Chronic Exposure to Chewing Tobacco Induces Metabolic Reprogramming and Cancer Stem Cell-Like Properties in Esophageal Epithelial Cells


 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Tobacco Treatment
2.2. Cell Culture-Based Assays
2.3. Western Blotting
2.4. SOD Assay
2.5. Sample Preparation for Exome Sequencing
2.6. Exome Data Analysis
2.7. Sample Preparation for Proteomic Analysis
2.8. Proteomic Data Analysis
2.9. Electron Microscopy
2.10. Statistical Analysis
3. Results
3.1. Chronic Exposure to Tobacco Extract Leads to Cancer Cell Phenotype in Esophageal Epithelial Cells
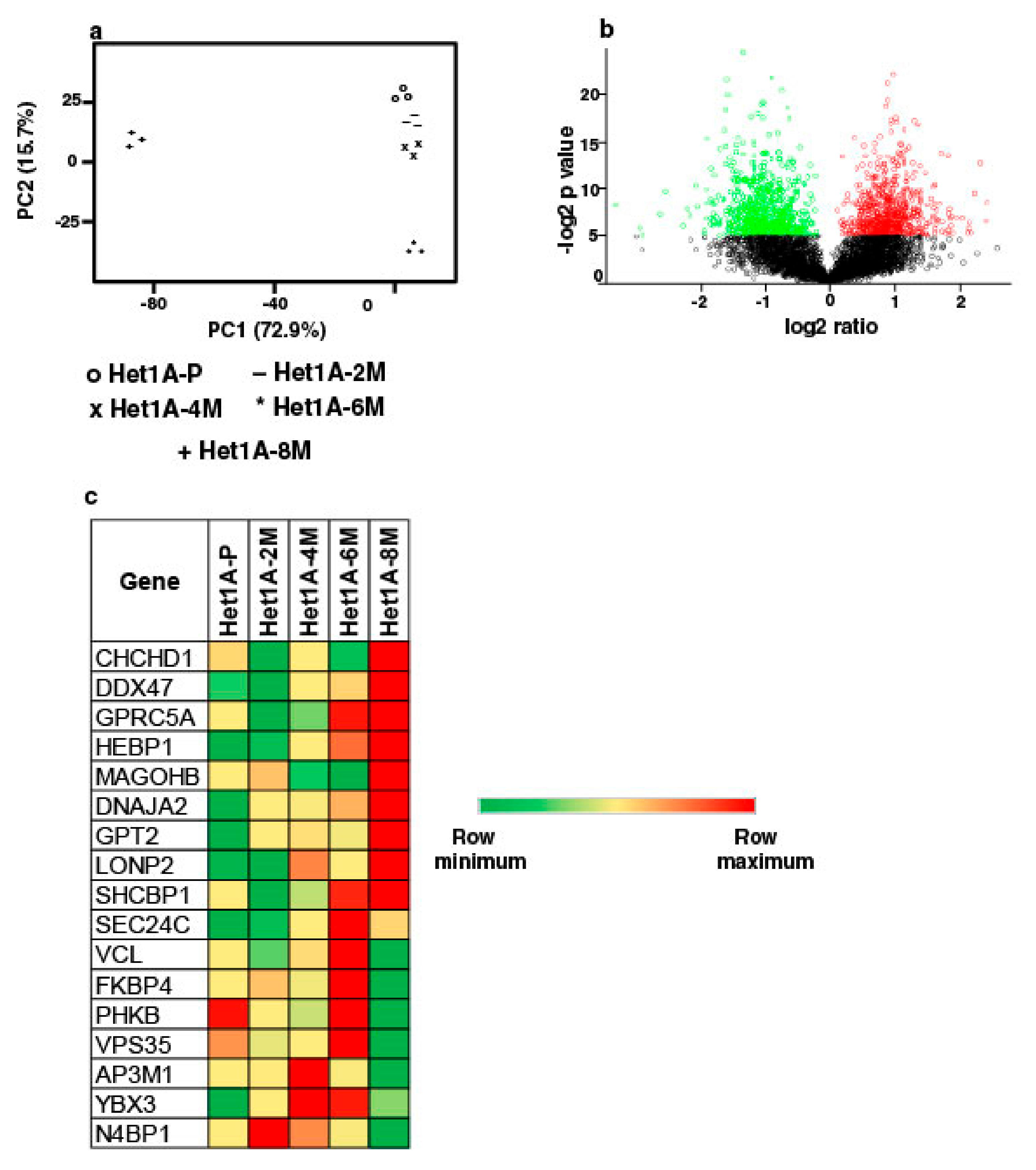
3.2. Genomic Alterations Associated with Esophageal Epithelial Cells Chronically Exposed to Tobacco Extract
3.3. Global Proteomic Profiling Shows Differentially Expressed Proteins Associated with Chronic Exposure to Chewing Tobacco
3.4. Protein Expression Pattern of Het1A-8M Shows Elevated Expression of Cancer Stem Cell (CSC) Markers
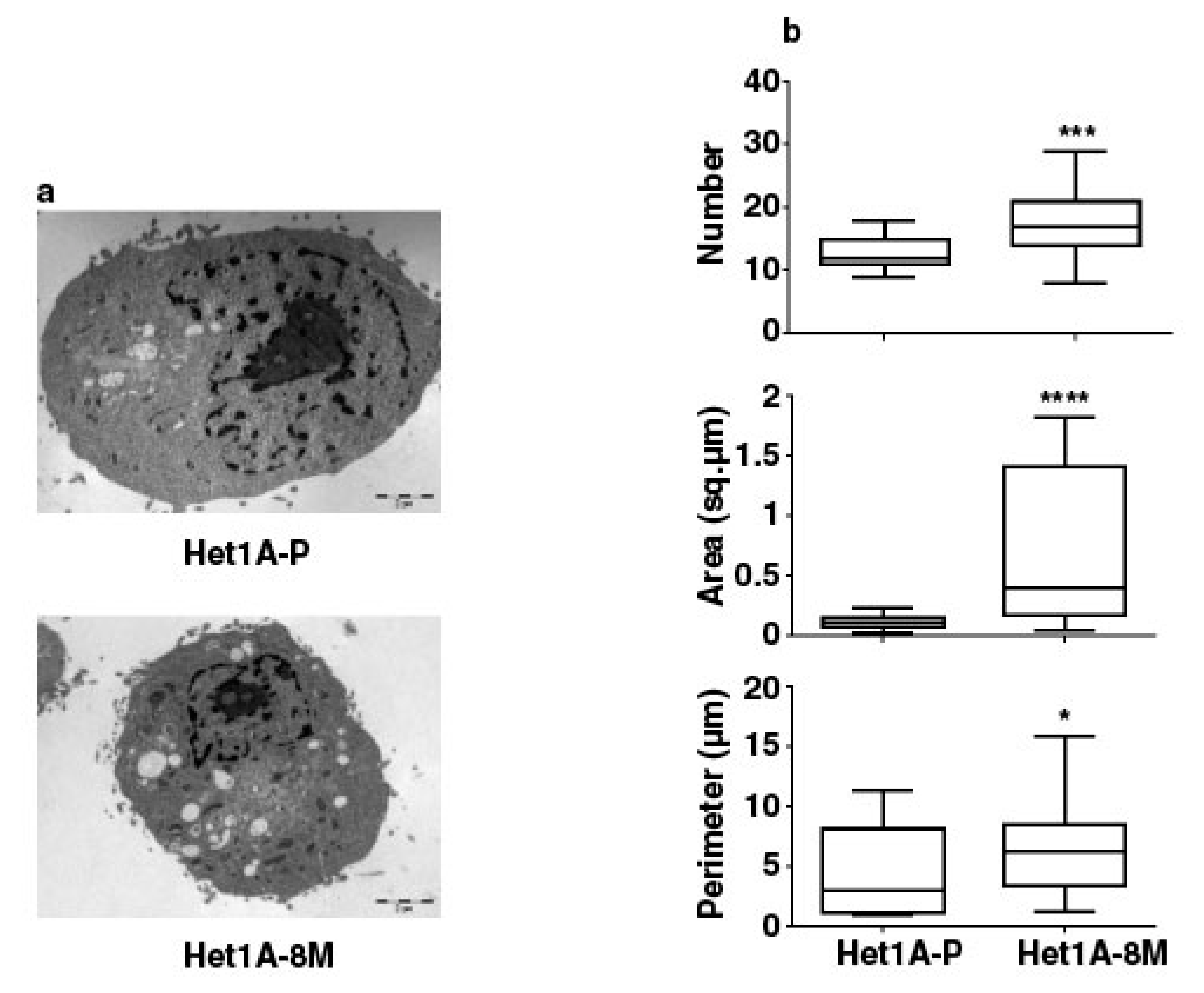
3.5. Esophageal Cells Chronically Exposed to Tobacco Extract Show Metabolic Reprogramming and Higher Mitochondrial Mass
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. GLOBOCAN. International Agency for Research on Cancer. 2012. Available online: http://gco.iarc.fr/today/fact-sheets-cancers?cancer=4&type=0&sex=0 (accessed on 23 April 2018).
- Pennathur, A.; Gibson, M.K.; Jobe, B.A.; Luketich, J.D. Oesophageal carcinoma. Lancet 2013, 381, 400–412. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Cancer Prevention. 2018. Available online: http://www.who.int/cancer/prevention/en/ (accessed on 23 April 2018).
- Eriksen, M.; Mackay, J.; Ross, H. The Tobacco Atlas; American Cancer Society: Atlanta, GA, USA, 2012. [Google Scholar]
- Phukan, R.K.; Ali, M.S.; Chetia, C.K.; Mahanta, J. Betel nut and tobacco chewing; potential risk factors of cancer of oesophagus in Assam, India. Br. J. Cancer. 2001, 85, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitra, S.; Ashok, L.; Anand, L.; Srinivasan, V.; Jayanthi, V. Risk factors for esophageal cancer in Coimbatore, southern India: A hospital-based case-control study. Indian J. Gastroenterol. 2004, 23, 19–21. [Google Scholar] [PubMed]
- Varmus, H.E. The molecular genetics of cellular oncogenes. Annu. Rev. Genet. 1984, 18, 553–612. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [Green Version]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Malpass, G.E.; Arimilli, S.; Prasad, G.L.; Howlett, A.C. Regulation of gene expression by tobacco product preparations in cultured human dermal fibroblasts. Toxicol. Appl. Pharmacol. 2014, 279, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanjappa, V.; Renuse, S.; Sathe, G.J.; Raja, R.; Syed, N.; Radhakrishnan, A.; Subbannayya, T.; Patil, A.; Marimuthu, A.; Sahasrabuddhe, N.A.; et al. Chronic exposure to chewing tobacco selects for overexpression of stearoyl-CoA desaturase in normal oral keratinocytes. Cancer Biol. Ther. 2015, 16, 1593–1603. [Google Scholar] [CrossRef]
- Subbannayya, Y.; Syed, N.; Barbhuiya, M.A.; Raja, R.; Marimuthu, A.; Sahasrabuddhe, N.; Pinto, S.M.; Manda, S.S.; Renuse, S.; Manju, H.; et al. Calcium calmodulin dependent kinase kinase 2—A novel therapeutic target for gastric adenocarcinoma. Cancer Boil. Ther. 2015, 16, 336–345. [Google Scholar] [CrossRef]
- Zhou, S.; Kachhap, S.; Sun, W.; Wu, G.; Chuang, A.; Poeta, L.; Grumbine, L.; Mithani, S.K.; Chatterjee, A.; Koch, W.; et al. Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc. Natl. Acad. Sci. USA 2007, 104, 7540–7545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, K.M.; Hawkins, A.S.; Santana-Cruz, I.; Adkins, R.S.; Shetty, A.C.; Nagaraj, S.; Sadzewicz, L.; Tallon, L.J.; Rasko, D.A.; Fraser, C.M.; et al. Aligner optimization increases accuracy and decreases compute times in multi-species sequence data. Microb. Genom. 2017, 3, e000122. [Google Scholar] [CrossRef] [Green Version]
- Saunders, C.T.; Wong, W.S.W.; Swamy, S.; Becq, J.; Murray, L.J.; Cheetham, R.K. Strelka: Accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics 2012, 28, 1811–1817. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Lienard, M.; Toffoli, S.; Kamal, M.; le Tourneau, C.; Gentien, D.; Servant, N.; Gestraud, P.; Frio, T.R.; et al. Multi-factor data normalization enables the detection of copy number aberrations in amplicon sequencing data. Bioinformatics 2014, 30, 3443–3450. [Google Scholar] [CrossRef] [Green Version]
- Manda, S.S.; Nirujogi, R.S.; Pinto, S.M.; Kim, M.-S.; Datta, K.K.; Sirdeshmukh, R.; Prasad, T.S.K.; Thongboonkerd, V.; Pandey, A.; Gowda, H. Identification and Characterization of Proteins Encoded by Chromosome 12 as Part of Chromosome-centric Human Proteome Project. J. Proteome Res. 2014, 13, 3166–3177. [Google Scholar] [CrossRef] [PubMed]
- Mol, P.; Kannegundla, U.; Dey, G.; Gopalakrishnan, L.; Dammalli, M.; Kumar, M.; Patil, A.H.; Basavaraju, M.; Rao, A.; Ramesha, K.P.; et al. Bovine Milk Comparative Proteome Analysis from Early, Mid, and Late Lactation in the Cattle Breed, Malnad Gidda (Bos indicus). OMICS 2018, 22, 223–235. [Google Scholar] [CrossRef]
- Carney, D.N.; Gazdar, A.F.; Minna, J.D. Positive correlation between histological tumor involvement and generation of tumor cell colonies in agarose in specimens taken directly from patients with small-cell carcinoma of the lung. Cancer Res. 1980, 40, 1820–1823. [Google Scholar] [PubMed]
- Jäättelä, M. Escaping Cell Death: Survival Proteins in Cancer. Exp. Cell Res. 1999, 248, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Niaz, K.; Maqbool, F.; Khan, F.; Bahadar, H.; Hassan, F.I.; Abdollahi, M. Smokeless tobacco (paan and gutkha) consumption, prevalence, and contribution to oral cancer. Epidemiol. Heal. 2017, 39, e2017009. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.C.; Hao, J.J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Kwong, D.; Lam, A.; Guan, X.; Law, S.; Tai, A.; Wong, J.; Sham, J. Chromosomal aberrations in esophageal squamous cell carcinoma among chinese: Gain of 12p predicts poor prognosis after surgery. Hum. Pathol. 2004, 35, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, Y.; Cheng, C.; Cui, H.; Cheng, L.; Kong, P.; Wang, J.; Li, Y.; Chen, W.; Song, B.; et al. Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. Am. J. Hum. Genet. 2015, 96, 597–611. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Su, H.; Hu, N.; Yang, H.H.; Wang, C.; Takikita, M.; Wang, Q.H.; Giffen, C.; Clifford, R.; Hewitt, S.M.; Shou, J.Z.; et al. Global gene expression profiling and validation in esophageal squamous cell carcinoma (ESCC) and its association with clinical phenotypes. Clin. Cancer Res. 2011, 17, 2955–2966. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Clifford, R.J.; Yang, H.H.; Wang, C.; Goldstein, A.M.; Ding, T.; Taylor, P.R.; Lee, M.P. Genome wide analysis of DNA copy number neutral loss of heterozygosity (CNNLOH) and its relation to gene expression in esophageal squamous cell carcinoma. BMC Genom. 2010, 11, 576. [Google Scholar] [CrossRef] [PubMed]
- Min, M.; Chen, X.B.; Wang, P.; Landeck, L.; Chen, J.Q.; Li, W.; Cai, S.Q.; Zheng, M.; Man, X.Y. Role of keratin 24 in human epidermal keratinocytes. PLoS ONE 2017, 12, e0174626. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 2016, 6, 87–99. [Google Scholar] [CrossRef]
- Ježek, P.; Hlavatá, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Boil. 2005, 37, 2478–2503. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Zhao, J.S.; Li, W.J.; Ge, D.; Zhang, P.J.; Li, J.J.; Lu, C.L.; Ji, X.D.; Guan, D.X.; Gao, H.; Xu, L.Y.; et al. Tumor Initiating Cells in Esophageal Squamous Cell Carcinomas Express High Levels of CD44. PLoS ONE 2011, 6, e21419. [Google Scholar] [CrossRef]
- Tsai, S.T.; Wang, P.J.; Liou, N.J.; Lin, P.S.; Chen, C.H.; Chang, W.C. ICAM1 Is a Potential Cancer Stem Cell Marker of Esophageal Squamous Cell Carcinoma. PLoS ONE 2015, 10, e0142834. [Google Scholar] [CrossRef]
- Huang, D.; Gao, Q.; Guo, L.; Zhang, C.; Jiang, W.; Li, H.; Wang, J.; Han, X.; Shi, Y.; Lu, S.H. Isolation and Identification of Cancer Stem-Like Cells in Esophageal Carcinoma Cell Lines. Stem Cells Dev. 2009, 18, 465–474. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and Mitochondrial Respiration: A Neglected Connection. Cancer Res. 2015, 75, 3687–3691. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Bonuccelli, G.; Ózsvári, B.; Peiris-Pagès, M.; Fiorillo, M.; Smith, D.L.; Bevilacqua, G.; Mazzanti, C.M.; McDonnell, L.A.; Naccarato, A.G.; et al. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget 2015, 6, 30453–30471. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Tsai et al. | Huang et al. | Het1A-8M/Het1A-P (Relative Fold Change) |
|---|---|---|---|
| HLA-B | + | Not reported | 2.9 |
| ICAM1 | + | Not reported | 3.7 |
| HLA-A | + | Not reported | 3.1 |
| RAB9A | + | Not reported | 2.2 |
| SLC3A2 | + | Not reported | 2.2 |
| SLC25A1 | + | Not reported | 1.7 |
| MMGT1 | + | Not reported | 1.6 |
| PI4K2A | + | Not reported | 1.7 |
| CYP1B1 | Not reported | + | 2.8 |
| CLK1 | Not reported | + | 2.2 |
| LEMD3 | Not reported | + | 1.8 |
| DDX10 | Not reported | + | 1.7 |
| RBM15 | Not reported | + | 1.4 |
| RANBP2 | Not reported | + | 1.7 |
| KIF14 | Not reported | + | 1.8 |
| CDCA2 | Not reported | + | 1.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Datta, K.K.; Patil, S.; Patel, K.; Babu, N.; Raja, R.; Nanjappa, V.; Mangalaparthi, K.K.; Dhaka, B.; Rajagopalan, P.; Deolankar, S.C.; et al. Chronic Exposure to Chewing Tobacco Induces Metabolic Reprogramming and Cancer Stem Cell-Like Properties in Esophageal Epithelial Cells. Cells 2019, 8, 949. https://doi.org/10.3390/cells8090949
Datta KK, Patil S, Patel K, Babu N, Raja R, Nanjappa V, Mangalaparthi KK, Dhaka B, Rajagopalan P, Deolankar SC, et al. Chronic Exposure to Chewing Tobacco Induces Metabolic Reprogramming and Cancer Stem Cell-Like Properties in Esophageal Epithelial Cells. Cells. 2019; 8(9):949. https://doi.org/10.3390/cells8090949
Chicago/Turabian StyleDatta, Keshava K., Shankargouda Patil, Krishna Patel, Niraj Babu, Remya Raja, Vishalakshi Nanjappa, Kiran Kumar Mangalaparthi, Bharti Dhaka, Pavithra Rajagopalan, Sayali Chandrashekhar Deolankar, and et al. 2019. "Chronic Exposure to Chewing Tobacco Induces Metabolic Reprogramming and Cancer Stem Cell-Like Properties in Esophageal Epithelial Cells" Cells 8, no. 9: 949. https://doi.org/10.3390/cells8090949
APA StyleDatta, K. K., Patil, S., Patel, K., Babu, N., Raja, R., Nanjappa, V., Mangalaparthi, K. K., Dhaka, B., Rajagopalan, P., Deolankar, S. C., Kannan, R., Kumar, P., Prasad, T. S. K., Mathur, P. P., Kumari, A., Manoharan, M., Coral, K., Murugan, S., Sidransky, D., ... Gowda, H. (2019). Chronic Exposure to Chewing Tobacco Induces Metabolic Reprogramming and Cancer Stem Cell-Like Properties in Esophageal Epithelial Cells. Cells, 8(9), 949. https://doi.org/10.3390/cells8090949