Impaired Nuclear Export of the Ribonucleoprotein Complex and Virus-Induced Cytotoxicity Combine to Restrict Propagation of the A/Duck/Malaysia/02/2001 (H9N2) Virus in Human Airway Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Specific Reagents
2.3. Immunoblotting Analysis of Whole-Cell Extracts
2.4. Nuclei Preparation
2.5. Construction of Recombinant Expression Vectors
2.6. Polymerase Reporter Assay
2.7. Immunofluorescence Microscopy
2.8. Propidium Iodide (PI) Staining
2.9. siRNA Design and Transfections
2.10. Real-Time Quantitative PCR (qPCR)
2.11. TUNNEL (TUN) Staining
3. Results
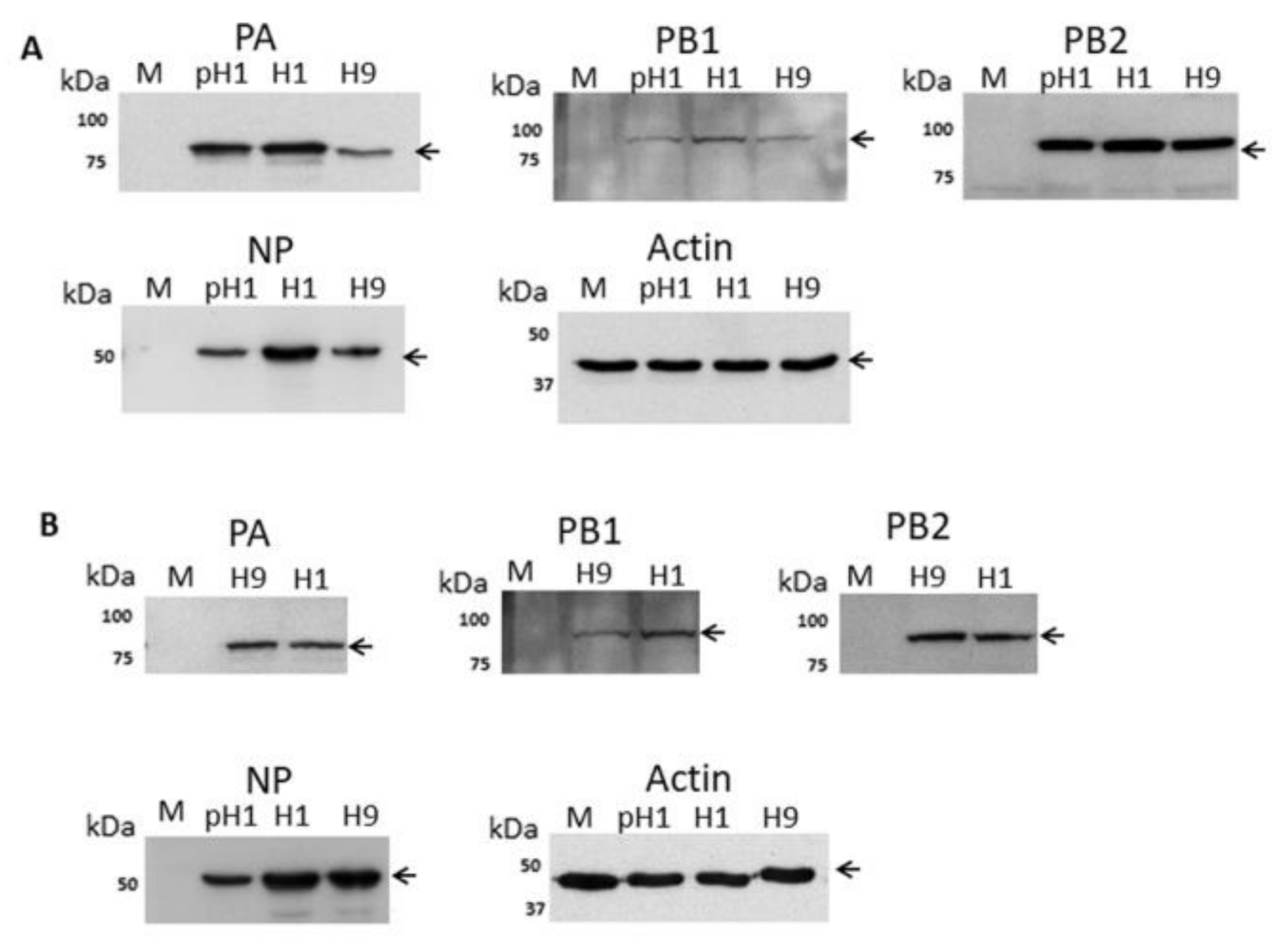
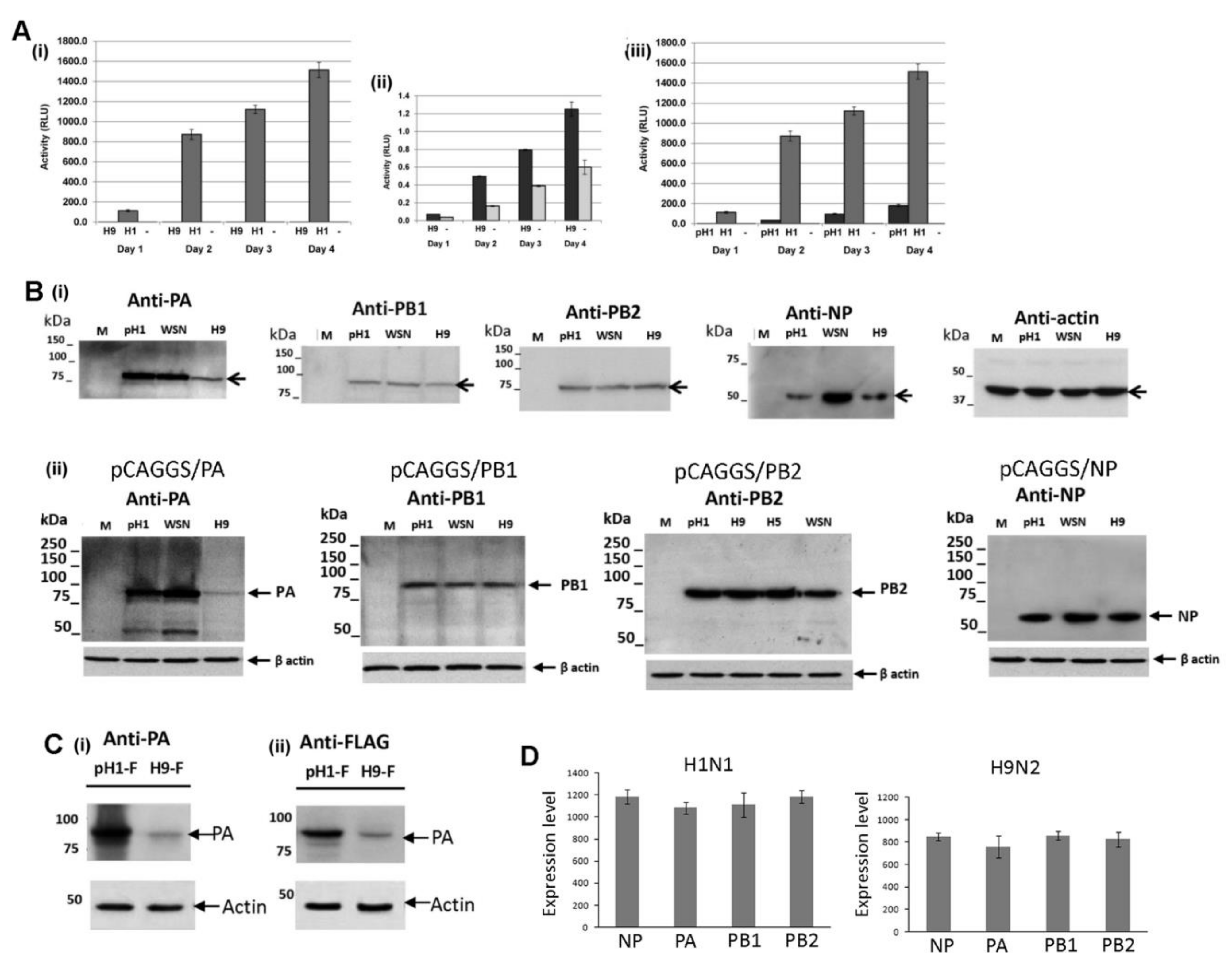
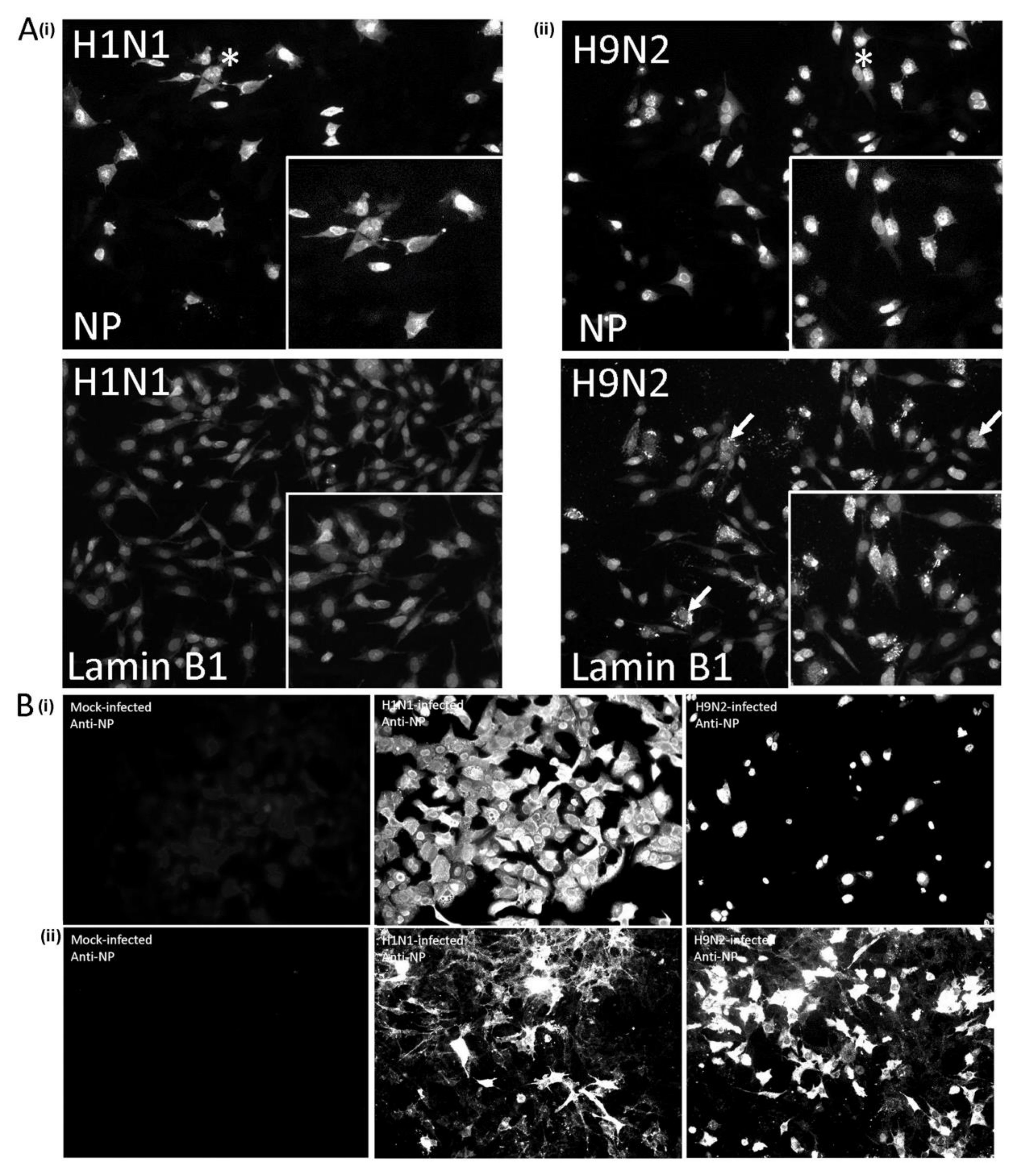
3.1. Expression of the H9N2 Virus Polymerase-Associated Proteins in A549 and CEF Cells
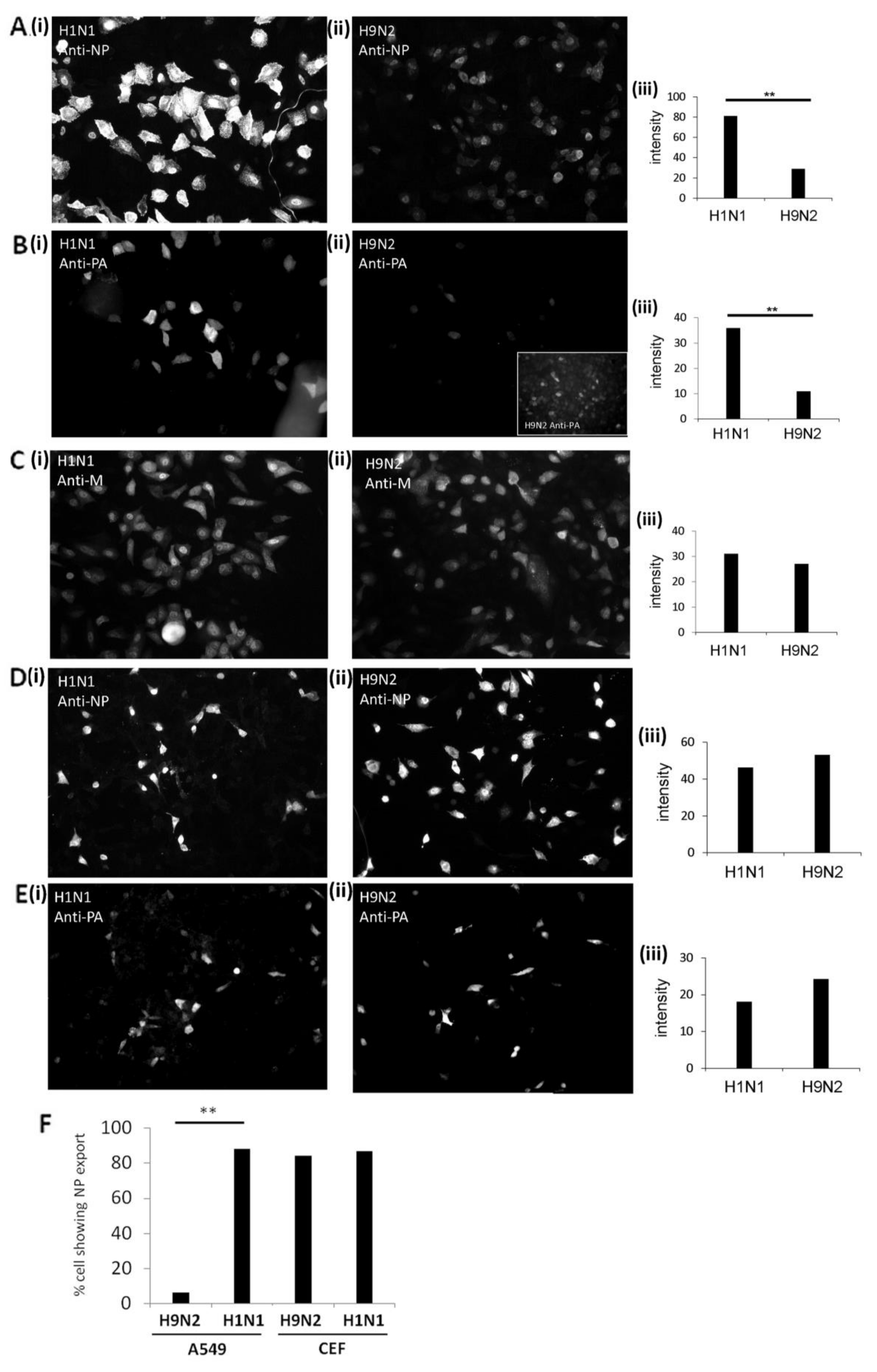
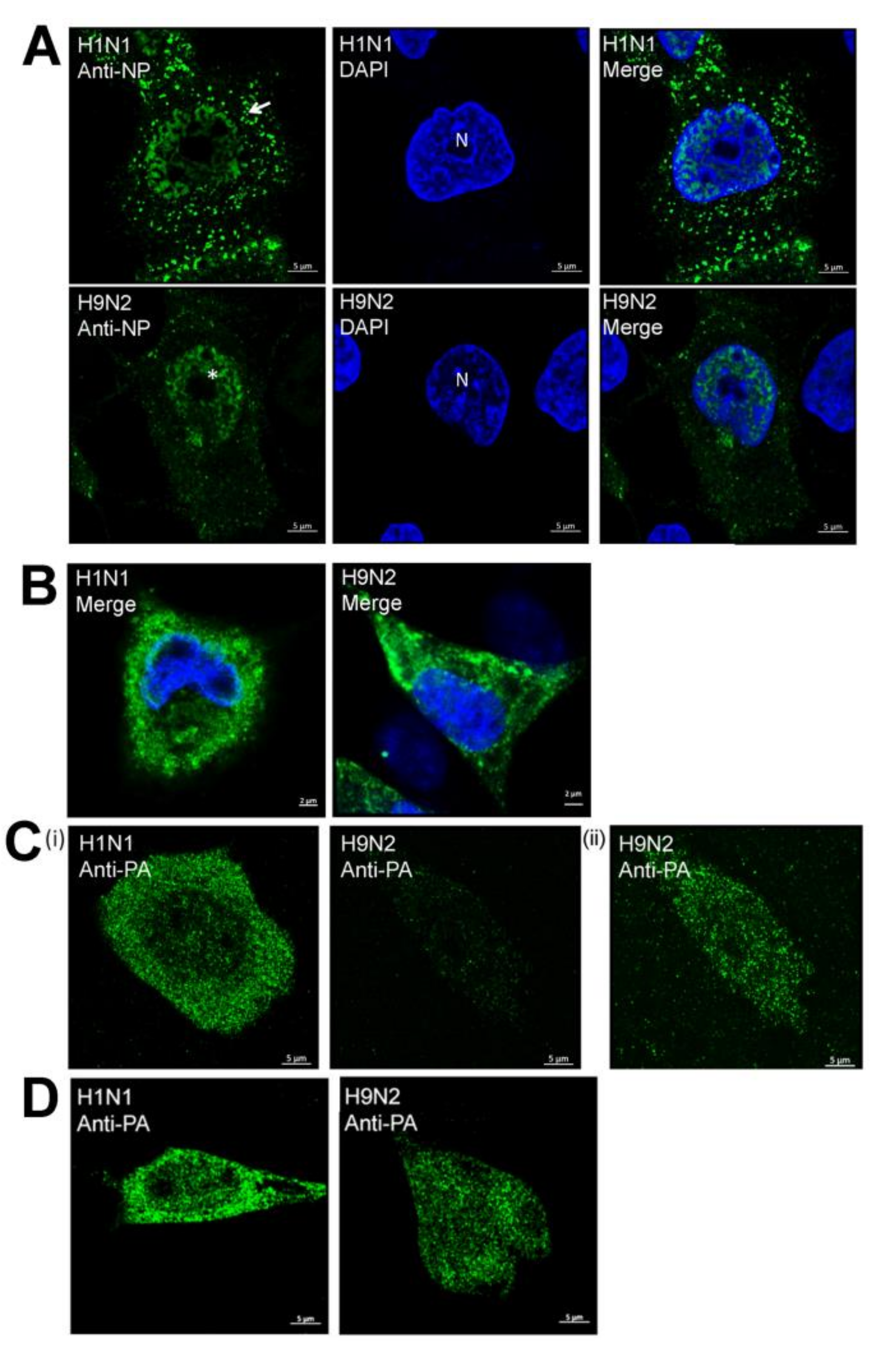
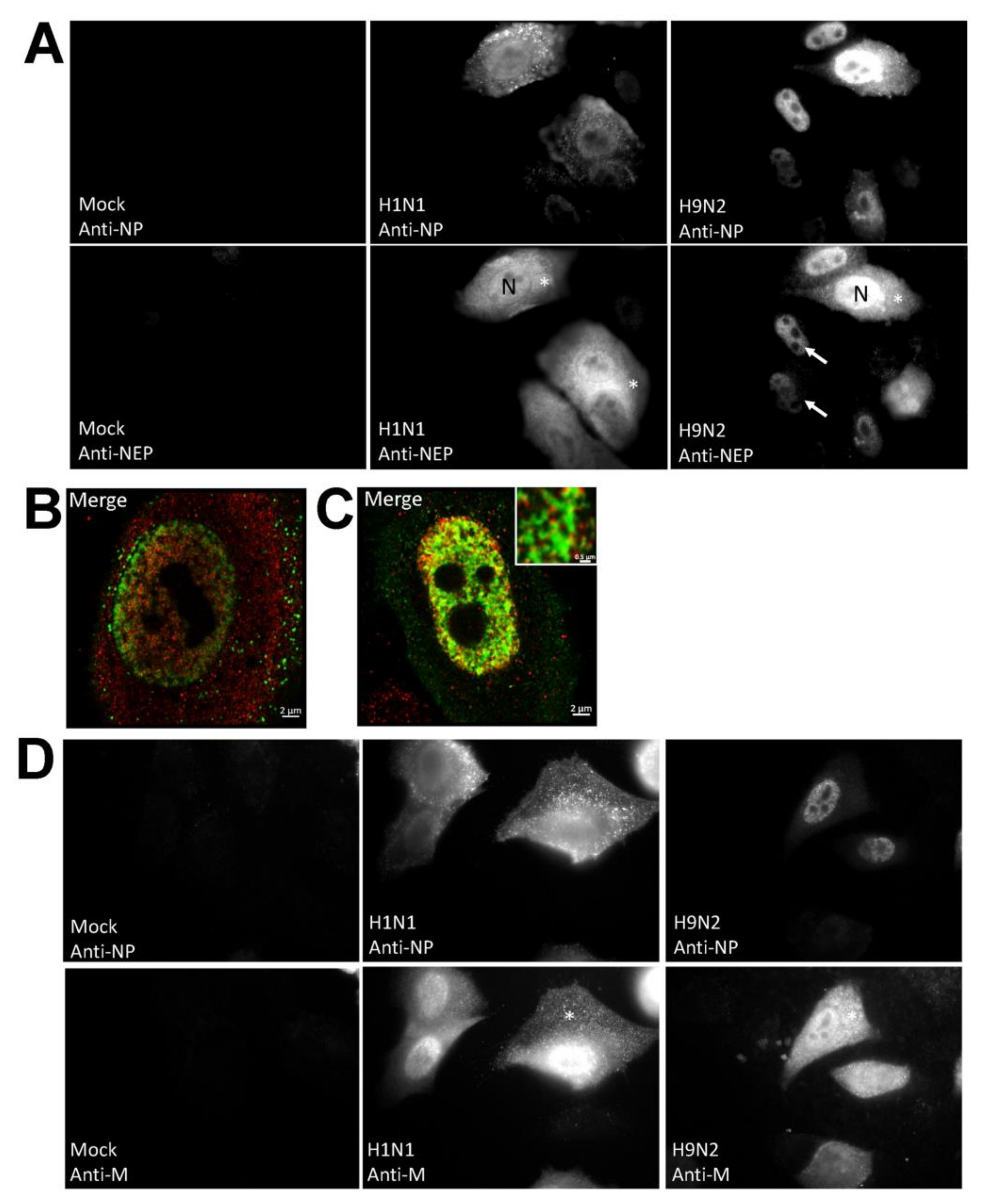
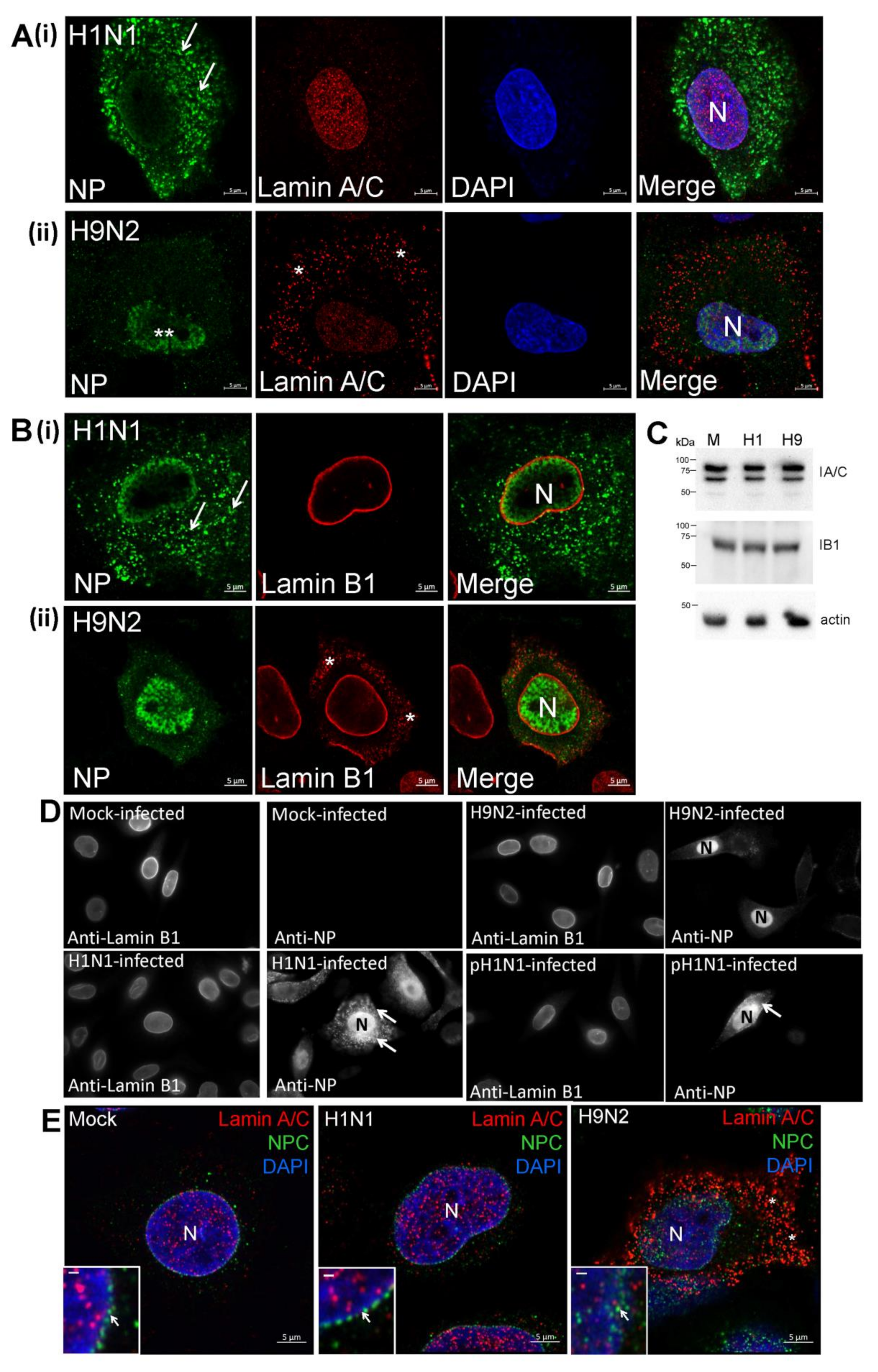
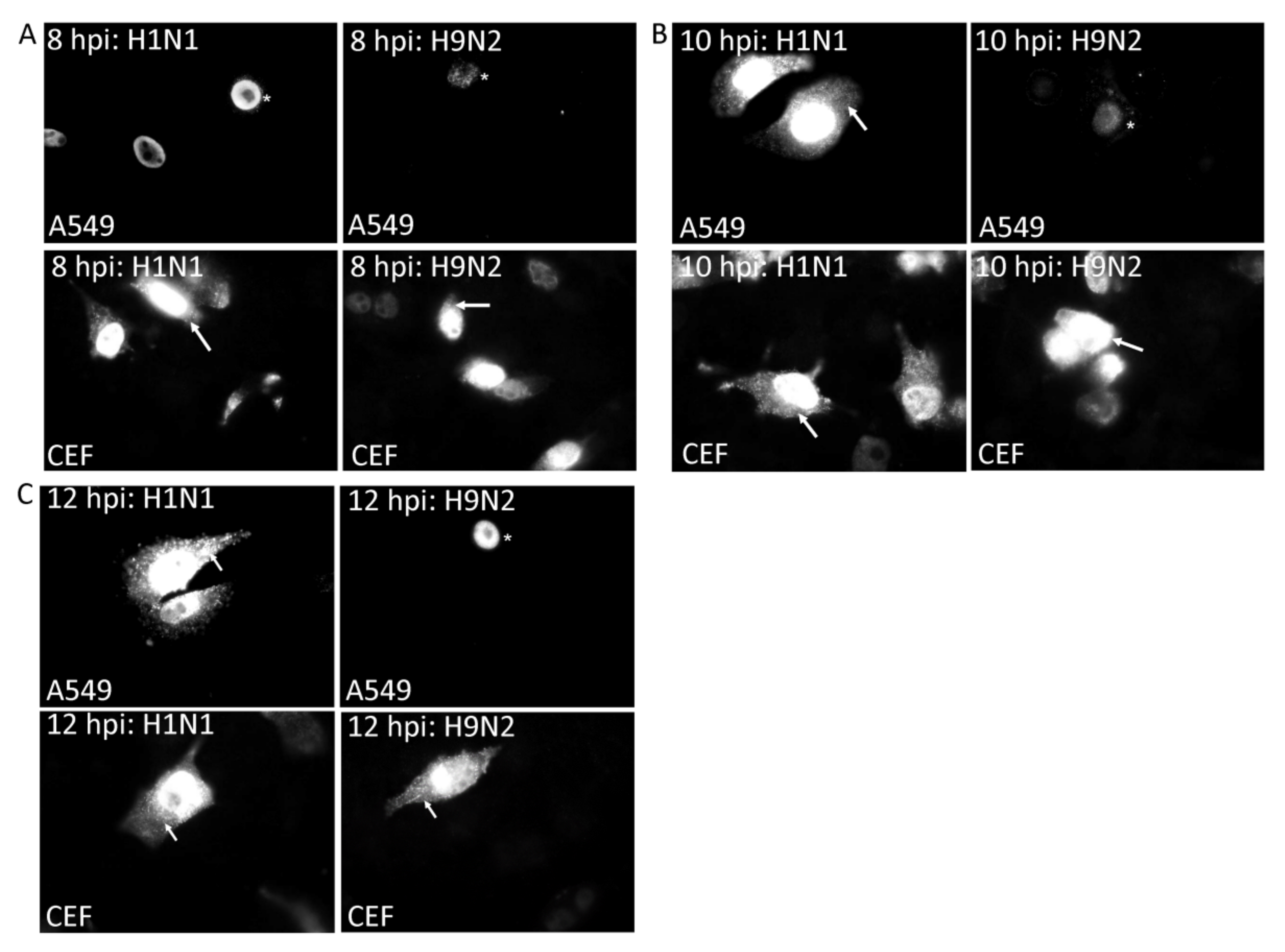
3.2. Impaired Nuclear Export of the RNP Complex Occurs in H9N2 Virus-Infected A549 Cells
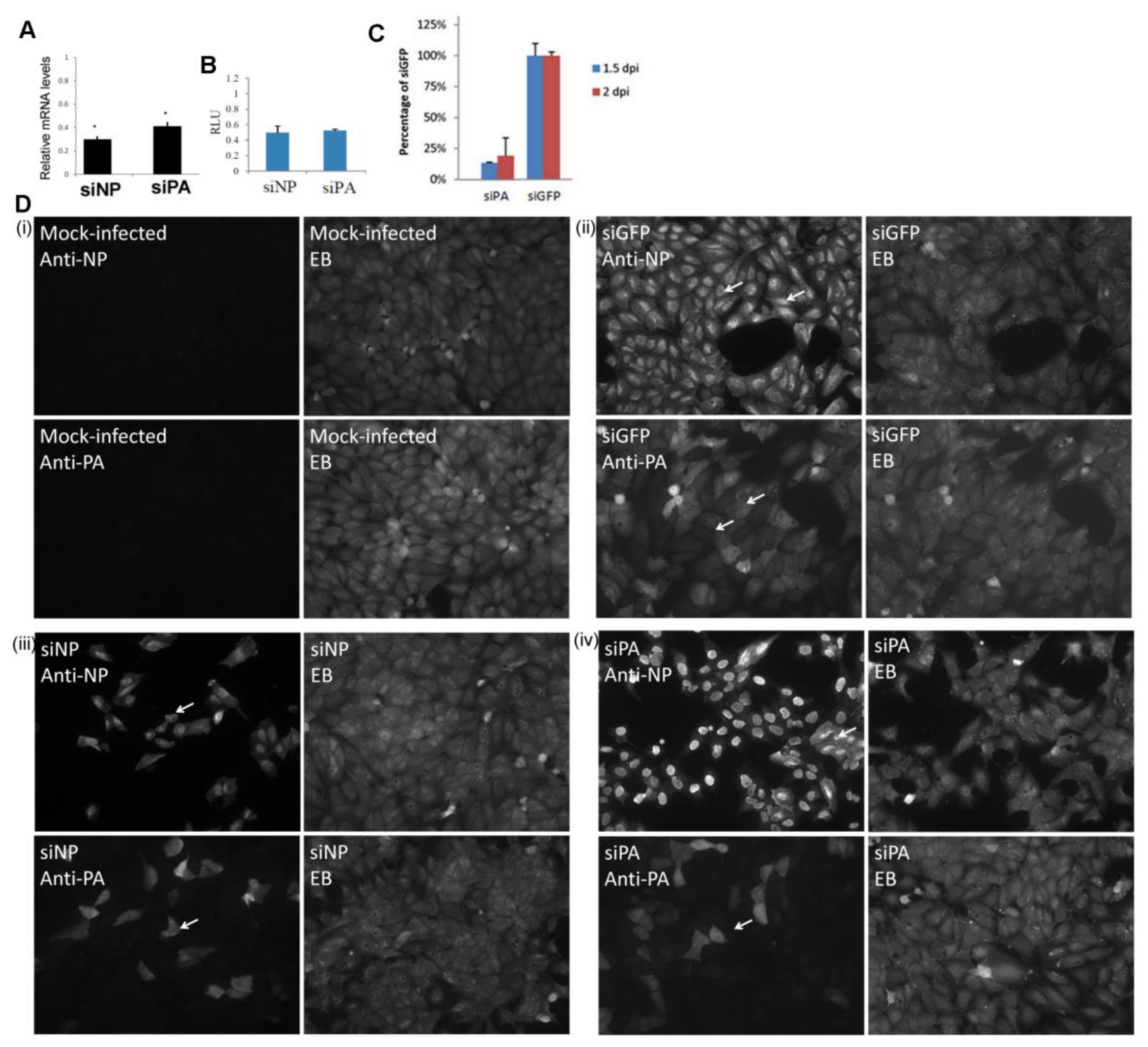
3.3. Reduced Expression of the H9N2 Virus NP and PA Proteins in Human Cells is Independent of Virus Infection
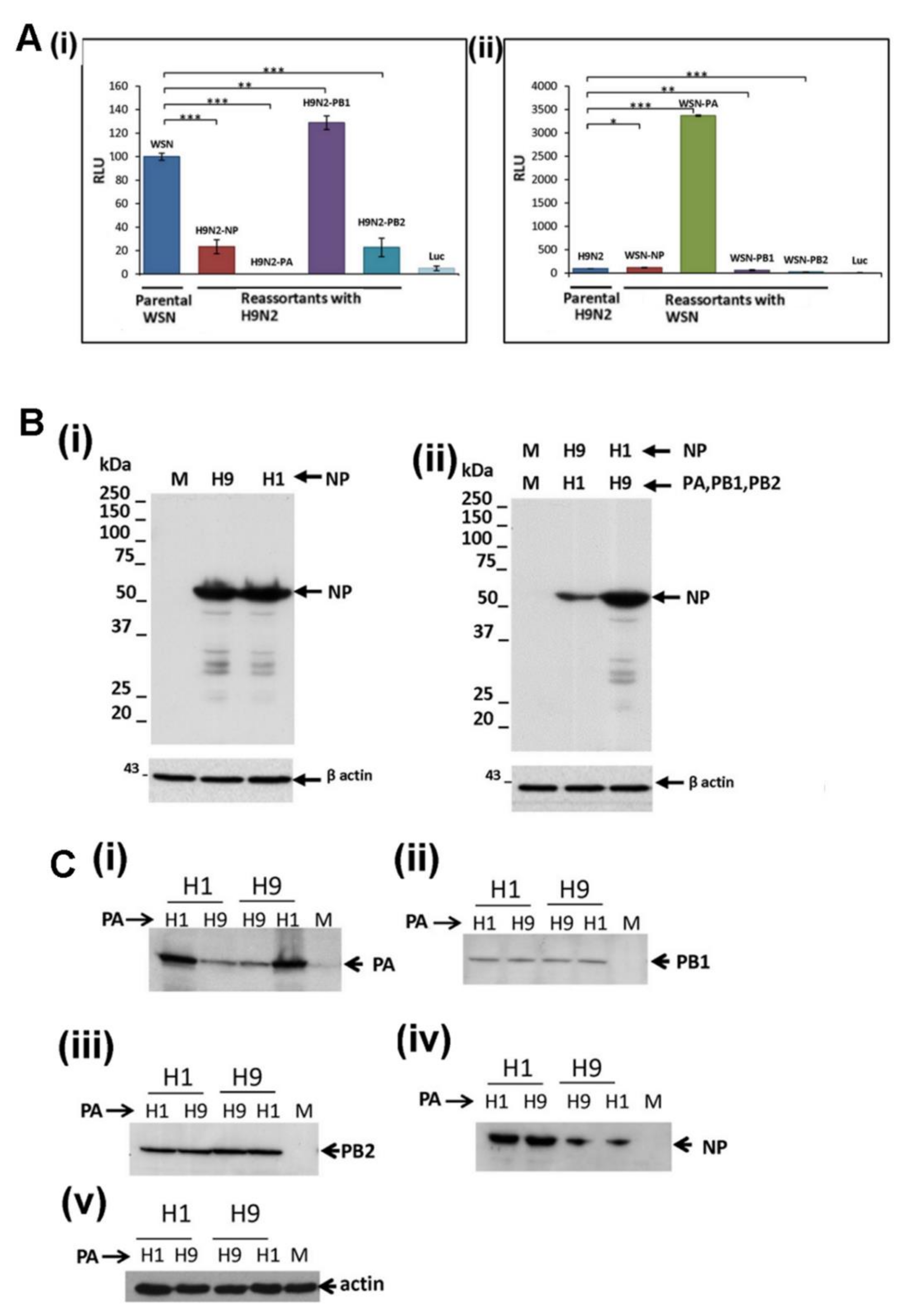
3.4. Reduced Expression of the H9N2 NP and PA Proteins is not Mitigated by Co-Expression with the Other H1N1/WSN Polymerase Proteins
3.5. Efficient Expression of the PA Protein Facilitates Nuclear Export of the RNP Complex
3.6. The H9N2 Virus RNP Complex is Exported from the Nucleus Via a Functional Nuclear Pore Complex
3.7. Efficient Export of the H9N2 Virus RNP Complex in Avian Cells Mitigates Virus-Induced Cytotoxicity in CEF Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boivin, S.; Cusack, S.; Ruigrok, R.W.; Hart, D.J. Influenza A virus polymerase: Structural insights into replication and host adaptation mechanisms. J. Biol. Chem. 2010, 285, 28411–28417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulmanen, I.; Broni, B.A.; Krug, R.M. Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc. Natl. Acad Sci. USA 1981, 78, 7355–7359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukaigawa, J.; Nayak, D.P. Two signals mediate nuclear localization of influenza virus (A/WSN/33) polymerase basic protein 2. J. Virol. 1991, 65, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaas, D.; Patzelt, E.; Kuechler, E. Identification of the cap binding protein of influenza virus. Nucleic Acids Res. 1982, 10, 4803–4812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, A.; Bouvier, D.; Crepin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [Google Scholar] [CrossRef]
- Gonzalez, S.; Zurcher, T.; Ortin, J. Identification of two separate domains in the influenza virus PB1 protein involved in the interaction with the PB2 and PA subunits: A model for the viral RNA polymerase structure. Nucleic Acids Res. 1996, 24, 4456–4463. [Google Scholar] [CrossRef] [Green Version]
- Perez, D.R.; Donis, R.O. Functional analysis of PA binding by influenza a virus PB1: Effects on polymerase activity and viral infectivity. J. Virol. 2001, 75, 8127–8136. [Google Scholar] [CrossRef] [Green Version]
- Ghanem, A.; Mayer, D.; Chase, G.; Tegge, W.; Frank, R.; Kochs, G.; García-Sastre, A.; Schwemmle, M. Peptide-mediated interference with influenza A virus polymerase. J. Virol. 2007, 81, 7801–7804. [Google Scholar] [CrossRef] [Green Version]
- Poole, E.L.; Medcalf, L.; Elton, D.; Digard, P. Evidence that the C-terminal PB2-binding region of the influenza A virus PB1 protein is a discrete alpha-helical domain. Febs Lett. 2007, 581, 5300–5306. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, K.; Obayashi, E.; Kawaguchi, A.; Suzuki, Y.; Tame, J.R.; Nagata, K.; Park, S.Y. Structural insight into the essential PB1-PB2 subunit contact of the influenza virus RNA polymerase. Embo. J. 2009, 28, 1803–1811. [Google Scholar] [CrossRef]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.; Guilligay, D.; Pflug, A.; Malet, H.; Berger, I.; Crépin, T.; Hart, D.; Lunardi, T.; Nanao, M.; Ruigrok, R.W.; et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 2014, 516, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Sun, D.; Liang, H.; Wang, J.; Li, J.; Guo, L.; Wang, X.; Guan, C.; Boruah, B.M.; Yuan, L.; et al. Cryo-EM structure of influenza virus RNA polymerase complex at 4.3 A resolution. Mol. Cell 2015, 57, 925–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, M.W.; Schulze, I.T.; Hirst, G.K.; Hauser, R. Isolation and characterization of the ribonucleoprotein of influenza virus. Virology 1969, 39, 250–259. [Google Scholar] [CrossRef]
- Compans, R.W.; Content, J.; Duesberg, P.H. Structure of the ribonucleoprotein of influenza virus. J. Virol. 1972, 10, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Murti, K.G.; Webster, R.G.; Jones, I.M. Localization of RNA polymerases on influenza viral ribonucleoproteins by immunogold labeling. Virology 1988, 164, 562–566. [Google Scholar] [CrossRef]
- Coloma, R.; Valpuesta, J.M.; Arranz, R.; Carrascosa, J.L.; Ortín, J.; Martín-Benito, J. The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog. 2009, 5, e1000491. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Boutz, P.L.; Nayak, D.P. Influenza virus nucleoprotein interacts with influenza virus polymerase proteins. J. Virol. 1998, 72, 5493–5501. [Google Scholar] [CrossRef] [Green Version]
- Paterson, D.; Fodor, E. Emerging roles for the influenza A virus nuclear export protein (NEP). PLoS Pathog 2012, 8, e1003019. [Google Scholar] [CrossRef] [Green Version]
- Schrauwen, E.J.; Fouchier, R.A. Host adaptation and transmission of influenza A viruses in mammals. Emerg. Microbes. Infect. 2014, 3, e9. [Google Scholar] [CrossRef] [Green Version]
- Manz, B.; Schwemmle, M.; Brunotte, L. Adaptation of avian influenza A virus polymerase in mammals to overcome the host species barrier. J. Virol 2013, 87, 7200–7209. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, T.M.; Te Velthuis, A.J. The RNA-dependent RNA polymerase of the influenza A virus. Future Virol. 2014, 9, 863–876. [Google Scholar] [CrossRef] [Green Version]
- Long, J.S.; Giotis, E.S.; Moncorge, O.; Frise, R.; Mistry, B.; James, J.; Morisson, M.; Iqbal, M.; Vignal, A.; Skinner, M.A.; et al. Species difference in ANP32A underlies influenza A virus polymerase host restriction. Nature 2016, 529, 101–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, G.; Klingel, K.; Otte, A.; Thiele, S.; Hudjetz, B.; Arman-Kalcek, G.; Sauter, M.; Shmidt, T.; Rother, F.; Baumgarte, S.; et al. Differential use of importin-alpha isoforms governs cell tropism and host adaptation of influenza virus. Nat. Commun. 2011, 2, 156. [Google Scholar] [CrossRef]
- Peiris, M.; Yuen, K.Y.; Leung, C.W.; Chan, K.H.; Ip, P.L.; Lai, R.W.; Orr, W.K.; Shortridge, K.F. Human infection with influenza H9N2. Lancet 1999, 354, 916–917. [Google Scholar] [CrossRef]
- Butt, K.M.; Smith, G.J.; Chen, H.; Zhang, L.J.; Leung, Y.H.; Xu, K.M.; Lim, W.; Webster, R.G.; Yuen, K.Y.; Peiris, J.S.; et al. Human infection with an avian H9N2 influenza A virus in Hong Kong in 2003. J. Clin. Microbiol 2005, 43, 5760–5767. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.U.; Anderson, B.D.; Heil, G.L.; Liang, S.; Gray, G.C. A Systematic Review and Meta-Analysis of the Seroprevalence of Influenza A(H9N2) Infection Among Humans. J. Infect Dis. 2015, 212, 562–569. [Google Scholar] [CrossRef]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Shi, W.; Shi, Y.; Wang, D.; Xiao, H.; Li, W.; Bi, Y.; Wu, Y.; Li, X.; Yan, W.; et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: Phylogenetic, structural, and coalescent analyses. Lancet 2013, 381, 1926–1932. [Google Scholar] [CrossRef]
- Cui, L.; Liu, D.; Shi, W.; Pan, J.; Qi, X.; Li, X.; Guo, X.; Zhou, M.; Li, W.; Li, J. Dynamic reassortments and genetic heterogeneity of the human-infecting influenza A (H7N9) virus. Nat. Commun. 2014, 5, 3142. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhou, L.; Zhou, M.; Chen, Z.; Li, F.; Wu, H.; Xiang, N.; Chen, E.; Tang, F.; Wang, D.; et al. Epidemiology of human infections with avian influenza A(H7N9) virus in China. N. Engl. J. Med. 2014, 370, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Yeo, D.S.; Ng, S.H.; Liaw, C.W.; Ng, L.M.; Wee, E.J.; Lim, E.A.; Seah, S.L.; Wong, W.K.; Lim, C.W.; Sugrue, R.J.; et al. Molecular characterization of low pathogenic avian influenza viruses, isolated from food products imported into Singapore. Vet. Microbiol. 2009, 138, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Sutejo, R.; Yeo, D.S.; Myaing, M.Z.; Hui, C.; Xia, J.; Ko, D.; Cheung, P.C.; Tan, B.H.; Sugrue, R.J. Activation of type I and III interferon signalling pathways occurs in lung epithelial cells infected with low pathogenic avian influenza viruses. PLoS ONE 2012, 7, e33732. [Google Scholar] [CrossRef] [PubMed]
- Myaing, M.; Jumat, R.; Huong, T.; Tan, B.H.; Sugrue, R.J. Truncated forms of the PA protein containing only the C-terminal domains are associated with the RNP complex within H1N1 influenza virus particles. J. Gen. Virol 2017, 98, 906–921. [Google Scholar] [CrossRef]
- Berkowitz, D.M.; Kakefuda, T.; Sporn, M. A simple and rapid method for the isolation of enzymatically active HeLa cell nuclei. J. Cell Biol 1969, 42, 851–854. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol 2001, 146, 2275–2289. [Google Scholar] [CrossRef]
- Ge, Q.; McManus, M.T.; Nguyen, T.; Shen, C.H.; Sharp, P.A.; Eisen, H.N.; Chen, J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc. Natl. Acad Sci. USA 2003, 100, 2718–2723. [Google Scholar] [CrossRef] [Green Version]
- Spackman, E.; Senne, D.A.; Myers, T.J.; Bulaga, L.L.; Garber, L.P.; Perdue, M.L.; Lohman, K.; Daum, L.T.; Suarez, D.L. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J. Clin Microbiol 2002, 40, 3256–3260. [Google Scholar] [CrossRef] [Green Version]
- Huong, T.N.; Iyer Ravi, L.; Tan, B.H.; Sugrue, R.J. Evidence for a biphasic mode of respiratory syncytial virus transmission in permissive HEp2 cell monolayers. Virol. J. 2016, 13, 12. [Google Scholar] [CrossRef] [Green Version]
- Shinde, V.; Bridges, C.B.; Uyeki, T.M.; Shu, B.; Balish, A.; Xu, X.; Lindstrom, S.; Gubareva, L.V.; Deyde, V.; Garten, R.J.; et al. Triple-reassortant swine influenza A (H1) in humans in the United States, 2005–2009. N. Engl. J. Med. 2009, 360, 2616–2625. [Google Scholar] [CrossRef] [Green Version]
- Pumroy, R.A.; Ke, S.; Hart, D.J.; Zachariae, U.; Cingolani, G. Molecular determinants for nuclear import of influenza A PB2 by importin alpha isoforms 3 and 7. Structure 2015, 23, 374–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bean, W.J., Jr.; Simpson, R.W. Primary transcription of the influenza virus genome in permissive cells. Virology 1973, 56, 646–651. [Google Scholar] [CrossRef]
- Ge, Q.; Filip, L.; Bai, A.; Nguyen, T.; Eisen, H.N.; Chen, J. Inhibition of influenza virus production in virus-infected mice by RNA interference. Proc. Natl. Acad. Sci. USA 2004, 101, 8676–8681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Z.; Harper, R.; Anunciacion, J.; Yang, Z.; Gao, W.; Qu, B.; Guan, Y.; Cardona, C.J. Host immune and apoptotic responses to avian influenza virus H9N2 in human tracheobronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 24–33. [Google Scholar] [CrossRef]
- Crompton, T.; Peitsch, M.C.; MacDonald, H.R.; Tschopp, J. Propidium iodide staining correlates with the extent of DNA degradation in isolated nuclei. Biochem. Biophys. Res. Commun. 1992, 183, 532–537. [Google Scholar] [CrossRef]
- Nishi, K.; Yoshida, M.; Fujiwara, D.; Nishikawa, M.; Horinouchi, S.; Beppu, T. Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J. Biol. Chem. 1994, 269, 6320–6324. [Google Scholar]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef] [Green Version]
- Liao, T.L.; Wu, C.Y.; Su, W.C.; Jeng, K.S.; Lai, M.M. Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. Embo J. 2010, 29, 3879–3890. [Google Scholar] [CrossRef]
- Chow, K.T.; Gale, M., Jr.; Loo, Y.M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef]
- Finkelstein, D.B.; Mukatira, S.; Mehta, P.K.; Obenauer, J.C.; Su, X.; Webster, R.G.; Naeve, C.W. Persistent host markers in pandemic and H5N1 influenza viruses. J. Virol. 2007, 81, 10292–10299. [Google Scholar] [CrossRef] [Green Version]
- Bussey, K.A.; Desmet, E.A.; Mattiacio, J.L.; Hamilton, A.; Bradel-Tretheway, B.; Bussey, H.E.; Kim, B.; Dewhurst, S.; Takimoto, T. PA residues in the 2009 H1N1 pandemic influenza virus enhance avian influenza virus polymerase activity in mammalian cells. J. Virol. 2011, 85, 7020–7028. [Google Scholar] [CrossRef] [Green Version]
- Miotto, O.; Heiny, A.T.; Albrecht, R.; Garcia-Sastre, A.; Tan, T.W.; August, J.T.; Brusic, V. Complete-proteome mapping of human influenza A adaptive mutations: Implications for human transmissibility of zoonotic strains. PLoS ONE 2010, 5, e9025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingold, H.; Pilpel, Y. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 2011, 7, 481. [Google Scholar] [CrossRef]
- Tuller, T.; Veksler-Lublinsky, I.; Gazit, N.; Kupiec, M.; Ruppin, E.; Ziv-Ukelson, M. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011, 12, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.; Mathews, M.B.; Mohr, I. Tinkering with translation: Protein synthesis in virus-infected cells. Cold Spring Harb Perspect Biol. 2013, 5, a012351. [Google Scholar] [CrossRef] [PubMed]
- Burgui, I.; Yanguez, E.; Sonenberg, N.; Nieto, A. Influenza virus mRNA translation revisited: Is the eIF4E cap-binding factor required for viral mRNA translation? J. Virol. 2007, 81, 12427–12438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanguez, E.; Castello, A.; Welnowska, E.; Carrasco, L.; Goodfellow, I.; Nieto, A. Functional impairment of eIF4A and eIF4G factors correlates with inhibition of influenza virus mRNA translation. Virology 2011, 413, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanguez, E.; Rodriguez, P.; Goodfellow, I.; Nieto, A. Influenza virus polymerase confers independence of the cellular cap-binding factor eIF4E for viral mRNA translation. Virology 2012, 422, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Fodor, E.; Smith, M. The PA subunit is required for efficient nuclear accumulation of the PB1 subunit of the influenza A virus RNA polymerase complex. J. Virol. 2004, 78, 9144–9153. [Google Scholar] [CrossRef] [Green Version]
- Nieto, A.; de la Luna, S.; Barcena, J.; Portela, A.; Ortin, J. Complex. structure of the nuclear translocation signal of influenza virus polymerase PA subunit. J. Gen. Virol 1994, 75, 29–36. [Google Scholar] [CrossRef]
- Wu, S.; Pan, S.; Zhang, L.; Baines, J.; Roller, R.; Ames, J.; Yang, M.; Wang, J.; Chen, D.; Liu, Y.; et al. Herpes Simplex Virus 1 Induces Phosphorylation and Reorganization of Lamin A/C through the gamma1345 Protein That Facilitates Nuclear Egress. J Virol 2016, 90, 10414–10422. [Google Scholar] [PubMed] [Green Version]
- Luo, M.; Zhao, X.; Song, Y.; Cheng, H.; Zhou, R. Nuclear autophagy: An. evolutionarily conserved mechanism of nuclear degradation in the cytoplasm. Autophagy 2016, 12, 1973–1983. [Google Scholar] [CrossRef] [Green Version]
- Gerace, L.; Blobel, G. The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell 1980, 19, 277–287. [Google Scholar] [CrossRef]
- Robijns, J.; Houthaeve, G.; Braeckmans, K.; De Vos, W.H. Loss of Nuclear Envelope Integrity in Aging and Disease. Int. Rev. Cell Mol. Biol. 2018, 336, 205–222. [Google Scholar] [PubMed]
- De Noronha, C.M.; Sherma, N.M.P.; Lin, H.W.; Cavrois, M.V.; Moir, R.D.; Goldman, R.D.; Greene, W.C. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science 2001, 294, 1105–1108. [Google Scholar] [CrossRef]
- De Vos, W.H.; Houben, F.; Kamps, M.; Malhas, A.; Verheyen, F.; Cox, J.; Manders, E.M.; Verstraeten, V.L.; van Steensel, M.A.; Marcelis, C.L.; et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum. Mol Genet. 2011, 20, 4175–4186. [Google Scholar] [CrossRef] [Green Version]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [Green Version]
- De Castro Martin, I.F.; Fournier, G.; Sachse, M.; Pizarro-Cerda, J.; Risco, C.; Naffakh, N. Influenza virus genome reaches the plasma membrane via a modified endoplasmic reticulum and Rab11-dependent vesicles. Nat. Commun 2017, 8, 1396. [Google Scholar] [CrossRef] [Green Version]
- Gerber, M.; Isel, C.; Moules, V.; Marquet, R. Selective packaging of the influenza A genome and consequences for genetic reassortment. Trends Microbiol. 2014, 22, 446–455. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Yeo, D.; Harur Muralidharan, N.; Lai, S.K.; Tong, C.; Tan, B.H.; Sugrue, R.J. Impaired Nuclear Export of the Ribonucleoprotein Complex and Virus-Induced Cytotoxicity Combine to Restrict Propagation of the A/Duck/Malaysia/02/2001 (H9N2) Virus in Human Airway Cells. Cells 2020, 9, 355. https://doi.org/10.3390/cells9020355
Kumar S, Yeo D, Harur Muralidharan N, Lai SK, Tong C, Tan BH, Sugrue RJ. Impaired Nuclear Export of the Ribonucleoprotein Complex and Virus-Induced Cytotoxicity Combine to Restrict Propagation of the A/Duck/Malaysia/02/2001 (H9N2) Virus in Human Airway Cells. Cells. 2020; 9(2):355. https://doi.org/10.3390/cells9020355
Chicago/Turabian StyleKumar, Sriram, Dawn Yeo, Nisha Harur Muralidharan, Soak Kuan Lai, Cathlyn Tong, Boon Huan Tan, and Richard J. Sugrue. 2020. "Impaired Nuclear Export of the Ribonucleoprotein Complex and Virus-Induced Cytotoxicity Combine to Restrict Propagation of the A/Duck/Malaysia/02/2001 (H9N2) Virus in Human Airway Cells" Cells 9, no. 2: 355. https://doi.org/10.3390/cells9020355
APA StyleKumar, S., Yeo, D., Harur Muralidharan, N., Lai, S. K., Tong, C., Tan, B. H., & Sugrue, R. J. (2020). Impaired Nuclear Export of the Ribonucleoprotein Complex and Virus-Induced Cytotoxicity Combine to Restrict Propagation of the A/Duck/Malaysia/02/2001 (H9N2) Virus in Human Airway Cells. Cells, 9(2), 355. https://doi.org/10.3390/cells9020355