; A Johnston, J.; Wilson, R.H.; Kay, E.W.; et al. The role of Cathepsin S as a marker of prognosis and predictor of chemotherapy benefit in adjuvant CRC: a pilot study. Br. J. Cancer 2011, 105, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Werle, B.; Lötterle, H.; Schanzenbächer, U.; Lah, T.T.; Kalman, E.; Kayser, K.; Bülzebruck, H.; Schirren, J.; Krasovec, M.; Kos, J.; et al. Immunochemical analysis of cathepsin B in lung tumours: an independent prognostic factor for squamous cell carcinoma patients. Br. J. Cancer 1999, 81, 510–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, S.-Y.; Kubo, H.; Shigemura, K.; Arnold, R.S.; Logani, S.; Wang, R.; Konaka, H.; Nakagawa, M.; Mousses, S.; Amin, M.; et al. Oxidative Stress Induces ADAM9 Protein Expression in Human Prostate Cancer Cells. Cancer Res. 2006, 66, 9519–9526. [Google Scholar] [CrossRef]
- Mazzocca, A.; Coppari, R.; De Franco, R.; Cho, J.-Y.; Libermann, T.A.; Pinzani, M.; Toker, A. A Secreted Form of ADAM9 Promotes Carcinoma Invasion through Tumor-Stromal Interactions. Cancer Res. 2005, 65, 4728–4738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, C.; McKie, N.; Buggy, Y.; Duggan, C.; Hill, A.D.; McDermott, E.; O’Higgins, N.; Duffy, M.J. Expression of ADAM-9 mRNA and protein in human breast cancer. Int. J. Cancer 2003, 105, 754–761. [Google Scholar] [PubMed]
- Peduto, L.; Reuter, V.E.; Shaffer, D.R.; Scher, H.I.; Blobel, C.P. Critical Function for ADAM9 in Mouse Prostate Cancer. Cancer Res. 2005, 65, 9312–9319. [Google Scholar] [PubMed] [Green Version]
- Chiang, K.-C.; Lai, C.-Y.; Chiou, H.-L.; Lin, C.-L.; Chen, Y.-S.; Kao, S.-H.; Hsieh, Y.-H. Timosaponin AIII inhibits metastasis of renal carcinoma cells through suppressing cathepsin C expression by AKT/miR-129-5p axis. J. Cell. Physiol. 2019, 234, 13332–13341. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; Linehan, W.M. The origin, evolution and route to metastasis of clear cell RCC. Nature reviews. Nephrology 2018, 14, 538–540. [Google Scholar] [PubMed]
- Hsieh, M.J.; Lin, C.W.; Chen, M.K.; Chien, S.Y.; Lo, Y.S.; Chuang, Y.C.; Hsi, Y.T.; Lin, C.C.; Chen, J.C.; Yang, S.F. Inhibition of cathepsin S confers sensitivity to methyl protodioscin in oral cancer cells via activation of p38 MAPK/JNK signaling pathways. Sci. Rep. 2017, 7, 45039. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Hsieh, Y.H.; Hwang, J.M.; Jan, H.J.; Hsieh, S.C.; Lin, S.H.; Lai, C.Y. Fisetin suppresses ADAM9 expression and inhibits invasion of glioma cancer cells through increased phosphorylation of ERK1/2. Tumour Biol. 2015, 36, 3407–3415. [Google Scholar]
- Heng, D.Y. The next 10 years: Challenges for the future and overcoming resistance to targeted therapies for renal cell carcinoma. Can. Urol. Assoc. J. 2016, 10, S256–S258. [Google Scholar]
- Moon, Y.J.; Wang, X.; Morris, M.E. Dietary flavonoids: Effects on xenobiotic and carcinogen metabolism. Toxicol. In Vitro 2006, 20, 187–210. [Google Scholar]
- Pal, H.C.; Sharma, S.; Elmets, C.A.; Athar, M.; Afaq, F. Fisetin inhibits growth, induces G2/M arrest and apoptosis of human epidermoid carcinoma A431 cells: Role of mitochondrial membrane potential disruption and consequent caspases activation. Exp. Dermatol. 2013, 22, 470–475. [Google Scholar]
- Min, K.-J.; Nam, J.-O.; Kwon, T.K. Fisetin Induces Apoptosis Through p53-Mediated Up-Regulation of DR5 Expression in Human Renal Carcinoma Caki Cells. Molecules 2017, 22, 1285. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Jung, J.I.; Chun, H.S.; Kwon, D.Y.; Park, J.H.; Cho, H.J.; Lim, D.Y.; Lee, H.S. Fisetin Inhibits the Activities of Cyclin-Dependent Kinases Leading to Cell Cycle Arrest in HT-29 Human Colon Cancer Cells. J. Nutr. 2005, 135, 2884–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Afaq, F.; Syed, D.N.; Mukhtar, H. Fisetin, a novel dietary flavonoid, causes apoptosis and cell cycle arrest in human prostate cancer LNCaP cells. Carcinogenesis 2008, 29, 1049–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cheng, Y.; Qu, W.; Sun, Y.; Wang, Z.; Wang, H.; Tian, B. Fisetin, a dietary flavonoid, induces cell cycle arrest and apoptosis through activation of p53 and inhibition of NF-kappa B pathways in bladder cancer cells. Basic Clin. Pharmacol. Toxicol. 2011, 108, 84–93. [Google Scholar] [CrossRef]
- Ferreira de Oliveira, J.M.P.; Pacheco, A.R.; Coutinho, L.; Oliveira, H.; Pinho, S.; Almeida, L.; Fernandes, E.; Santos, C. Combination of etoposide and fisetin results in anti-cancer efficiency against osteosarcoma cell models. Arch. Toxicol. 2018, 92, 1205–1214. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Hewage, S.R.K.M.; Ryu, Y.S.; Oh, M.C.; Kwon, T.K.; Chae, S.; Hyun, J.W. Fisetin induces apoptosis and endoplasmic reticulum stress in human non-small cell lung cancer through inhibition of the MAPK signaling pathway. Tumor Boil. 2016, 37, 9615–9624. [Google Scholar] [CrossRef]
- Sevenich, L.; Bowman, R.L.; Mason, S.D.; Quail, D.F.; Rapaport, F.; Elie, B.T.; Brogi, E.; Brastianos, P.K.; Hahn, W.C.; Holsinger, L.J.; et al. Analysis of tumor- and stroma-supplied proteolytic networks reveals a brain metastasis-promoting role for cathepsin S. Nat. Cell Bio. 2014, 16, 876–888. [Google Scholar] [CrossRef]
- Gocheva, V.; Wang, H.-W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzanakakis, G.N.; Margioris, A.N.; Tsatsakis, A.M.; Vezeridis, M.P. The metastatic potential of human pancreatic cell lines in the liver of nude mice correlates well with cathepsin B activity. Int. J. Gastrointest. Cancer 2003, 34, 27–38. [Google Scholar]
- Lei, D.; Zhang, F.; Yao, D.; Xiong, N.; Jiang, X.; Zhao, H. Galangin increases ERK1/2 phosphorylation to decrease ADAM9 expression and prevents invasion in A172 glioma cells. Mol. Med. Rep. 2018, 17, 667–673. [Google Scholar]
- Huang, C.-F.; Yang, S.-F.; Chiou, H.-L.; Hsu, W.-H.; Hsu, J.-C.; Liu, C.-J.; Hsieh, Y.-H. Licochalcone A inhibits the invasive potential of human glioma cells by targeting the MEK/ERK and ADAM9 signaling pathways. Food Funct. 2018, 9, 6196–6204. [Google Scholar]
- Fritzsche, F.R.; Wassermann, K.; Jung, M.; Tölle, A.; Kristiansen, I.; Lein, M.; Johannsen, M.; Dietel, M.; Jung, K.; Kristiansen, G. ADAM9 is highly expressed in renal cell cancer and is associated with tumour progression. BMC Cancer 2008, 8, 179. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-T.; Hsieh, M.-J.; Hsieh, Y.-H.; Hsin, M.-C.; Chuang, Y.-T.; Yang, S.-F.; Yang, J.-S.; Lin, C.-W. Sulforaphane suppresses oral cancer cell migration by regulating cathepsin S expression. Oncotarget 2018, 9, 17564–17575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsin, M.-C.; Hsieh, Y.-H.; Wang, P.-H.; Ko, J.-L.; Hsin, I.-L.; Yang, S.-F. Hispolon suppresses metastasis via autophagic degradation of cathepsin S in cervical cancer cells. Cell Death Dis. 2017, 8, e3089. [Google Scholar] [CrossRef] [PubMed]
- Syed, D.N.; Chamcheu, J.-C.; Khan, M.I.; Sechi, M.; Lall, R.K.; Adhami, V.M.; Mukhtar, H. Fisetin inhibits human melanoma cell growth through direct binding to p70S6K and mTOR: findings from 3-D melanoma skin equivalents and computational modeling. Biochem. Pharmacol. 2014, 89, 349–360. [Google Scholar] [CrossRef]
- Khan, M.I.; Adhami, V.M.; Lall, R.K.; Sechi, M.; Joshi, D.C.; Haidar, O.M.; Syed, D.N.; Siddiqui, I.A.; Chiu, S.-Y.; Mukhtar, H. YB-1 expression promotes epithelial-to-mesenchymal transition in prostate cancer that is inhibited by a small molecule fisetin. Oncotarget 2014, 5, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gong, X.; Jiang, R.; Lin, D.; Zhou, T.; Zhang, A.; Li, H.; Zhang, X.; Wan, J.; Kuang, G.; et al. Fisetin Inhibited Growth and Metastasis of Triple-Negative Breast Cancer by Reversing Epithelial-to-Mesenchymal Transition via PTEN/Akt/GSK3beta Signal Pathway. Front. Pharmacol. 2018, 9, 772. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, M.-H.; Tsai, J.-P.; Yang, S.-F.; Chiou, H.-L.; Lin, C.-L.; Hsieh, Y.-H.; Chang, H.-R. Fisetin Suppresses the Proliferation and Metastasis of Renal Cell Carcinoma through Upregulation of MEK/ERK-Targeting CTSS and ADAM9. Cells 2019, 8, 948. https://doi.org/10.3390/cells8090948
Hsieh M-H, Tsai J-P, Yang S-F, Chiou H-L, Lin C-L, Hsieh Y-H, Chang H-R. Fisetin Suppresses the Proliferation and Metastasis of Renal Cell Carcinoma through Upregulation of MEK/ERK-Targeting CTSS and ADAM9. Cells. 2019; 8(9):948. https://doi.org/10.3390/cells8090948
Chicago/Turabian StyleHsieh, Min-Hong, Jen-Pi Tsai, Shun-Fa Yang, Hui-Ling Chiou, Chia-Liang Lin, Yi-Hsien Hsieh, and Horng-Rong Chang. 2019. "Fisetin Suppresses the Proliferation and Metastasis of Renal Cell Carcinoma through Upregulation of MEK/ERK-Targeting CTSS and ADAM9" Cells 8, no. 9: 948. https://doi.org/10.3390/cells8090948
APA StyleHsieh, M. -H., Tsai, J. -P., Yang, S. -F., Chiou, H. -L., Lin, C. -L., Hsieh, Y. -H., & Chang, H. -R. (2019). Fisetin Suppresses the Proliferation and Metastasis of Renal Cell Carcinoma through Upregulation of MEK/ERK-Targeting CTSS and ADAM9. Cells, 8(9), 948. https://doi.org/10.3390/cells8090948