Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases



Abstract
:1. Introduction
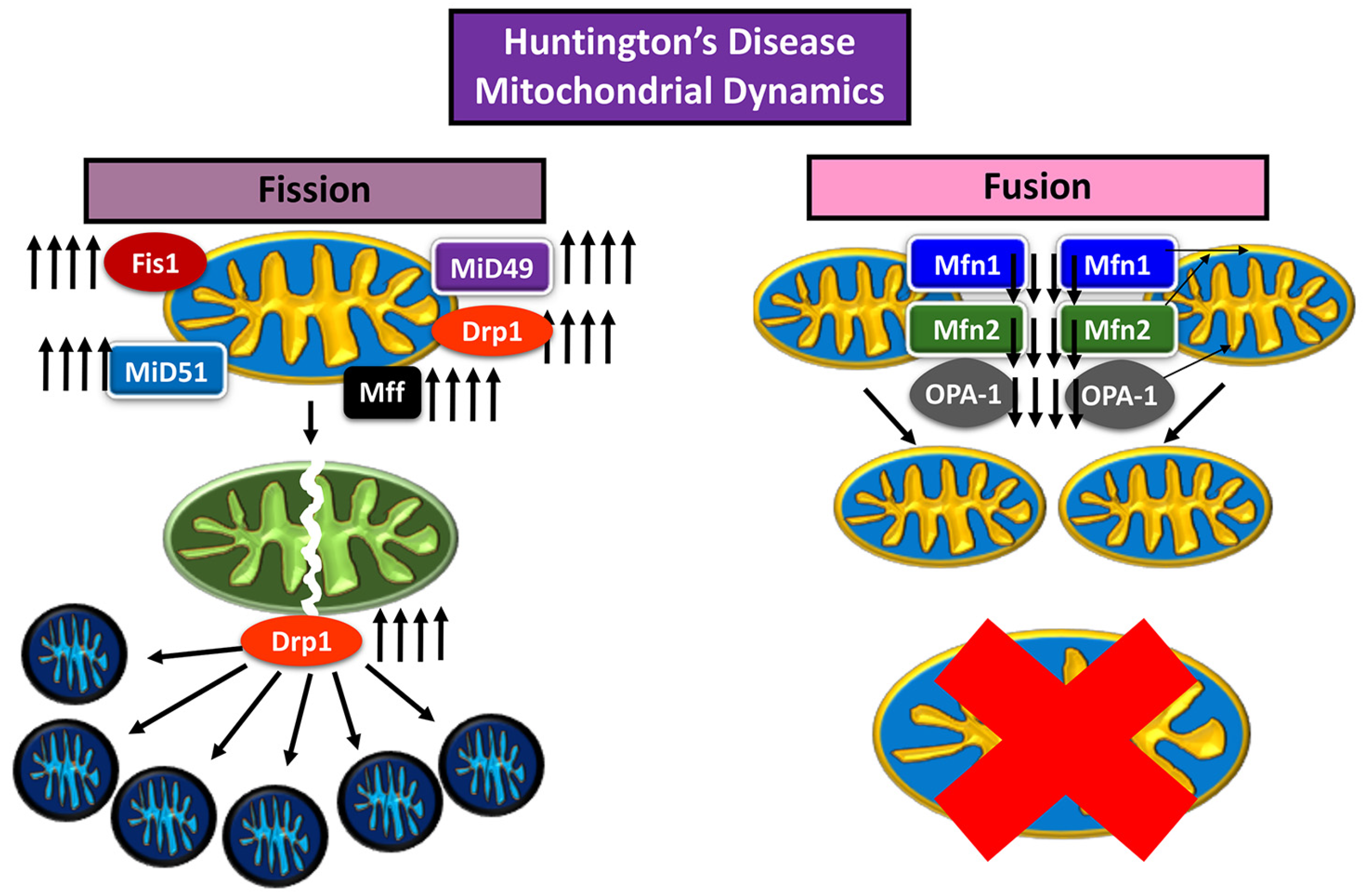
2. Mitochondrial Fusion and Fission
3. Dynamin-Related Protein 1
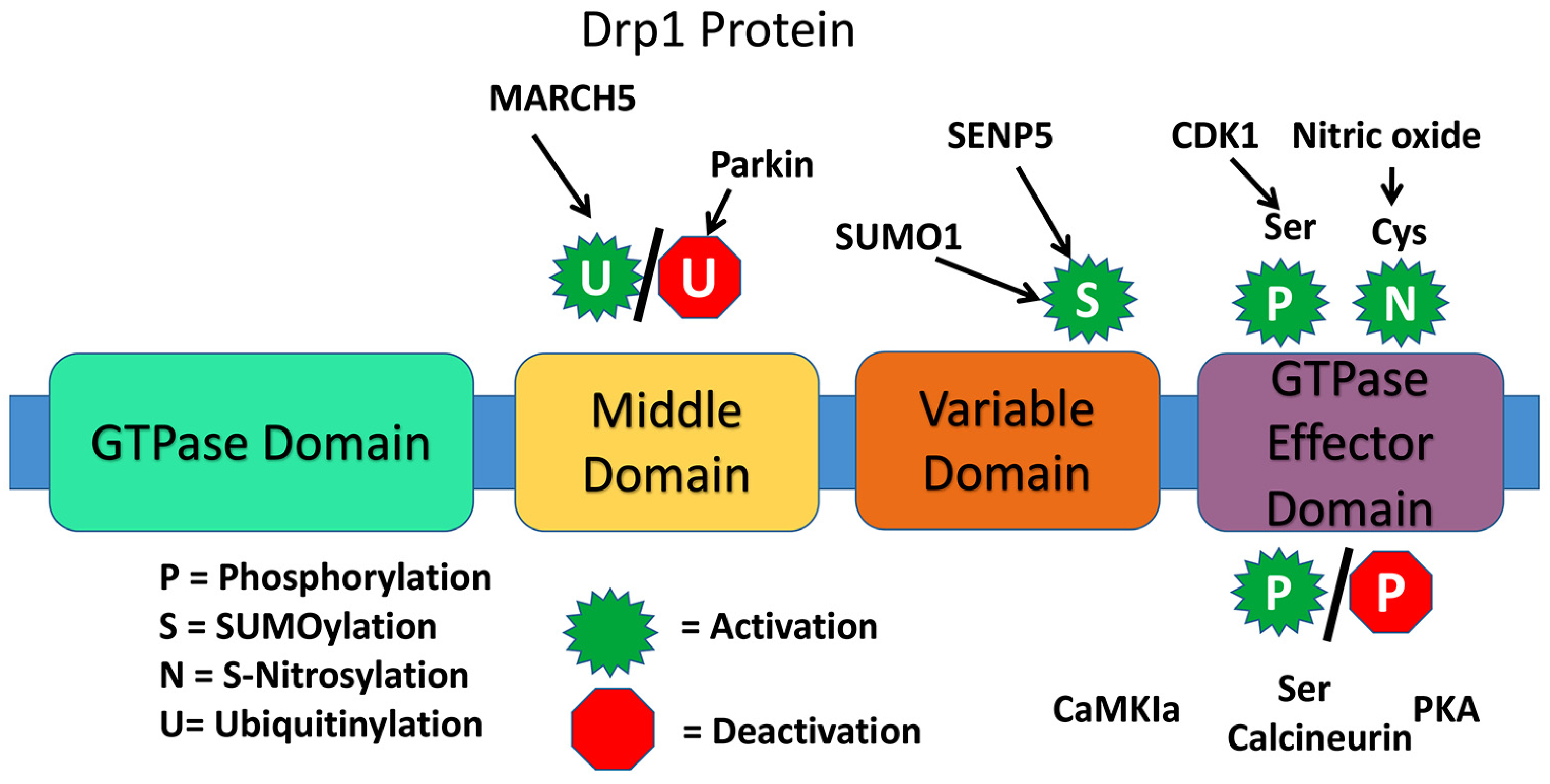
4. Drp1 Structure
5. Drp1 Functions
Drp1 Knockout Studies
6. Drp1 and Abnormal Mitochondrial Dynamics
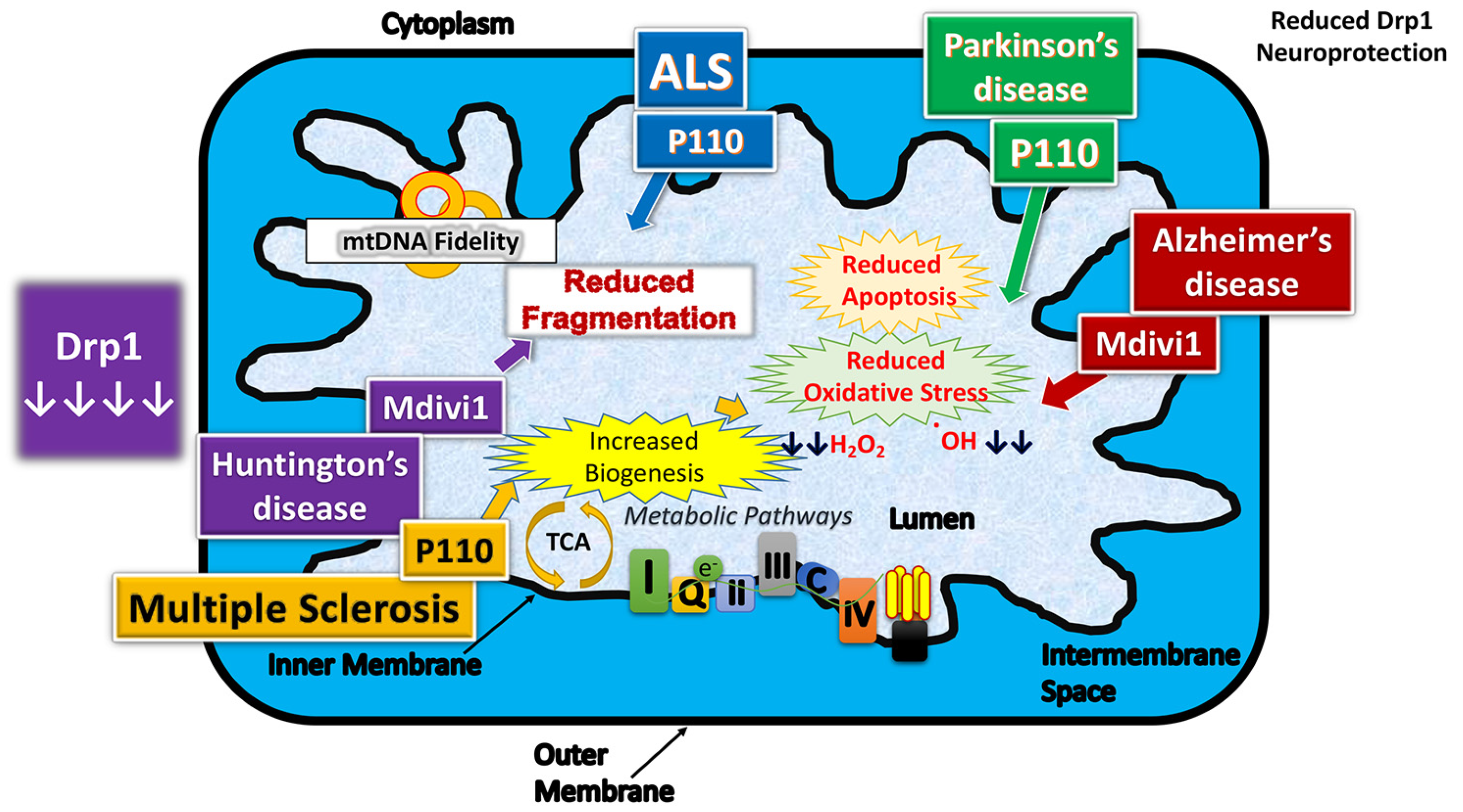
6.1. Drp1 and Alzheimer’s Disease
6.2. Drp1 and Huntington’s Disease
6.3. Drp1 and Amyotrophic Lateral Sclerosis
6.4. Drp1 and Parkinson’s Disease
6.5. Drp1 and Multiple Sclerosis
6.6. Drp1 and Down Syndrome
6.7. Drp1 and Diabetes/Obesity
7. Mechanism of Mitochondrial Dysfunction via Drp1 Phosphorylation
8. Mitochondrial Division Inhibitors and their Protective Effects
8.1. Mdivi 1
8.1.1. Genetic Studies
8.1.2. Pharmacological Studies
8.2. P110
8.3. Dynasore
8.4. DDQ
9. Is Mitochondrial Division Inhibitor 1 a Drp1 Inhibitor?
10. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Reddy, P.H.; Oliver, D.M.A. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Saxton, W.M.; Hollenbeck, P.J. The axonal transport of mitochondria. J. Cell Sci. 2012, 125, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef]
- Devoto, V.M.P.; Falzone, T.L. Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? Dis. Model. Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Odagiri, K.; Katoh, H.; Kawashima, H.; Tanaka, T.; Ohtani, H.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J. Mol. Cell. Cardiol. 2009, 46, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boncompagni, S.; Rossi, A.E.; Micaroni, M.; Beznoussenko, G.V.; Polishchuk, R.S.; Dirksen, R.T.; Protasi, F. Mitochondria Are Linked to Calcium Stores in Striated Muscle by Developmentally Regulated Tethering Structures. Mol. Biol. Cell 2009, 20, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Zhivotovsky, B.; Orrenius, S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007, 14, 1243–1247. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Bukowiecki, R.; Adjaye, J.; Prigione, A. Mitochondrial Function in Pluripotent Stem Cells and Cellular Reprogramming. Gerontology 2014, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.G.; Gardner, D.K.; Harvey, A.J. Pluripotent Stem Cell Metabolism and Mitochondria: Beyond ATP. Stem Cells Int. 2017, 2017, 2874283. [Google Scholar] [CrossRef] [PubMed]
- Gorman, A.M. Neuronal cell death in neurodegenerative diseases: Recurring themes around protein handling. J. Cell. Mol. Med. 2008, 12, 2263–2280. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Sokol, R.J. Mitochondrial hepatopathies: Advances in genetics and pathogenesis. Hepatology 2007, 45, 1555–1565. [Google Scholar] [CrossRef]
- Lee, W.S.; Sokol, R.J. Liver Disease in Mitochondrial Disorders. Semin. Liver Dis. 2007, 27, 259–273. [Google Scholar] [CrossRef] [Green Version]
- Honda, H.M.; Korge, P.; Weiss, J.N. Mitochondria and ischemia/reperfusion injury. Ann. N. Y. Acad. Sci. 2006, 1047, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J. Mitochondria and heart disease. Adv. Exp. Med. Biol. 2012, 942, 249–267. [Google Scholar]
- Karamanlidis, G.; Bautista-Hernandez, V.; Fynn-Thompson, F.; del Nido, P.; Tian, R. Impaired Mitochondrial Biogenesis Precedes Heart Failure in Right Ventricular Hypertrophy in Congenital Heart Disease. Circ. Heart Fail. 2011, 4, 707–713. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Mitochondrial Medicine for Aging and Neurodegenerative Diseases. Neuromol. Med. 2008, 10, 291–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, D.M.A.; Reddy, P.H. Molecular Basis of Alzheimer’s Disease: Focus on Mitochondria. J. Alzheimers Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Huang, Z.; Xie, F.; Chen, L. Dynamin-related protein1: A critical protein in the pathogenesis of neural system dysfunctions and neurodegenerative diseases. J. Cell. Physiol. 2018, 234, 10032–10046. [Google Scholar] [CrossRef] [PubMed]
- Osman, C.; Noriega, T.R.; Okreglak, V.; Fung, J.C.; Walter, P. Integrity of the yeast mitochondrial genome, but not its distribution and inheritance, relies on mitochondrial fission and fusion. Proc. Natl. Acad. Sci. USA 2015, 112, E947–E956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenger, J.; Klinglmayr, E.; Frohlich, C.; Eibl, C.; Gimeno, A.; Hessenberger, M.; Puehringer, S.; Daumke, O.; Goettig, P. Functional Mapping of Human Dynamin-1-Like GTPase Domain Based on X-ray Structure Analyses. PLoS ONE 2013, 8, e71835. [Google Scholar] [CrossRef]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [Green Version]
- Ho, D.H.; Je, A.R.; Lee, H.; Son, I.; Kweon, H.-S.; Kim, H.-G.; Seol, W. LRRK2 Kinase Activity Induces Mitochondrial Fission in Microglia via Drp1 and Modulates Neuroinflammation. Exp. Neurobiol. 2018, 27, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Smith, H.L.; Chau, K.-Y.; Schapira, A.H.V. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 2016, 213, 163–171. [Google Scholar] [CrossRef]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 448. [Google Scholar] [CrossRef]
- Shin, H.W. Identification and subcellular localization of a novel mammalian dynamin-related protein homologous to yeast Vps1p and Dnm1p. J. Biochem. 1997, 122, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Reddy, T.P.; Manczak, M.; Calkins, M.J.; Shirendeb, U.; Mao, P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 2011, 67, 103–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandimalla, R.; Reddy, P.H. Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim. Biophys. Acta 2016, 1862, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Ishira, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Seasaki, H.; Kageyama, Y.; Reddy, P.H. Dynamin-related protein 1 heterozygote knockout mice do not have synaptic and mitochondrial deficiencies. Biochim. Biophys. Acta 2012, 1822, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via di_erential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 109, 153–159. [Google Scholar] [CrossRef]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J. Alzheimers Dis. 2010, 20, S609–S631. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Silva-Ayala, D.; Lopez, T.; Gutierrez, M.; Perrimon, N.; Lopez, S.; Arias, C.F. Genome-wide RNAi screen reveals a role for the ESCRT complex in rotavirus cell entry. Proc. Natl. Acad. Sci. USA 2013, 110, 10270–10275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Huang, Y.; Li, L. Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int. J. Mol. Sci. 2017, 18, 144. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.-H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 Ameliorates Synaptic Depression, Aβ Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-Division Inhibitor 1 Protects Against Amyloid-β induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 147–162. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar] [CrossRef]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poguiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunningham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much More than Mitophagy. Trends Neurosci. 2015, 37, 315–324. [Google Scholar] [CrossRef]
- Dagda, R.K.; Gusdon, A.M.; Pien, I.; Strack, S.; Green, S.; Li, C.; Van Housten, B.; Cherra III, S.J.; Chu, C.T. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ. 2011, 18, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Finkelsztejn, A. Multiple Sclerosis: Overview of Disease-Modifying Agents. Perspect. Med. Chem. 2014, 6, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.; Herrup, K.; Qi, X.; Yang, Y. Inhibition of Drp1 hyper-activation is protective in animal models of experimental multiple sclerosis. Exp. Neurol. 2017, 292, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; Rossi, L.; Marzulli, D.; Bellomo, F.; De Rasmo, D.; Signorile, A.; Vacca, R.A. Inhibition of Drp1-mediated mitochondrial fission improves mitochondrial dynamics and bioenergetics stimulating neurogenesis in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3117–3127. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Xu, J.; Zou, X.; Li, Y.; Chen, C.; Zheng, A.; Li, H.; Li, H.; Szeto, I.M.; Shi, Y.; et al. Hydroxytyrosol prevents diet-induced metabolic syndrome and attenuates mitochondrial abnormalities in obese mice. Free Radic. Biol. Med. 2014, 67, 396–407. [Google Scholar] [CrossRef]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Wang, Y.; Gan, X.; Fang, D.; Zhong, C.; Wu, L.; Hu, G.; Sosunov, A.A.; McKhann, G.M.; Yu, H.; et al. Drp1-mediated mitochondrial abnormalities link to synaptic injury in diabetes model. Diabetes 2015, 64, 1728–1742. [Google Scholar] [CrossRef]
- Makino, A.; Scott, B.T.; Dillmann, W.H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010, 53, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Guo, M.Y.; Shang, L.; Hu, Y.Y.; Jiang, L.P.; Wan, Y.Y.; Zhou, Q.Q.; Zhang, K.; Liao, H.F.; Yi, J.L.; Han, X.J. The role of Cdk5-mediated Drp1 phosphorylation in Aβ1-42 induced mitochondrial fission and neuronal apoptosis. J. Cell. Biochem. 2018, 119, 4815–4825. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-J.; Lu, Y.-F.; Li, S.-A.; Kaitsuka, T.; Sato, Y.; Tomizawa, K.; Nairn, A.C.; Takei, K.; Matsui, H.; Matsushita, M. CaM kinase Iα-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 2008, 182, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Din, S.; Mason, M.; Volkers, M.; Johnson, B.; Cottage, C.T.; Wang, Z.; Joyo, A.Y.; Quijada, P.; Erhardt, P.; Magnuson, N.S.; et al. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc. Natl. Acad. Sci. USA 2013, 110, 5969–5974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, M.; Kageyama, Y.; Iijima, M.; Sesaki, H. PARK2/Parkin becomes critical when DNM1L/Drp1 is absent. Autophagy 2015, 11, 573–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, A.J.; Qi, X. Drp1 phosphorylation by MAPK1 causes mitochondrial dysfunction in cell culture model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2018, 496, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Slupe, A.M.; Merrill, R.A.; Flippo, K.H.; Lobas, M.A.; Houtman, J.C.; Strack, S. A calcineurin docking motif (LXVP) in dynamin-related protein 1 contributes to mitochondrial fragmentation and ischemic neuronal injury. J. Biol. Chem. 2013, 288, 12353–12365. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Qvit, N.; Su, Y.C.; Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Kuruva, C.S.; Manczak, M.; Yin, X.; Ogunmokun, G.; Reddy, A.P.; Reddy, P.H. Aqua-soluble DDQ reduces the levels of Drp1 and Aβ and inhibits abnormal interactions between Aβ and Drp1 and protects Alzheimer’s disease neurons from Aβ- and Drp1-induced mitochondrial and synaptic toxicities. Hum. Mol. Genet. 2017, 26, 3375–3395. [Google Scholar] [PubMed]
- Mallat, A.; Uchiyama, L.F.; Lewis, S.C.; Fredenburg, R.A.; Terada, Y.; Ji, N.; Nunnari, J.; Tseng, C.C. Discovery and characterization of selective small molecule inhibitors of the mammalian mitochondrial division dynamin, DRP1. Biochem. Biophys. Res. Commun. 2018, 499, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Yin, X.; Reddy, A.P. Synergistic Protective Effects of Mitochondrial Division Inhibitor 1 and Mitochondria-Targeted Small Peptide SS31 in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1549–1565. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Manczak, M.; Reddy, P.H. Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Bido, S.; Soria, F.N.; Fan, R.Z.; Bezard, E.; Tieu, K. Mitochondrial division inhibitor-1 is neuroprotective in the A53T-α-synuclein rat model of Parkinson’s disease. Sci. Rep. 2017, 7, 7495. [Google Scholar] [CrossRef]
- Rappold, P.M.; Cui, M.; Grima, J.C.; Fan, R.Z.; de Mesy-Bentley, K.L.; Chen, L.; Zhuang, X.; Bowers, W.J.; Tieu, K. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat. Commun. 2014, 5, 5244. [Google Scholar] [CrossRef]
- Cui, M.; Tang, X.; Christian, W.V.; Yoon, Y.; Tieu, K. Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J. Biol. Chem. 2010, 285, 11740–11752. [Google Scholar] [CrossRef] [PubMed]
- Felichia, E.; Hoffer, B.; Qi, X.; Luo, Y. Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP. Sci. Rep. 2016, 13, 32656. [Google Scholar] [CrossRef] [PubMed]
- Sian, J.; Youdim, M.B.H.; Riederer, P. MPTP-Induced Parkinsonian Syndrome. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Siegel, G.J., Agranoff, B.W., Albers, R.W., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999. [Google Scholar]
- Guo, X.; Disatnik, M.H.; Monbureau, M.; Shamloo, M.; Mochly-Rosen, D.; Qi, X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Investig. 2013, 123, 5371–5388. [Google Scholar] [CrossRef] [PubMed]
- Seil, M.; El Ouaaliti, M.; Dehaye, J.P. Secretion of IL-1β triggered by dynasore in murine peritoneal macrophages. Innate Immun. 2012, 18, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zhang, L.; Dhillon, R.; Hong, T.T.; Shaw, R.M.; Zhu, J. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS ONE 2013, 8, e60967. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum. Mol. Genet. 2019, 28, 177–199. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Regulator Enzyme | Phosphoryl Group Addition Site | Effect | Citation |
|---|---|---|---|---|
| Mouse cortical neurons | Cdk5-mediated Drp1 phosphorylation at Ser579 is required for Aβ1-42 induced mitochondrial fission—GTPase domain activity is increased | Cdk5 could phosphorylate the recombinant Drp1 at Ser579. Aβ42 stimulation increased the phosphorylation level of Drp1. Cdk5 inhibitor roscovitine and knockdown of Cdk5 efficiently prevented Aβ42 induced Drp1 phosphorylation in neurons | Drp1 phosphorylation is activated by Ser579 | Guo et al. (2018) [61] |
| HeLa (cervical cancer cells derived from Henrietta Lacks) Cyclin-dependent kinase 1 (Cdk1) | Ser385 (splice variant 1 of rat Drp1 Ser616), does not affect GTPase domain activity | Drp1 is activated during cell division when it accepts phosphoryl group from Cdk1 | Drp1 is activated during cell division when it accepts phosphoryl group from Cdk1 | Taguchi et al. (2007) [62] |
| HeLa | Protein kinase A (PKA) | Ser637 of Drp1’s GTPase effector domain | Drp1 GTPase domain activity is inhibited when phosphorylated by PKA | Chang and Blackstone (2007) [63] |
| HeLa neurons | Ca2+ calmodulin dependent PKI 1 (CaMKIα) | Ser600 (the Drp1 splice variant of Ser637) | Phosphorylation by CaMKIα activates Drp1 | Han et al. (2008) [64] |
| Neonatal cardiomyocytes | Pim-1 | Ser637 | Drp1 phosphorylation by Pim-1 stops fission | Din et al. (2013) [65] |
| PC12 | PKA, Calcineurin, and calcium coupling | Ser656 (splice variant 1 of rat Drp1 Ser637), does not affect GTPase domain activity | Phosphoryl group is transferred from PKA to Drp1, which stops GTPase activity, and inhibits apoptosis Calcineurin and calcium work to dephosphorylate and activate Drp1, which upregulates apoptosis | Cribbs and Strack (2007) [66] |
| Calcineurin and calcium coupling | Ser637 | Calcineurin and calcium work to dephosphorylate and activate Drp1, which upregulates apoptosis | Cereghetti et al. (2008) [67] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliver, D.; Reddy, P.H. Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells 2019, 8, 961. https://doi.org/10.3390/cells8090961
Oliver D, Reddy PH. Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells. 2019; 8(9):961. https://doi.org/10.3390/cells8090961
Chicago/Turabian StyleOliver, Darryll, and P. Hemachandra Reddy. 2019. "Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases" Cells 8, no. 9: 961. https://doi.org/10.3390/cells8090961
APA StyleOliver, D., & Reddy, P. H. (2019). Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells, 8(9), 961. https://doi.org/10.3390/cells8090961