RNA Sequencing of H3N2 Influenza Virus-Infected Human Nasal Epithelial Cells from Multiple Subjects Reveals Molecular Pathways Associated with Tissue Injury and Complications

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Derivation of Human Nasal Epithelial Stem/Progenitor Cells (hNESPCs) and In Vitro Differentiation of hNECs
2.2. Inoculation of Human Influenza H3N2 Virus in Fully Differentiated hNECs and Viral Quantification
2.3. Viral Plaque Assay
2.4. Total RNA Extraction and Real-Time Quantitative PCR
2.5. Library Preparation for RNAseq
2.6. RNAseq Analysis
2.7. Geneset Enrichment Analysis
2.8. Luminex Assay for Cytokines and Chemokines
3. Results
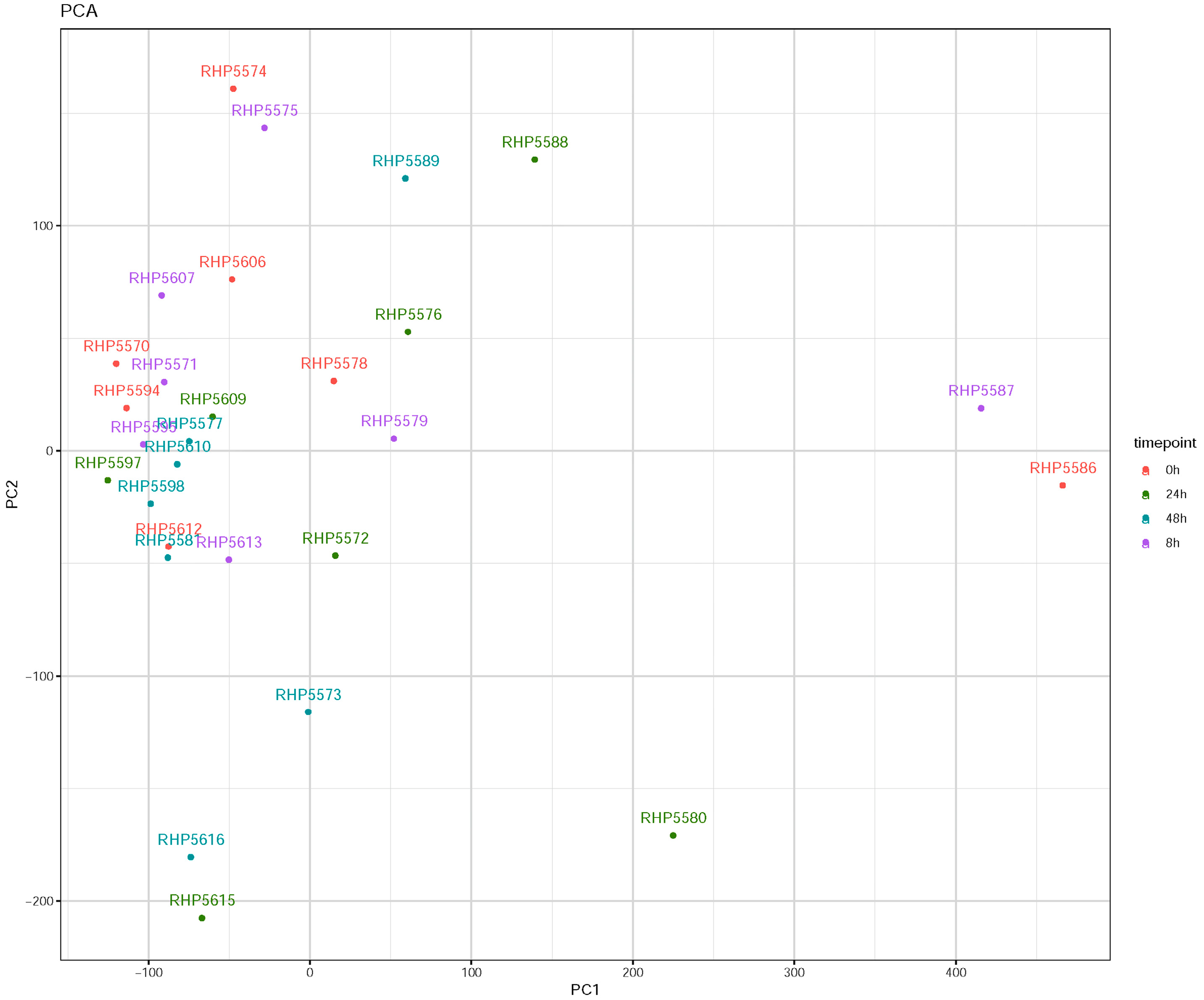
3.1. Influenza Virus Infection Induces Similar Responses in hNECs from Multiple Donors
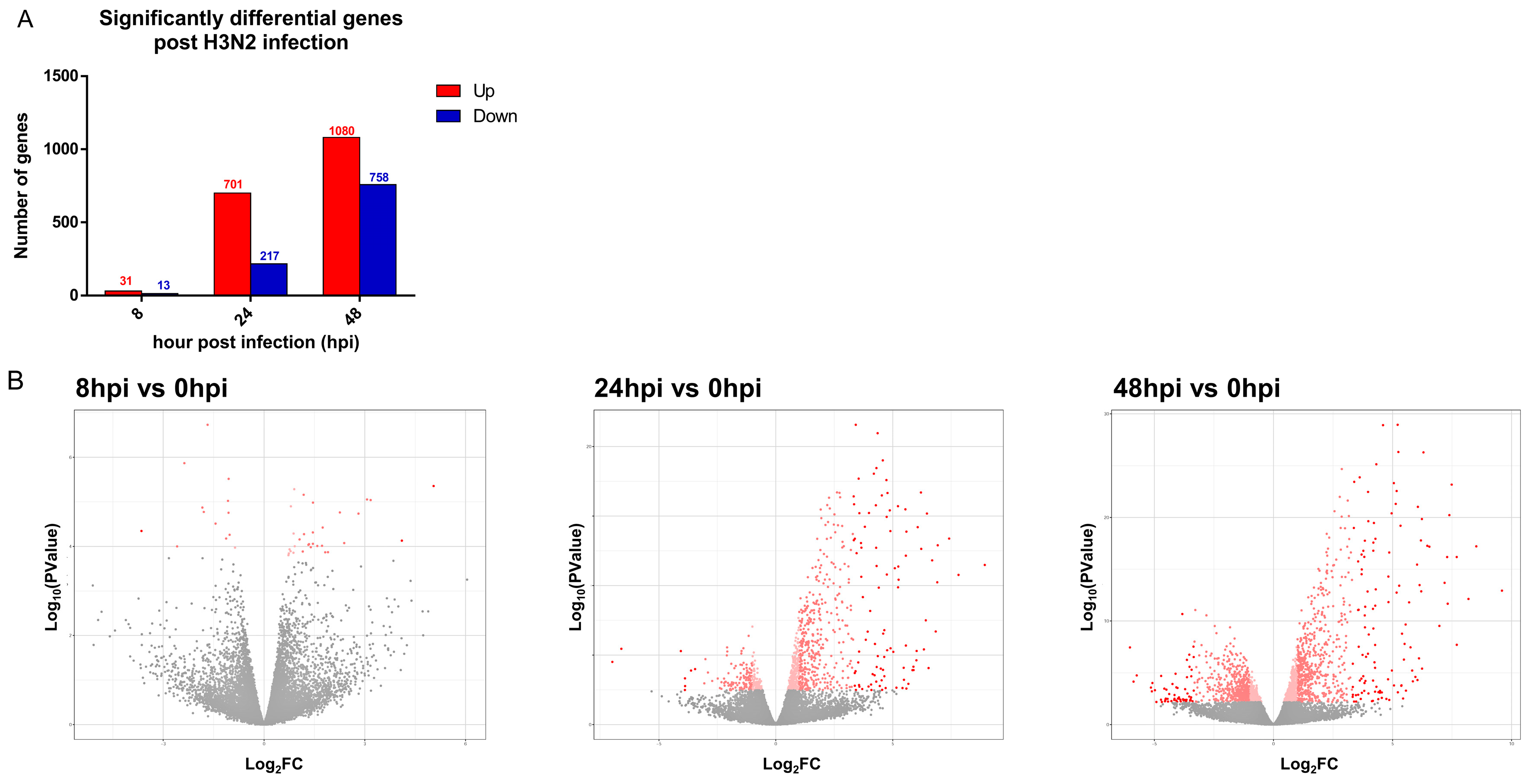
3.2. Transcriptomic Changes Are Detected at 8 hpi, and Peak at 48 hpi
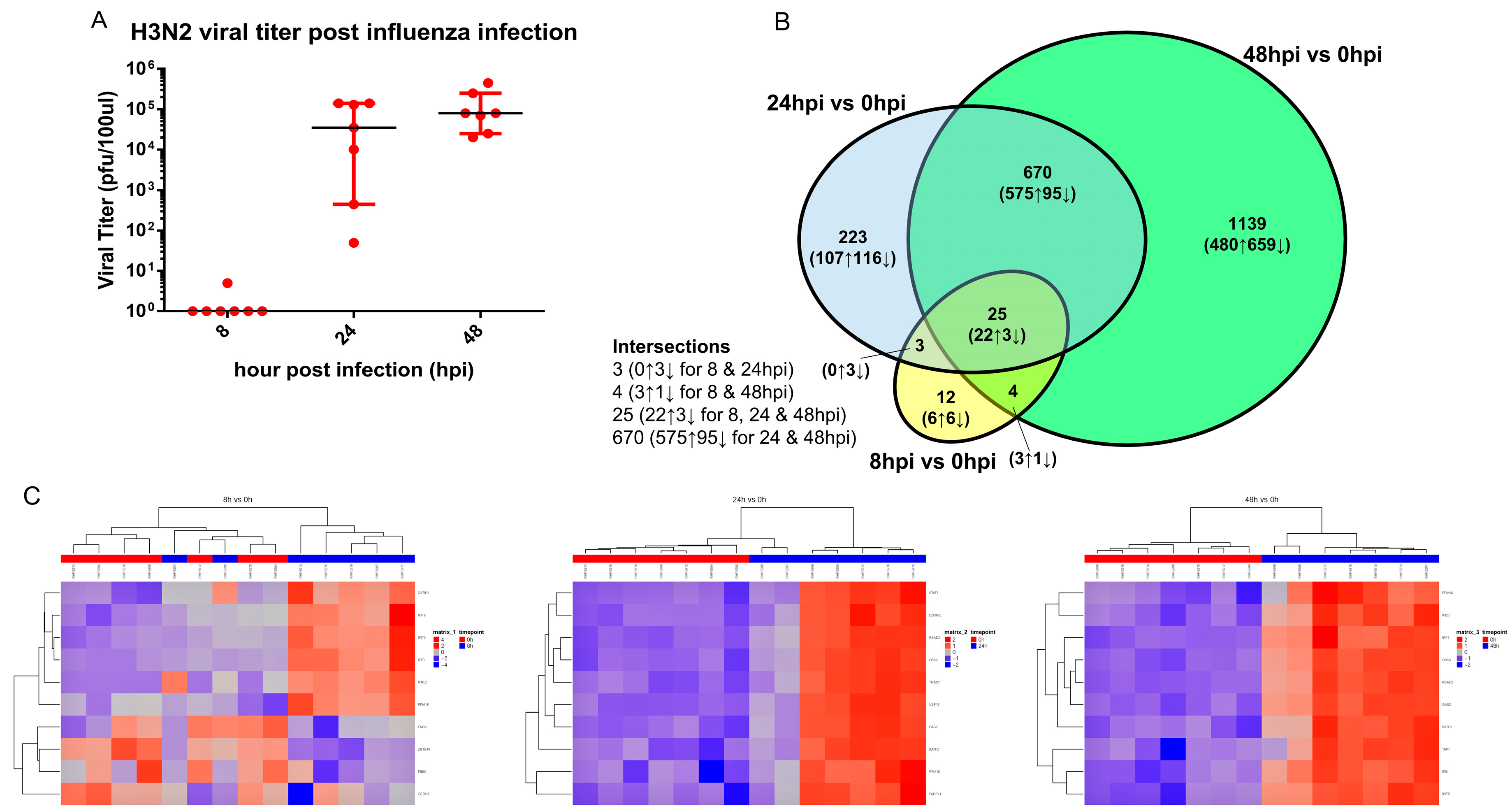
3.3. Transcriptomic Change Alterations Correlate with Viral Titer, with Intersecting Genes Remaining Consistent in Directional Changes of Expression
3.4. Pathway Enrichment of Significant Gene Changes Revealed Potential Epithelium-Initiated Pathways Contributing to Influenza Pathology and Pathogenesis
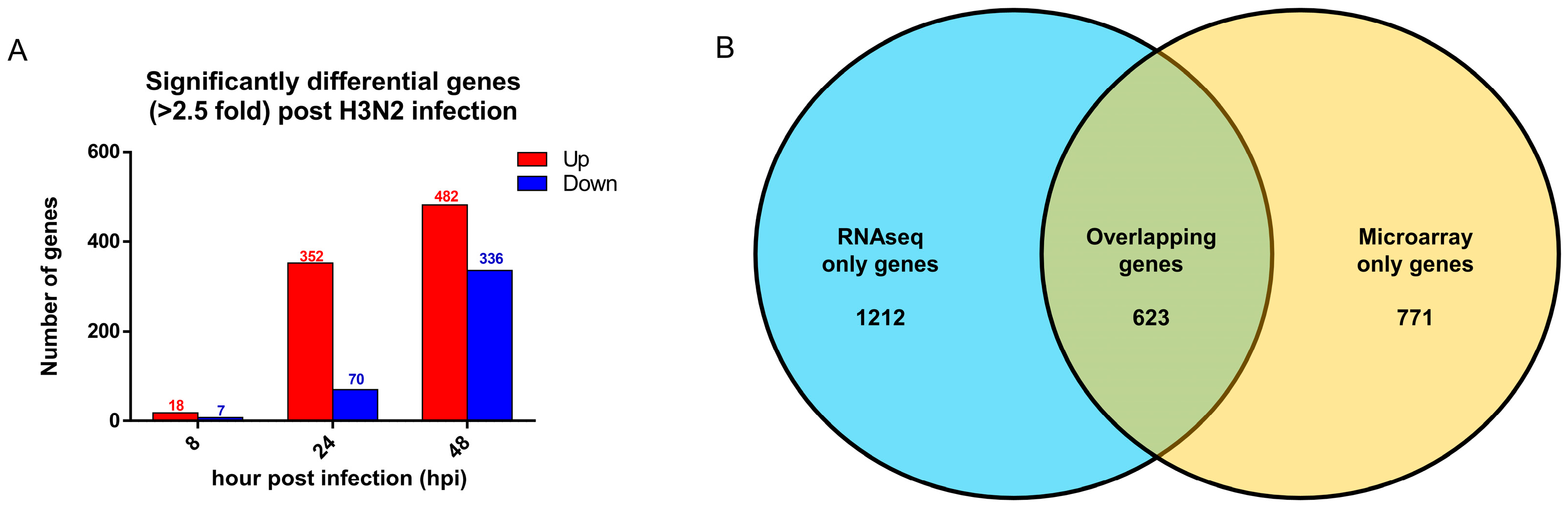
3.5. Comparison with Established Influenza Gene Expression Signatures and Transcriptomes Revealed Consistent Upregulation of Influenza Response Genes
3.6. Secretory Cytokine Profiles During Influenza Virus Infection of hNECs Reveal Early Suppression of Cytokines in Apical Secretion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Available online: http://www.who.int/mediacentre/factsheets/fs211/en/. (accessed on 15 May 2019).
- Moscona, A. Oseltamivir resistance--disabling our influenza defenses. N. Engl. J. Med. 2005, 353, 2633–2636. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.Y.; Andiappan, A.K.; Yan, Y.; Tan, K.S.; Ong, H.H.; Thong, K.T.; Ong, Y.K.; Yu, F.G.; Low, H.B.; et al. Role of IL-13Ralpha2 in modulating IL-13-induced MUC5AC and ciliary changes in healthy and CRSwNP mucosa. Allergy 2018, 73, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T. Epithelial damage and response. Clin. Exp. Allergy 2000, 30 (Suppl. 1), 37–41. [Google Scholar] [CrossRef]
- Yan, Y.; Tan, K.S.; Li, C.; Tran, T.; Chao, S.S.; Sugrue, R.J.; Shi, L.; Chow, V.; Wang de, Y. Human nasal epithelial cells derived from multiple individuals exhibit differential responses to H3N2 influenza virus infection in vitro. J. Allergy Clin. Immunol. 2016, 138, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.S.; Yan, Y.; Koh, W.H.; Li, L.; Choi, H.; Tran, T.; Sugrue, R.J.; Wang, D.Y.; Chow, V.T. Comparative Transcriptomic and Metagenomic Analyses of Influenza Virus-Infected Nasal Epithelial Cells from Multiple Individuals Reveal Specific Nasal-Initiated Signatures. Front. Microbiol. 2018, 9, 2685. [Google Scholar] [CrossRef] [PubMed]
- Münk, C.F.S.A.; König, R. Systems-biology approaches to discover anti-viral effectors of the human innate immune response. Viruses 2011, 3, 1112–1130. [Google Scholar] [CrossRef] [PubMed]
- Stertz, S.; Shaw, M.L. Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes Infect. 2011, 13, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, T.; Watanabe, S.; Kawaoka, Y. Cellular Networks Involved in the Influenza Virus Life Cycle. Cell Host Microbe 2010, 7, 427–439. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Y.; Yan, Y.; Tan, K.S.; Tan, S.S.L.; Seet, J.E.; Arumugam, T.V.; Chow, V.T.K.; Wang, Y.; Tran, T. CD151, a novel host factor of nuclear export signaling in influenza virus infection. J. Allergy Clin. Immunol. 2018, 141, 1799–1817. [Google Scholar] [CrossRef] [PubMed]
- Essaidi-Laziosi, M.; Brito, F.; Benaoudia, S.; Royston, L.; Cagno, V.; Fernandes-Rocha, M.; Piuz, I.; Zdobnov, E.; Huang, S.; Constant, S.; et al. Propagation of respiratory viruses in human airway epithelia reveals persistent virus-specific signatures. J. Allergy Clin. Immunol. 2018, 141, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Guibas, G.V.; Tsolia, M.; Christodoulou, I.; Stripeli, F.; Sakkou, Z.; Papadopoulos, N.G. Distinction between rhinovirus-induced acute asthma and asthma-augmented influenza infection. Clin. Exp. Allergy: J. Br. Soc. Allergy Clin. Immunol. 2018, 48, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Koban, R.; Neumann, M.; Daugs, A.; Bloch, O.; Nitsche, A.; Langhammer, S.; Ellerbrok, H. A novel three-dimensional cell culture method enhances antiviral drug screening in primary human cells. Antivir. Res. 2018, 150, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Mantione, K.J.; Kream, R.M.; Kuzelova, H.; Ptacek, R.; Raboch, J.; Samuel, J.M.; Stefano, G.B. Comparing bioinformatic gene expression profiling methods: Microarray and RNA-Seq. Med. Sci. Monit. Basic Res. 2014, 20, 138–142. [Google Scholar] [PubMed]
- Zhao, X.N.; Yu, F.G.; Li, C.W.; Li, Y.Y.; Chao, S.S.; Loh, W.S.; Pan, X.L.; Shi, L.; Wang, D.Y. The use of nasal epithelial stem/progenitor cells to produce functioning ciliated cells in vitro. Am. J. Rhinol. Allergy 2012, 26, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S.; Faridani, O.R.; Bjorklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Tang, J.W.; Loh, T.P.; Oon, L.L.; Koay, E.S. Predicting clinical severity based on substitutions near epitope A of influenza A/H3N2. Infect. Genet. Evol. 2015, 34, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.P.; Zhao, X.; Pang, J.; Chen, M.I.; Lee, V.J.; Ang, L.W.; Lin, R.V.; Gao, C.Q.; Hsu, L.Y.; Cook, A.R. Effectiveness of seasonal influenza vaccinations against laboratory-confirmed influenza-associated infections among Singapore military personnel in 2010-2013. Influenza Other Respir. Viruses 2014, 8, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.S.; Ong, H.H.; Yan, Y.; Liu, J.; Li, C.; Ong, Y.K.; Thong, K.T.; Choi, H.W.; Wang, D.Y.; Chow, V.T. In Vitro Model of Fully Differentiated Human Nasal Epithelial Cells Infected With Rhinovirus Reveals Epithelium-Initiated Immune Responses. J. Infect. Dis. 2018, 217, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; McCabe, T.M.; Crotta, S.; Gad, H.H.; Hessel, E.M.; Beinke, S.; Hartmann, R.; Wack, A. IFNlambda is a potent anti-influenza therapeutic without the inflammatory side effects of IFNalpha treatment. EMBO. Mol. Med. 2016, 8, 1099–1112. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-lambda Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890. [Google Scholar] [CrossRef]
- Li, N.; Parrish, M.; Chan, T.K.; Yin, L.; Rai, P.; Yoshiyuki, Y.; Abolhassani, N.; Tan, K.B.; Kiraly, O.; Chow, V.T.; et al. Influenza infection induces host DNA damage and dynamic DNA damage responses during tissue regeneration. Cell. Mol. Life Sci. 2015, 72, 2973–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stairiker, C.; Mueller, Y.; van Meurs, M.; Brouwers-Haspels, I.; Erkeland, S.; Katsikis, P.D. Heatr9 is upregulated during influenza virus infection in lung alveolar epithelial cells. J. Immunol. 2018, 200 (Suppl. 1), 109–116. [Google Scholar]
- Molinier-Frenkel, V.; Prevost-Blondel, A.; Castellano, F. The IL4I1 Enzyme: A New Player in the Immunosuppressive Tumor Microenvironment. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Q.; Xie, J.; Xu, Y. Cigarette smoke inhibits BAFF expression and mucosal immunoglobulin A responses in the lung during influenza virus infection. Respir. Res. 2015, 16, 37. [Google Scholar] [CrossRef] [PubMed]
- Rutigliano, J.A.; Sharma, S.; Morris, M.Y.; Oguin, T.H., 3rd; McClaren, J.L.; Doherty, P.C.; Thomas, P.G. Highly pathological influenza A virus infection is associated with augmented expression of PD-1 by functionally compromised virus-specific CD8+ T cells. J. Virol. 2014, 88, 1636–1651. [Google Scholar] [CrossRef]
- Guo, X.J.; Thomas, P.G. New fronts emerge in the influenza cytokine storm. Semin. Immunopathol. 2017, 39, 541–550. [Google Scholar] [CrossRef]
- Ramana, C.V.; DeBerge, M.P.; Kumar, A.; Alia, C.S.; Durbin, J.E.; Enelow, R.I. Inflammatory impact of IFN-gamma in CD8+ T cell-mediated lung injury is mediated by both Stat1-dependent and -independent pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L650–L657. [Google Scholar] [CrossRef]
- Wiley, J.A.; Cerwenka, A.; Harkema, J.R.; Dutton, R.W.; Harmsen, A.G. Production of interferon-gamma by influenza hemagglutinin-specific CD8 effector T cells influences the development of pulmonary immunopathology. Am. J. Pathol. 2001, 158, 119–130. [Google Scholar] [CrossRef]
- Tian, T.; Zi, X.; Peng, Y.; Wang, Z.; Hong, H.; Yan, Y.; Guan, W.; Tan, K.S.; Liu, J.; Ong, H.H.; et al. H3N2 influenza virus infection enhances oncostatin M expression in human nasal epithelium. Exp. Cell. Res. 2018, 371, 322–329. [Google Scholar] [CrossRef]
- Wurzer, W.J.; Ehrhardt, C.; Pleschka, S.; Berberich-Siebelt, F.; Wolff, T.; Walczak, H.; Planz, O.; Ludwig, S. NF-kappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J. Biol. Chem. 2004, 279, 30931–30937. [Google Scholar] [CrossRef]
- Mayank, A.K.; Sharma, S.; Nailwal, H.; Lal, S.K. Nucleoprotein of influenza A virus negatively impacts antiapoptotic protein API5 to enhance E2F1-dependent apoptosis and virus replication. Cell Death Dis. 2015, 6, e2018. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Lim, W.; Bist, P.; Perumalsamy, R.; Lukman, H.M.; Li, F.; Welker, L.B.; Yan, B.; Sethi, G.; Tambyah, P.A.; et al. Influenza A virus enhances its propagation through the modulation of Annexin-A1 dependent endosomal trafficking and apoptosis. Cell Death Differ. 2016, 23, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Fujikura, D.; Miyazaki, T. Programmed Cell Death in the Pathogenesis of Influenza. Int. J. Mol. Sci. 2018, 19, 2065. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.H.; Baumgarth, N. The Multifaceted B Cell Response to Influenza Virus. J. Immunol. 2019, 202, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.; Monneaux, F.; Soulas-Sprauel, P.; Muller, S.; Martin, T.; Korganow, A.S.; Pasquali, J.L. Influenza virus-induced type I interferon leads to polyclonal B-cell activation but does not break down B-cell tolerance. J. Virol. 2007, 81, 12525–12534. [Google Scholar] [CrossRef]
- Ogawa, H.; Azuma, M.; Tsunematsu, T.; Morimoto, Y.; Kondo, M.; Tezuka, T.; Nishioka, Y.; Tsuneyama, K. Neutrophils induce smooth muscle hyperplasia via neutrophil elastase-induced FGF-2 in a mouse model of asthma with mixed inflammation. Clin. Exp. Allergy: J. Br. Soc. Allergy Clin. Immunol. 2018, 48, 1715–1725. [Google Scholar] [CrossRef]
- Tourdot, S.; Mathie, S.; Hussell, T.; Edwards, L.; Wang, H.; Openshaw, P.J.; Schwarze, J.; Lloyd, C.M. Respiratory syncytial virus infection provokes airway remodelling in allergen-exposed mice in absence of prior allergen sensitization. Clin. Exp. Allergy: J. Br. Soc. Allergy Clin. Immunol. 2008, 38, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Y.; Tam, J.K.C.; Tan, S.L.S.; Tai, Y.K.; Chin, C.Y.; Steward, A.G.; Ashman, L.K.; Sekiguchi, K.; Langenbach, S.Y.; Stelmack, G.; et al. CD151, a laminin receptor showing increased expression in asthma, contributes to airway hyperresponsiveness through calcium signaling. J. Allergy Clin. Immunol. 2016, 139, 82–92. [Google Scholar] [CrossRef]
- Salter, B.; Pray, C.; Radford, K.; Martin, J.G.; Nair, P. Regulation of human airway smooth muscle cell migration and relevance to asthma. Respir. Res. 2017, 18, 156. [Google Scholar] [CrossRef] [PubMed]
- Hirota, J.A.; Nguyen, T.T.; Schaafsma, D.; Sharma, P.; Tran, T. Airway smooth muscle in asthma: Phenotype plasticity and function. Pulm Pharm. 2009, 22, 370–378. [Google Scholar] [CrossRef] [PubMed]
- De Chassey, B.; Aublin-Gex, A.; Ruggieri, A.; Meyniel-Schicklin, L.; Pradezynski, F.; Davoust, N.; Chantier, T.; Tafforeau, L.; Mangeot, P.E.; Ciancia, C.; et al. The interactomes of influenza virus NS1 and NS2 proteins identify new host factors and provide insights for ADAR1 playing a supportive role in virus replication. PLoS Pathog. 2013, 9, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Mashima, R.; Saeki, K.; Aki, D.; Minoda, Y.; Takaki, H.; Sanada, T.; Kobayashi, T.; Aburatani, H.; Yamanashi, Y.; Yoshimura, A. FLN29, a novel interferon- and LPS-inducible gene acting as a negative regulator of toll-like receptor signaling. J. Biol. Chem. 2005, 280, 41289–41297. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Horvath, C.M. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J. Virol. 2006, 80, 12332–12342. [Google Scholar] [CrossRef] [PubMed]
- Horst, A.K.; Najjar, S.M.; Wagener, C.; Tiegs, G. CEACAM1 in Liver Injury, Metabolic and Immune Regulation. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Vitenshtein, A.; Weisblum, Y.; Hauka, S.; Halenius, A.; Oiknine-Djian, E.; Tsukerman, P.; Bauman, Y.; Bar-On, Y.; Stern-Ginossar, N.; Enk, J.; et al. CEACAM1-Mediated Inhibition of Virus Production. Cell. Rep. 2016, 15, 2331–2339. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wang, S.; Yu, Y.; Zhang, J.; Sun, Z.; Yan, Y.; Zhou, J. Subcellular proteomic analysis of human host cells infected with H3N2 swine influenza virus. Proteomics 2013, 13, 3309–3326. [Google Scholar] [CrossRef]
- Wang, J.; Yang, B.; Hu, Y.; Zheng, Y.; Zhou, H.; Wang, Y.; Ma, Y.; Mao, K.; Yang, L.; Lin, G.; et al. Negative regulation of Nmi on virus-triggered type I IFN production by targeting IRF7. J. Immunol. 2013, 191, 3393–3399. [Google Scholar] [CrossRef]
- Graham, A.C.; Temple, R.M.; Obar, J.J. Mast cells and influenza a virus: Association with allergic responses and beyond. Front. Immunol. 2015, 6, 238. [Google Scholar] [CrossRef]
- Le Bel, M.; Gosselin, J. Leukotriene B4 Enhances NOD2-Dependent Innate Response against Influenza Virus Infection. PLoS ONE 2015, 10, e0139856. [Google Scholar] [CrossRef]
- Andres-Terre, M.; McGuire, H.M.; Pouliot, Y.; Bongen, E.; Sweeney, T.E.; Tato, C.M.; Khatri, P. Integrated, Multi-cohort Analysis Identifies Conserved Transcriptional Signatures across Multiple Respiratory Viruses. Immunity 2015, 43, 1199–1211. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, R.; Maeda, N.; Shibata, K.; Yamada, H.; Kase, T.; Yoshikai, Y. Interleukin-15 is critical in the pathogenesis of influenza a virus-induced acute lung injury. J. Virol. 2010, 84, 5574–5582. [Google Scholar] [CrossRef]
- Ito, Y.; Correll, K.; Zemans, R.L.; Leslie, C.C.; Murphy, R.C.; Mason, R.J. Influenza induces IL-8 and GM-CSF secretion by human alveolar epithelial cells through HGF/c-Met and TGF-alpha/EGFR signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1178–L1188. [Google Scholar] [CrossRef]
- Venkataraman, T.; Coleman, C.M.; Frieman, M.B. Overactive Epidermal Growth Factor Receptor Signaling Leads to Increased Fibrosis after Severe Acute Respiratory Syndrome Coronavirus Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Morichi, S.; Morishita, N.; Takeshita, M.; Ishida, Y.; Oana, S.; Yamanaka, G.; Kashiwagi, Y.; Kawashima, H. Vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) levels in the cerebrospinal fluid of children with influenza-associated encephalopathy. J. Infect. Chemother. 2017, 23, 80–84. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time, Hours Post Infection (hpi) | 8 | 24 | 48 |
|---|---|---|---|
| Number of significant genes | 44 (31↑; 13↓) | 921 (704↑; 217↓) | 1838 (1080↑; 758↓) |
| Number of significant genes over 10-fold (>Log2FC of 3.4) | 3 (2↑; 1↓) | 99 (91↑; 8↓) | 167 (112↑; 55↓) |
| Highest upregulated gene | IFNL2 | IFNL1 | IFNL1 |
| Highest downregulated gene | KLK12 | NAP1L3 | CYP26A1 |
| GOid | Description | Annotated | Significant | Expected | p-Value_adj | Genes |
|---|---|---|---|---|---|---|
| GO:0060333 | Interferon-gamma-mediated signaling pathway | 72 | 36 | 10.35 | 9.184 × 10−11 | TRIM22 ↑; IRF9 ↑; TRIM38 ↑; GBP1 ↑; GBP2 ↑; HCK ↓; HLA-A ↑; HLA-B ↑; HLA-C ↑; HLA-E ↑; HLA-F ↑; HLA-G ↑; ICAM1 ↑; IRF1 ↑; IRF2 ↑; IRF6 ↑; IRF7 ↑; JAK2 ↑; MT2A ↑; OAS1 ↑; OAS2 ↑; OAS3 ↑; PML ↑; B2M ↑; PTAFR ↑; SP100 ↑; TRIM21 ↑; STAT1 ↑; TRIM25 ↑; TRIM26 ↑; CAMK2D ↑; NLRC5 ↑; TRIM5 ↑; OASL ↑; NMI ↑; CD44 ↑ |
| GO:0008637 | Apoptotic mitochondrial changes | 81 | 22 | 11.64 | 0.015 | CLU ↓; ERBB4 ↓; EYA2 ↓; IFI6 ↑; GCLC ↓; GCLM ↓; SFN ↑; SLC25A4 ↓; HK2 ↑; IFIT2 ↑; MCL1 ↑; MMP9 ↑; PLAUR ↑; PMAIP1 ↑; BAK1 ↑; SOD2 ↑; BNIP3L ↓; PLA2G6 ↓; CASP8 ↑; AIFM2 ↑; TNFSF10 ↑; NOL3 ↓ |
| GO:0048661 | Positive regulation of smooth muscle cell proliferation | 58 | 19 | 8.34 | 0.036 | NAMPT ↑; ABCC4 ↓; PPARGC1A ↓; HBEGF ↑; EREG ↑; FGFR2 ↓; ID2 ↓; IRAK1 ↑; JAK2 ↑; MMP9 ↑; NOTCH3 ↓; IRAK4 ↑; SERPINF2 ↓; PTAFR ↑; PTGS2 ↑; CCL5 ↑; STAT1 ↑; THBS1 ↑; CAMK2D ↑ |
| GO:0045824 | Negative regulation of innate immune response | 28 | 13 | 4.02 | 0.015 | ADAR ↑; TRAFD1 ↑; A2M ↓; HLA-E ↑; IFI16 ↑; LGALS9 ↑; SERPINB9 ↑; CEACAM1 ↑; SERPING1 ↑; TNFAIP3 ↑; DHX58 ↑; NLRC5 ↑; NMI ↑ |
| GO:0070542 | Response to fatty acid | 32 | 13 | 4.6 | 0.015 | PPARGC1A ↓; CREB1 ↑; CTGF ↓; ALAD ↓; GIPR ↓; PDK4 ↓; PTAFR ↑; PTGS2 ↑; TLR2 ↑; UCP2 ↓; ZC3H12A ↑; CAT ↓; CD36 ↓ |
| GO:0060544 | Regulation of necroptotic process | 11 | 7 | 1.58 | 0.043 | RBCK1 ↑; RIPK3 ↑; SLC25A4 ↓; BIRC2 ↑; BIRC3 ↑; CFLAR ↑; ARHGEF2 ↑ |
| ID | Description | Gene Ratio | Bg Ratio | p-value | p.adjust | q-value | geneID |
|---|---|---|---|---|---|---|---|
| 877300 | Interferon gamma signaling | 33/776 | 65/4637 | 2.081 × 10−10 | 3.83 × 10−8 | 3.32 × 10−8 | OAS2 ↑; OAS3 ↑;IRF7 ↑; OAS1 ↑; STAT1 ↑; TRIM21 ↑; TRIM5 ↑; PML ↑; HLA-F ↑; HLA-E ↑; SP100 ↑; OASL ↑; TRIM22 ↑; TRIM25 ↑; HLA-B ↑; GBP1 ↑; HLA-C ↑; B2M ↑; TRIM38 ↑; IRF6 ↑; HLA-A ↑; IRF9 ↑; JAK2 ↑; TRIM26 ↑; IRF1 ↑; GBP2 ↑; HLA-G ↑; IRF2 ↑; CAMK2D ↑; PTAFR ↑; CD44 ↑; MT2A ↑; ICAM1 ↑ |
| 983705 | Signaling by the B Cell Receptor (BCR) | 38/776 | 119/4637 | 2.72153 × 10−5 | 0.001440597 | 0.001249039 | PSME2 ↑; BTC ↑; PSMB9 ↑; DAPP1 ↑; PSMB8 ↑; RASGRP3 ↑; RICTOR ↑; EREG ↑; PSMA6 ↑; PSMA4 ↑; MOV10 ↑; HBEGF ↑; ITPR3 ↑; PSMF1 ↑; FGF2 ↑; CARD11 ↑; PSME1 ↑; CREB1 ↑; LYN ↑; PSMB7 ↑; FGFR3 ↓; MALT1 ↑; CDKN1A ↑; PIK3AP1 ↑; AGO1 ↓; FGF1 ↓; FGFR2 ↓; FGF5 ↑; ERBB4 ↓; ITPR1 ↓; UBC ↑; NCK1 ↑; ERBB2 ↓; SH3KBP1 ↑; PSMA3 ↑; PSMA5 ↑; TRIB3 ↑; GAB1 ↑ |
| 109581 | Apoptosis | 30/776 | 88/4637 | 4.88082 × 10−5 | 0.00227963 | 0.001976505 | PSME2 ↑; PSMB9 ↑; PMAIP1 ↑; TNFSF10 ↑; DSG3 ↑; PSMB8 ↑; BAK1 ↑; TICAM1 ↑; H1F0 ↑; CASP7 ↑; TLR3 ↑; CFLAR ↑; FAS ↑; PSMA6 ↑; PSMA4 ↑; PSMF1 ↑; OCLN ↑; TRAF2 ↑; XIAP ↑; PSME1 ↑; CASP8 ↑; MAGED1 ↓; SFN ↑; PSMB7 ↑; TJP1 ↑; BIRC2 ↑; UBC ↑; PLEC ↑; PSMA3 ↑; PSMA5 ↑ |
| 71291 | Metabolism of amino acids and derivatives | 40/776 | 134/4637 | 9.37168 × 10−5 | 0.004025804 | 0.003490489 | IL4I1 ↑; IDO1 ↑; PSME2 ↑; SQRDL ↑; PSMB9 ↑; PSMB8 ↑; GAMT ↓; PSMA6 ↑; SMS ↑; PSMA4 ↑; KYNU ↑; SLC5A5 ↓; PSMF1 ↑; SHMT1 ↓; ALDH7A1 ↓; AZIN2 ↑; PSME1 ↑; AFMID ↓; BBOX1 ↓; GCLC ↓; IYD ↓; ALDH4A1 ↓; FAH ↓; PSMB7 ↑; HIBCH ↓; MCCC1 ↓; ALDH18A1 ↓; TST ↓; GCLM ↓; BCAT1 ↑; PSAT1 ↑; GPT ↓; ASNS ↑; PSMA3 ↑; ADI1 ↓; PSMA5 ↑; SLC25A10 ↓; ALDH6A1 ↓ |
| 1168372 | Downstream signaling events of B Cell Receptor (BCR) | 31/776 | 98/4637 | 0.000180538 | 0.004865597 | 0.004218614 | PSME2 ↑; BTC ↑; PSMB9 ↑; PSMB8 ↑; RASGRP3 ↑; RICTOR ↑; EREG ↑; PSMA6 ↑; PSMA4 ↑; MOV10 ↑; HBEGF ↑; PSMF1 ↑; FGF2 ↑; CARD11 ↑; PSME1 ↑; CREB1 ↑; PSMB7 ↑; FGFR3 ↓; MALT1 ↑; CDKN1A ↑; AGO1 ↓; FGF1 ↓; FGFR2 ↓; FGF5 ↑; ERBB4 ↓; UBC ↑; ERBB2 ↓; PSMA3 ↑; PSMA5 ↑; TRIB3 ↑; GAB1 ↑ |
| Gene_Name | logCPM | FDR 8 h | LogFC 8 h | FDR 24 h | LogFC 24 h | FDR 48 h | LogFC 48 h |
|---|---|---|---|---|---|---|---|
| ZBP1 | 5.467096 | N.S. | 2.71 × 10−6 | 6.419275 | 2.70 × 10−8 | 6.97086 | |
| IFI6 | 7.740724 | N.S. | 2.77 × 10−14 | 4.780717 | 1.80 × 10−23 | 6.30199 | |
| CD38 | 5.090161 | N.S. | 1.91 × 10−8 | 5.232278 | 2.10 × 10−10 | 5.699259 | |
| MX1 | 10.7197 | N.S. | 2.36 × 10−13 | 4.87848 | 3.48 × 10−17 | 5.339009 | |
| HERC5 | 6.743117 | N.S. | 1.05 × 10−9 | 5.093646 | 6.89 × 10−12 | 5.28321 | |
| IFIH1 | 7.876294 | N.S. | 2.71 × 10−4 | 3.566556 | 7.51 × 10−6 | 3.972719 | |
| HERC6 | 8.376077 | N.S. | 1.66 × 10−11 | 3.339444 | 7.15 × 10−6 | 3.843258 | |
| RTP4 | 4.998082 | N.S. | 4.97 × 10−11 | 2.846709 | 7.28 × 10−15 | 3.25471 | |
| PARP12 | 5.511677 | N.S. | 3.12 × 10−2 | 2.203849 | 1.99 × 10−2 | 2.11728 | |
| LY6E | 8.624757 | N.S. | 2.10 × 10−6 | 1.451684 | 8.74 × 10−12 | 1.989543 | |
| LGALS3BP | 11.0223 | N.S. | 1.30 × 10−3 | 0.855231 | 1.57 × 10−8 | 1.336304 | |
| Gene name | Microarray LogFC 48 h | Microarray FDR 48 h | RNAseq LogFC 48 h | RNAseq FDR 48 h |
|---|---|---|---|---|
| ZBP1 | 1.30628 | 1.04 × 10−6 | 6.97086 | 2.70 × 10−8 |
| IFI6 | 3.78685 | 6.67 × 10−6 | 6.30199 | 1.80 × 10−23 |
| CD38 | 3.61175 | 5.88 × 10−7 | 5.699259 | 2.10 × 10−10 |
| MX1 | 3.17584 | 1.57 × 10−8 | 5.339009 | 3.48 × 10−17 |
| HERC5 | 3.53433 | 1.65 × 10−8 | 5.28321 | 6.89 × 10−12 |
| IFIH1 | 4.08252 | 5.01 × 10−8 | 3.972719 | 7.51 × 10−6 |
| HERC6 | 3.10932 | 9.16 × 10−8 | 3.843258 | 7.15 × 10−16 |
| RTP4 | 2.41668 | 6.25 × 10−7 | 3.25471 | 7.28 × 10−15 |
| PARP12 | 2.02756 | 4.75 × 10−8 | 2.11728 | 1.99 × 10−2 |
| LY6E | 1.11967 | 6.02 × 10−5 | 1.989543 | 8.74 × 10−12 |
| LGALS3BP | 0.863641 | 7.59 × 10−4 | 1.336304 | 1.57 × 10−8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, K.S.; Andiappan, A.K.; Lee, B.; Yan, Y.; Liu, J.; Tang, S.A.; Lum, J.; He, T.T.; Ong, Y.K.; Thong, M.; et al. RNA Sequencing of H3N2 Influenza Virus-Infected Human Nasal Epithelial Cells from Multiple Subjects Reveals Molecular Pathways Associated with Tissue Injury and Complications. Cells 2019, 8, 986. https://doi.org/10.3390/cells8090986
Tan KS, Andiappan AK, Lee B, Yan Y, Liu J, Tang SA, Lum J, He TT, Ong YK, Thong M, et al. RNA Sequencing of H3N2 Influenza Virus-Infected Human Nasal Epithelial Cells from Multiple Subjects Reveals Molecular Pathways Associated with Tissue Injury and Complications. Cells. 2019; 8(9):986. https://doi.org/10.3390/cells8090986
Chicago/Turabian StyleTan, Kai Sen, Anand Kumar Andiappan, Bernett Lee, Yan Yan, Jing Liu, See Aik Tang, Josephine Lum, Ting Ting He, Yew Kwang Ong, Mark Thong, and et al. 2019. "RNA Sequencing of H3N2 Influenza Virus-Infected Human Nasal Epithelial Cells from Multiple Subjects Reveals Molecular Pathways Associated with Tissue Injury and Complications" Cells 8, no. 9: 986. https://doi.org/10.3390/cells8090986
APA StyleTan, K. S., Andiappan, A. K., Lee, B., Yan, Y., Liu, J., Tang, S. A., Lum, J., He, T. T., Ong, Y. K., Thong, M., Lim, H. F., Choi, H. W., Rotzschke, O., Chow, V. T., & Wang, D. Y. (2019). RNA Sequencing of H3N2 Influenza Virus-Infected Human Nasal Epithelial Cells from Multiple Subjects Reveals Molecular Pathways Associated with Tissue Injury and Complications. Cells, 8(9), 986. https://doi.org/10.3390/cells8090986