The Immuno-Modulatory Effects of Inhibitor of Apoptosis Protein Antagonists in Cancer Immunotherapy

 and
and
Abstract
:1. Inhibitor of Apoptosis Proteins
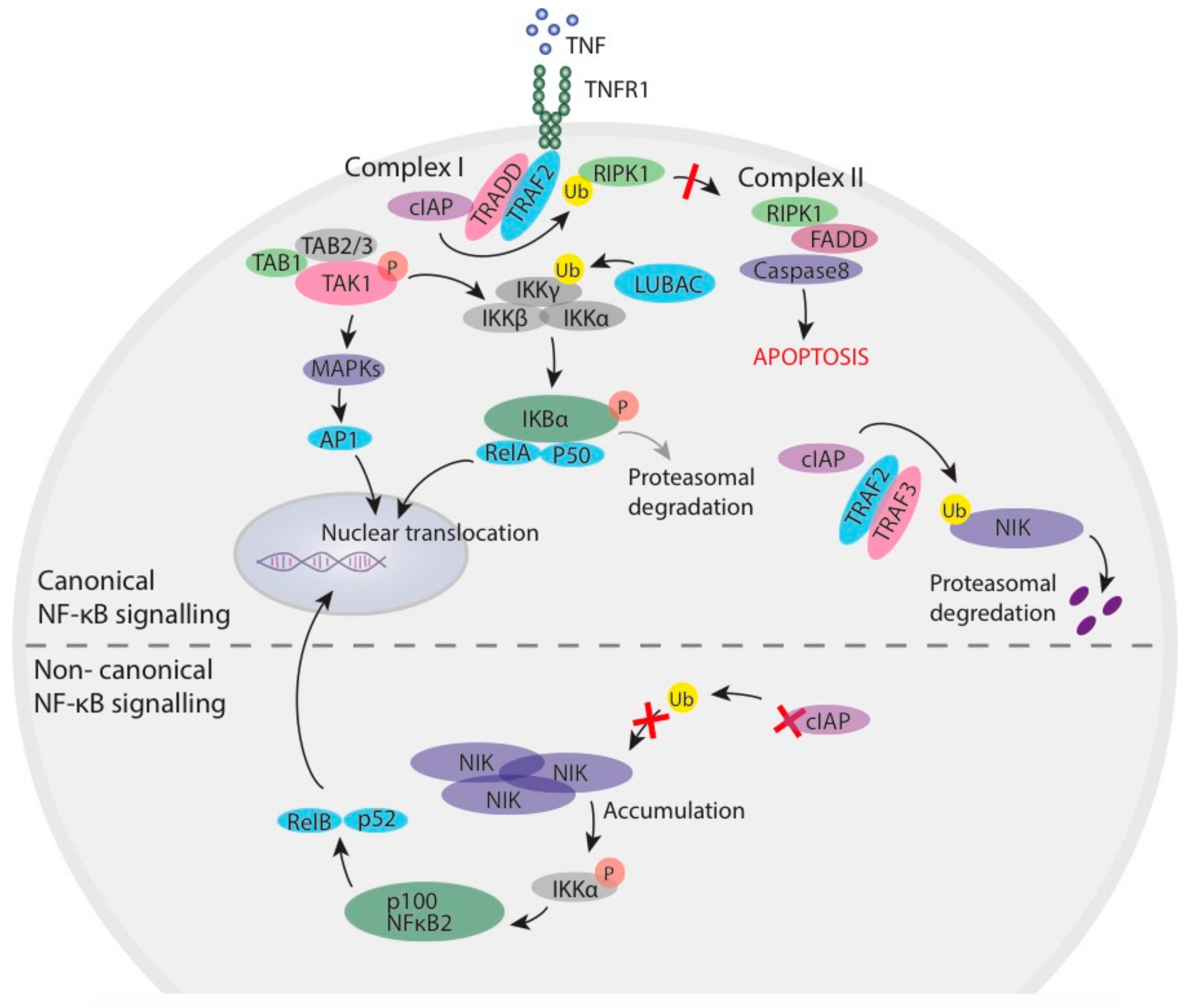
1.1. Inhibitor of Apoptosis Proteins in NF-κB Signalling
1.2. IAPs and Smac-Mimetics in Cancer
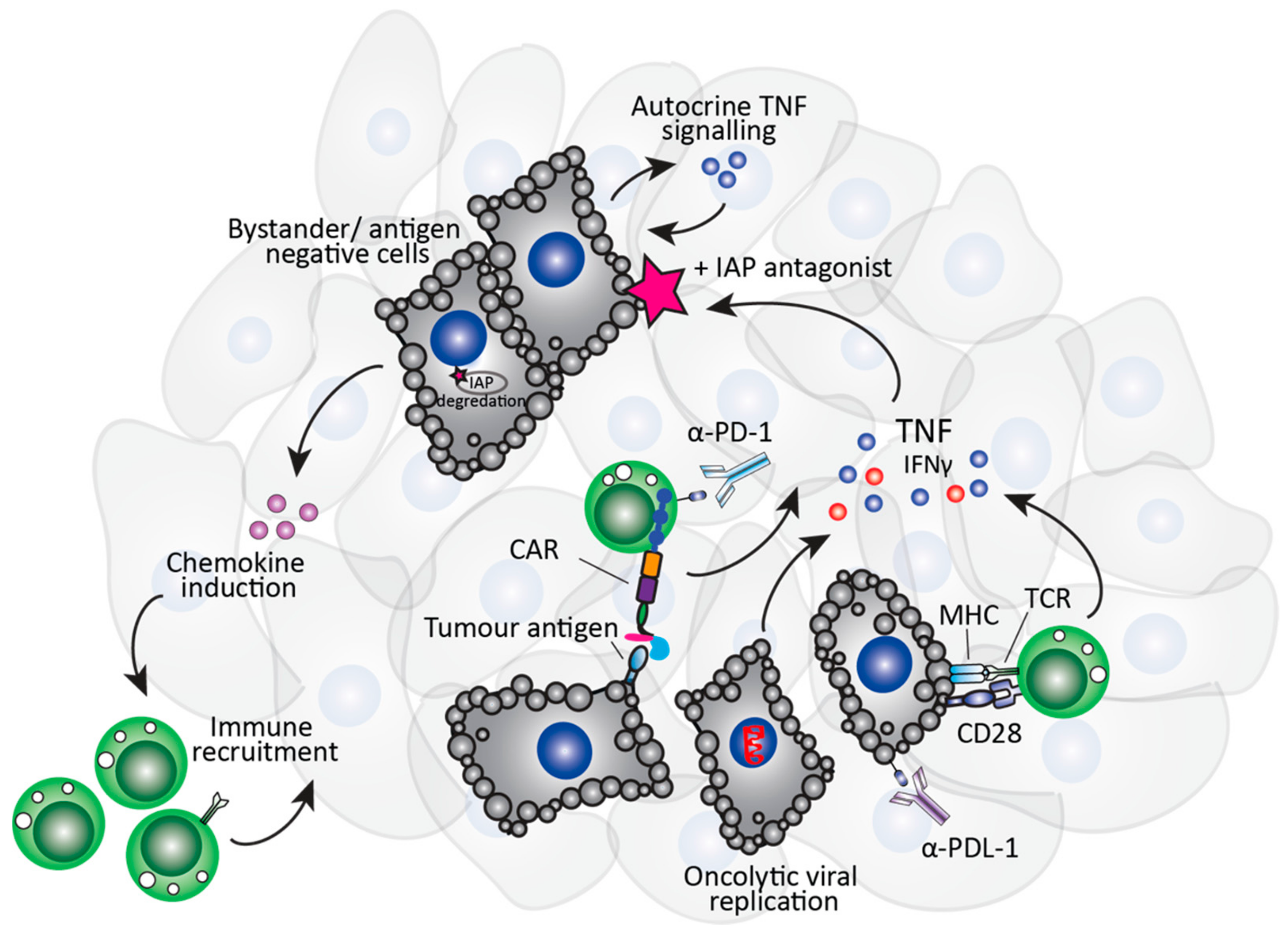
1.3. The Immuno-Modulatory Properties of Smac-Mimetics in Cancer
2. IAP Antagonists and Immunotherapy Approaches
2.1. Role of Smac-Mimetics in T Cell-Specific Immunotherapies
2.2. Role of Smac-Mimetics in NK Cell-Specific Immunotherapies
2.3. Role of Smac-Mimetics in Oncolytic Virus Immunotherapy
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Silke, J.; Meier, P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Silke, J.; Vucic, D. IAP family of cell death and signaling regulators. Methods Enzymol. 2014, 545, 35–65. [Google Scholar] [CrossRef]
- Sun, C.; Cai, M.; Gunasekera, A.H.; Meadows, R.P.; Wang, H.; Chen, J.; Zhang, H.; Wu, W.; Xu, N.; Ng, S.C.; et al. NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature 1999, 401, 818–822. [Google Scholar] [CrossRef]
- Harlin, H.; Reffey, S.B.; Duckett, C.S.; Lindsten, T.; Thompson, C.B. Characterization of XIAP-deficient mice. Mol. Cell Biol. 2001, 21, 3604–3608. [Google Scholar] [CrossRef] [Green Version]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Yang, Y.; Fang, S.; Jensen, J.P.; Weissman, A.M.; Ashwell, J.D. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 2000, 288, 874–877. [Google Scholar] [CrossRef]
- Damgaard, R.B.; Nachbur, U.; Yabal, M.; Wong, W.W.; Fiil, B.K.; Kastirr, M.; Rieser, E.; Rickard, J.A.; Bankovacki, A.; Peschel, C.; et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol. Cell 2012, 46, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Stafford, C.A.; Lawlor, K.E.; Heim, V.J.; Bankovacki, A.; Bernardini, J.P.; Silke, J.; Nachbur, U. IAPs Regulate Distinct Innate Immune Pathways to Co-ordinate the Response to Bacterial Peptidoglycans. Cell Rep. 2018, 22, 1496–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vince, J.E.; Pantaki, D.; Feltham, R.; Mace, P.D.; Cordier, S.M.; Schmukle, A.C.; Davidson, A.J.; Callus, B.A.; Wong, W.W.; Gentle, I.E.; et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-{kappa}b and to prevent tnf-induced apoptosis. J. Biol. Chem. 2009, 284, 35906–35915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.; Kabaleeswaran, V.; Wang, Y.; Cheng, G.; Wu, H. Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: Affinity, specificity, and regulation. Mol. Cell 2010, 38, 101–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwell, K.; Zhang, L.; Workman, L.M.; Ting, A.T.; Iwai, K.; Habelhah, H. Two coordinated mechanisms underlie tumor necrosis factor alpha-induced immediate and delayed IkappaB kinase activation. Mol. Cell Biol. 2013, 33, 1901–1915. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.W.; Gentle, I.E.; Nachbur, U.; Anderton, H.; Vaux, D.L.; Silke, J. RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Differ. 2010, 17, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.J.; Conze, D.B.; Li, T.; Srinivasula, S.M.; Ashwell, J.D. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected]. Nat. Cell Biol. 2006, 8, 398–406. [Google Scholar] [CrossRef]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Dynek, J.N.; Goncharov, T.; Dueber, E.C.; Fedorova, A.V.; Izrael-Tomasevic, A.; Phu, L.; Helgason, E.; Fairbrother, W.J.; Deshayes, K.; Kirkpatrick, D.S.; et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 2010, 29, 4198–4209. [Google Scholar] [CrossRef]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef]
- Tokunaga, F. Linear ubiquitination-mediated NF-kappaB regulation and its related disorders. J. Biochem. 2013, 154, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Iwai, K. LUBAC, a novel ubiquitin ligase for linear ubiquitination, is crucial for inflammation and immune responses. Microbes Infect. 2012, 14, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef]
- Darding, M.; Meier, P. IAPs: Guardians of RIPK1. Cell Death Differ. 2012, 19, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Wong, W.W.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M.; et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Thomas, R.M.; Suzuki, H.; De Brabander, J.K.; Wang, X.; Harran, P.G. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science 2004, 305, 1471–1474. [Google Scholar] [CrossRef] [Green Version]
- Geserick, P.; Hupe, M.; Moulin, M.; Wong, W.W.; Feoktistova, M.; Kellert, B.; Gollnick, H.; Silke, J.; Leverkus, M. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 2009, 187, 1037–1054. [Google Scholar] [CrossRef] [Green Version]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Mirza, N.N.; Garbitt, V.M.; Sharik, M.E.; Dueck, A.C.; Asmann, Y.W.; Akhmetzyanova, I.; Kosiorek, H.E.; Calcinotto, A.; Riggs, D.L.; et al. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat. Med. 2016, 22, 1411–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.M.; Busselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017, 22, 898–919. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Feng, Q.; Kim, J.M.; Schneiderman, D.; Liston, P.; Li, M.; Vanderhyden, B.; Faught, W.; Fung, M.F.; Senterman, M.; et al. Human ovarian cancer and cisplatin resistance: Possible role of inhibitor of apoptosis proteins. Endocrinology 2001, 142, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Tamm, I.; Kornblau, S.M.; Segall, H.; Krajewski, S.; Welsh, K.; Kitada, S.; Scudiero, D.A.; Tudor, G.; Qui, Y.H.; Monks, A.; et al. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin. Cancer Res. 2000, 6, 1796–1803. [Google Scholar] [PubMed]
- Ramp, U.; Krieg, T.; Caliskan, E.; Mahotka, C.; Ebert, T.; Willers, R.; Gabbert, H.E.; Gerharz, C.D. XIAP expression is an independent prognostic marker in clear-cell renal carcinomas. Hum. Pathol. 2004, 35, 1022–1028. [Google Scholar] [CrossRef]
- Oost, T.K.; Sun, C.; Armstrong, R.C.; Al-Assaad, A.S.; Betz, S.F.; Deckwerth, T.L.; Ding, H.; Elmore, S.W.; Meadows, R.P.; Olejniczak, E.T.; et al. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J. Med. Chem. 2004, 47, 4417–4426. [Google Scholar] [CrossRef]
- Condon, S.M.; Mitsuuchi, Y.; Deng, Y.; LaPorte, M.G.; Rippin, S.R.; Haimowitz, T.; Alexander, M.D.; Kumar, P.T.; Hendi, M.S.; Lee, Y.H.; et al. Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J. Med. Chem. 2014, 57, 3666–3677. [Google Scholar] [CrossRef]
- Sharma, S.K.; Straub, C.; Zawel, L. Development of Peptidomimetics Targeting IAPs. Int. J. Pept. Res. Ther. 2006, 12, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Kipp, R.A.; Case, M.A.; Wist, A.D.; Cresson, C.M.; Carrell, M.; Griner, E.; Wiita, A.; Albiniak, P.A.; Chai, J.; Shi, Y.; et al. Molecular targeting of inhibitor of apoptosis proteins based on small molecule mimics of natural binding partners. Biochemistry 2002, 41, 7344–7349. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.J.; Huerta, S. Smac mimetics as new cancer therapeutics. Anticancer Drugs 2009, 20, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Gaither, A.; Porter, D.; Yao, Y.; Borawski, J.; Yang, G.; Donovan, J.; Sage, D.; Slisz, J.; Tran, M.; Straub, C.; et al. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-alpha signaling. Cancer Res. 2007, 67, 11493–11498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dueber, E.C.; Schoeffler, A.J.; Lingel, A.; Elliott, J.M.; Fedorova, A.V.; Giannetti, A.M.; Zobel, K.; Maurer, B.; Varfolomeev, E.; Wu, P.; et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science 2011, 334, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Feltham, R.; Bettjeman, B.; Budhidarmo, R.; Mace, P.D.; Shirley, S.; Condon, S.M.; Chunduru, S.K.; McKinlay, M.A.; Vaux, D.L.; Silke, J.; et al. Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J. Biol. Chem. 2011, 286, 17015–17028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatani, Y.; Kleffmann, T.; Linke, K.; Condon, S.M.; Hinds, M.G.; Day, C.L. Regulation of ubiquitin transfer by XIAP, a dimeric RING E3 ligase. Biochem. J. 2013, 450, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Flygare, J.A.; Fairbrother, W.J. Small-molecule pan-IAP antagonists: A patent review. Expert Opin. Ther. Pat. 2010, 20, 251–267. [Google Scholar] [CrossRef]
- Mitsuuchi, Y.; Benetatos, C.A.; Deng, Y.; Haimowitz, T.; Beck, S.C.; Arnone, M.R.; Kapoor, G.S.; Seipel, M.E.; Chunduru, S.K.; McKinlay, M.A.; et al. Bivalent IAP antagonists, but not monovalent IAP antagonists, inhibit TNF-mediated NF-kappaB signaling by degrading TRAF2-associated cIAP1 in cancer cells. Cell Death Discov. 2017, 3, 16046. [Google Scholar] [CrossRef] [Green Version]
- Petersen, S.L.; Wang, L.; Yalcin-Chin, A.; Li, L.; Peyton, M.; Minna, J.; Harran, P.; Wang, X. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 2007, 12, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Beug, S.T.; Tang, V.A.; LaCasse, E.C.; Cheung, H.H.; Beauregard, C.E.; Brun, J.; Nuyens, J.P.; Earl, N.; St-Jean, M.; Holbrook, J.; et al. Smac mimetics and innate immune stimuli synergize to promote tumor death. Nat. Biotechnol. 2014, 32, 182–190. [Google Scholar] [CrossRef] [Green Version]
- McComb, S.; Aguade-Gorgorio, J.; Harder, L.; Marovca, B.; Cario, G.; Eckert, C.; Schrappe, M.; Stanulla, M.; von Stackelberg, A.; Bourquin, J.P.; et al. Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci. Transl. Med. 2016, 8, 339–370. [Google Scholar] [CrossRef] [PubMed]
- Richmond, J.; Robbins, A.; Evans, K.; Beck, D.; Kurmasheva, R.T.; Billups, C.A.; Carol, H.; Heatley, S.; Sutton, R.; Marshall, G.M.; et al. Acute Sensitivity of Ph-like Acute Lymphoblastic Leukemia to the SMAC-Mimetic Birinapant. Cancer Res. 2016, 76, 4579–4591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brumatti, G.; Ma, C.; Lalaoui, N.; Nguyen, N.Y.; Navarro, M.; Tanzer, M.C.; Richmond, J.; Ghisi, M.; Salmon, J.M.; Silke, N.; et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci. Transl. Med. 2016, 8, 339ra369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, C.; Tromp, J.M.; van Laar, J.; Thijssen, R.; Elias, J.A.; Malara, A.; Krippner-Heidenreich, A.; Silke, J.; van Oers, M.H.; Eldering, E. CLL cells are resistant to smac mimetics because of an inability to form a ripoptosome complex. Cell Death Dis. 2013, 4, e782. [Google Scholar] [CrossRef] [Green Version]
- Noonan, A.M.; Bunch, K.P.; Chen, J.Q.; Herrmann, M.A.; Lee, J.M.; Kohn, E.C.; O’Sullivan, C.C.; Jordan, E.; Houston, N.; Takebe, N.; et al. Pharmacodynamic markers and clinical results from the phase 2 study of the SMAC mimetic birinapant in women with relapsed platinum-resistant or -refractory epithelial ovarian cancer. Cancer 2016, 122, 588–597. [Google Scholar] [CrossRef]
- Moulin, M.; Anderton, H.; Voss, A.K.; Thomas, T.; Wong, W.W.; Bankovacki, A.; Feltham, R.; Chau, D.; Cook, W.D.; Silke, J.; et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 2012, 31, 1679–1691. [Google Scholar] [CrossRef] [Green Version]
- Gardam, S.; Turner, V.M.; Anderton, H.; Limaye, S.; Basten, A.; Koentgen, F.; Vaux, D.L.; Silke, J.; Brink, R. Deletion of cIAP1 and cIAP2 in murine B lymphocytes constitutively activates cell survival pathways and inactivates the germinal center response. Blood 2011, 117, 4041–4051. [Google Scholar] [CrossRef] [Green Version]
- Conze, D.B.; Zhao, Y.; Ashwell, J.D. Non-canonical NF-kappaB activation and abnormal B cell accumulation in mice expressing ubiquitin protein ligase-inactive c-IAP2. PLoS Biol. 2010, 8, e1000518. [Google Scholar] [CrossRef]
- Giardino Torchia, M.L.; Conze, D.B.; Ashwell, J.D. c-IAP1 and c-IAP2 redundancy differs between T and B cells. PLoS ONE 2013, 8, e66161. [Google Scholar] [CrossRef]
- Ebert, G.; Preston, S.; Allison, C.; Cooney, J.; Toe, J.G.; Stutz, M.D.; Ojaimi, S.; Scott, H.W.; Baschuk, N.; Nachbur, U.; et al. Cellular inhibitor of apoptosis proteins prevent clearance of hepatitis B virus. Proc. Natl. Acad. Sci. USA 2015, 112, 5797–5802. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Casola, S.; Kutok, J.L.; Rajewsky, K.; Schmidt-Supprian, M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J. Immunol. 2004, 173, 2245–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.W.; Vince, J.E.; Lalaoui, N.; Lawlor, K.E.; Chau, D.; Bankovacki, A.; Anderton, H.; Metcalf, D.; O’Reilly, L.; Jost, P.J.; et al. cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood 2014, 123, 2562–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinesh, G.G.; Chunduru, S.; Kamat, A.M. Smac mimetic enables the anticancer action of BCG-stimulated neutrophils through TNF-alpha but not through TRAIL and FasL. J. Leukoc. Biol. 2012, 92, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecis, D.; De Cesare, M.; Perego, P.; Conti, A.; Corna, E.; Drago, C.; Seneci, P.; Walczak, H.; Colombo, M.P.; Delia, D.; et al. Smac mimetics induce inflammation and necrotic tumour cell death by modulating macrophage activity. Cell Death Dis. 2013, 4, e920. [Google Scholar] [CrossRef] [Green Version]
- Rettinger, E.; Glatthaar, A.; Abhari, B.A.; Oelsner, S.; Pfirrmann, V.; Huenecke, S.; Kuci, S.; Kreyenberg, H.; Willasch, A.M.; Klingebiel, T.; et al. SMAC Mimetic BV6 Enables Sensitization of Resistant Tumor Cells but also Affects Cytokine-Induced Killer (CIK) Cells: A Potential Challenge for Combination Therapy. Front. Pediatr. 2014, 2, 75. [Google Scholar] [CrossRef] [Green Version]
- Bake, V.; Roesler, S.; Eckhardt, I.; Belz, K.; Fulda, S. Synergistic interaction of Smac mimetic and IFNalpha to trigger apoptosis in acute myeloid leukemia cells. Cancer Lett. 2014, 355, 224–231. [Google Scholar] [CrossRef]
- Hao, Q.; Tang, H. Interferon-gamma and Smac mimetics synergize to induce apoptosis of lung cancer cells in a TNFalpha-independent manner. Cancer Cell Int. 2018, 18, 84. [Google Scholar] [CrossRef]
- Tanzer, M.C.; Khan, N.; Rickard, J.A.; Etemadi, N.; Lalaoui, N.; Spall, S.K.; Hildebrand, J.M.; Segal, D.; Miasari, M.; Chau, D.; et al. Combination of IAP antagonist and IFNgamma activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell Death Differ. 2017, 24, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, V.; Painuly, U.; Kimlinger, T.; Haug, J.; Rajkumar, S.V.; Kumar, S. Inhibitor of apoptosis proteins as therapeutic targets in multiple myeloma. Leukemia 2014, 28, 1519–1528. [Google Scholar] [CrossRef] [Green Version]
- Shekhar, T.M.; Miles, M.A.; Gupte, A.; Taylor, S.; Tascone, B.; Walkley, C.R.; Hawkins, C.J. IAP antagonists sensitize murine osteosarcoma cells to killing by TNFalpha. Oncotarget 2016, 7, 33866–33886. [Google Scholar] [CrossRef] [Green Version]
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 3103–3110. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Schilder, R.J.; Martin, L.P.; Levin, M.; Graham, M.A.; Weng, D.E.; Adjei, A.A. A Phase I Study of the SMAC-Mimetic Birinapant in Adults with Refractory Solid Tumors or Lymphoma. Mol. Cancer Ther. 2015, 14, 2569–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beug, S.T.; LaCasse, E.C.; Korneluk, R.G. Smac mimetics combined with innate immune stimuli create the perfect cytokine storm to kill tumor cells. Oncoimmunology 2014, 3, e28541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carretero-Gonzalez, A.; Lora, D.; Ghanem, I.; Zugazagoitia, J.; Castellano, D.; Sepulveda, J.M.; Lopez-Martin, J.A.; Paz-Ares, L.; de Velasco, G. Analysis of response rate with ANTI PD1/PD-L1 monoclonal antibodies in advanced solid tumors: A meta-analysis of randomized clinical trials. Oncotarget 2018, 9, 8706–8715. [Google Scholar] [CrossRef] [Green Version]
- Acuto, O.; Michel, F. CD28-mediated co-stimulation: A quantitative support for TCR signalling. Nat. Rev. Immunol. 2003, 3, 939–951. [Google Scholar] [CrossRef]
- Khoshnan, A.; Tindell, C.; Laux, I.; Bae, D.; Bennett, B.; Nel, A.E. The NF-kappa B cascade is important in Bcl-xL expression and for the anti-apoptotic effects of the CD28 receptor in primary human CD4+ lymphocytes. J. Immunol. 2000, 165, 1743–1754. [Google Scholar] [CrossRef] [Green Version]
- Varfolomeev, E.; Goncharov, T.; Maecker, H.; Zobel, K.; Komuves, L.G.; Deshayes, K.; Vucic, D. Cellular inhibitors of apoptosis are global regulators of NF-kappaB and MAPK activation by members of the TNF family of receptors. Sci. Signal. 2012, 5, 22. [Google Scholar] [CrossRef]
- Knights, A.J.; Fucikova, J.; Pasam, A.; Koernig, S.; Cebon, J. Inhibitor of apoptosis protein (IAP) antagonists demonstrate divergent immunomodulatory properties in human immune subsets with implications for combination therapy. Cancer Immunol. Immunother. 2013, 62, 321–335. [Google Scholar] [CrossRef]
- Dougan, M.; Dougan, S.; Slisz, J.; Firestone, B.; Vanneman, M.; Draganov, D.; Goyal, G.; Li, W.; Neuberg, D.; Blumberg, R.; et al. IAP inhibitors enhance co-stimulation to promote tumor immunity. J. Exp. Med. 2010, 207, 2195–2206. [Google Scholar] [CrossRef]
- Pan, W.; Luo, Q.; Yan, X.; Yuan, L.; Yi, H.; Zhang, L.; Li, B.; Zhang, Y.; Sun, J.; Qiu, M.Z.; et al. A novel SMAC mimetic APG-1387 exhibits dual antitumor effect on HBV-positive hepatocellular carcinoma with high expression of cIAP2 by inducing apoptosis and enhancing innate anti-tumor immunity. Biochem. Pharmacol. 2018, 154, 127–135. [Google Scholar] [CrossRef]
- Rizk, J.; Kaplinsky, J.; Agerholm, R.; Kadekar, D.; Ivars, F.; Agace, W.W.; Wong, W.W.; Szucs, M.J.; Myers, S.A.; Carr, S.A.; et al. SMAC mimetics promote NIK-dependent inhibition of CD4(+) TH17 cell differentiation. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emeagi, P.U.; Van Lint, S.; Goyvaerts, C.; Maenhout, S.; Cauwels, A.; McNeish, I.A.; Bos, T.; Heirman, C.; Thielemans, K.; Aerts, J.L.; et al. Proinflammatory characteristics of SMAC/DIABLO-induced cell death in antitumor therapy. Cancer Res. 2012, 72, 1342–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearney, C.J.; Vervoort, S.J.; Hogg, S.J.; Ramsbottom, K.M.; Freeman, A.J.; Lalaoui, N.; Pijpers, L.; Michie, J.; Brown, K.K.; Knight, D.A.; et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vredevoogd, D.W.; Kuilman, T.; Ligtenberg, M.A.; Boshuizen, J.; Stecker, K.E.; de Bruijn, B.; Krijgsman, O.; Huang, X.; Kenski, J.C.N.; Lacroix, R.; et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell 2019, 178, 585–599e515. [Google Scholar] [CrossRef]
- Kearney, C.J.; Lalaoui, N.; Freeman, A.J.; Ramsbottom, K.M.; Silke, J.; Oliaro, J. PD-L1 and IAPs co-operate to protect tumors from cytotoxic lymphocyte-derived TNF. Cell Death Differ. 2017, 24, 1705–1716. [Google Scholar] [CrossRef] [Green Version]
- Beug, S.T.; Beauregard, C.E.; Healy, C.; Sanda, T.; St-Jean, M.; Chabot, J.; Walker, D.E.; Mohan, A.; Earl, N.; Lun, X.; et al. Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Michie, J.; Beavis, P.A.; Freeman, A.J.; Vervoort, S.J.; Ramsbottom, K.M.; Narasimhan, V.; Lelliott, E.J.; Lalaoui, N.; Ramsay, R.G.; Johnstone, R.W.; et al. Antagonism of IAPs Enhances CAR T-cell Efficacy. Cancer Immunol. Res. 2019, 7, 183–192. [Google Scholar] [CrossRef]
- Mardiana, S.; Lai, J.; House, I.G.; Beavis, P.A.; Darcy, P.K. Switching on the green light for chimeric antigen receptor T-cell therapy. Clin. Transl. Immunol. 2019, 8, e1046. [Google Scholar] [CrossRef]
- Beavis, P.A.; Henderson, M.A.; Giuffrida, L.; Mills, J.K.; Sek, K.; Cross, R.S.; Davenport, A.J.; John, L.B.; Mardiana, S.; Slaney, C.Y.; et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Invest. 2017, 127, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Kearney, C.J.; Sheridan, C.; Cullen, S.P.; Tynan, G.A.; Logue, S.E.; Afonina, I.S.; Vucic, D.; Lavelle, E.C.; Martin, S.J. Inhibitor of apoptosis proteins (IAPs) and their antagonists regulate spontaneous and tumor necrosis factor (TNF)-induced proinflammatory cytokine and chemokine production. J. Biol. Chem. 2013, 288, 4878–4890. [Google Scholar] [CrossRef] [Green Version]
- Dobson, C.C.; Naing, T.; Beug, S.T.; Faye, M.D.; Chabot, J.; St-Jean, M.; Walker, D.E.; LaCasse, E.C.; Stojdl, D.F.; Korneluk, R.G.; et al. Oncolytic virus synergizes with Smac mimetic compounds to induce rhabdomyosarcoma cell death in a syngeneic murine model. Oncotarget 2017, 8, 3495–3508. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Tao, Z.; Blanc, J.M.; Zaorsky, N.G.; Sun, Y.; Vuagniaux, G.; Dicker, A.P.; Lu, B. Debio 1143, an antagonist of multiple inhibitor-of-apoptosis proteins, activates apoptosis and enhances radiosensitization of non-small cell lung cancer cells in vitro. Am. J. Cancer Res. 2014, 4, 943–951. [Google Scholar] [PubMed]
- Matzinger, O.; Viertl, D.; Tsoutsou, P.; Kadi, L.; Rigotti, S.; Zanna, C.; Wiedemann, N.; Vozenin, M.C.; Vuagniaux, G.; Bourhis, J. The radiosensitizing activity of the SMAC-mimetic, Debio 1143, is TNFalpha-mediated in head and neck squamous cell carcinoma. Radiother. Oncol. 2015, 116, 495–503. [Google Scholar] [CrossRef]
- Tao, Z.; McCall, N.S.; Wiedemann, N.; Vuagniaux, G.; Yuan, Z.; Lu, B. SMAC Mimetic Debio 1143 and Ablative Radiation Therapy Synergize to Enhance Antitumor Immunity against Lung Cancer. Clin. Cancer Res. 2019, 25, 1113–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743e1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, A.J.; Vervoort, S.J.; Ramsbottom, K.M.; Kelly, M.J.; Michie, J.; Pijpers, L.; Johnstone, R.W.; Kearney, C.J.; Oliaro, J. Natural Killer Cells Suppress T Cell-Associated Tumor Immune Evasion. Cell Rep. 2019, 28, 2784–2794e2785. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, K.; Hombach, A.; Seeger, J.M.; Wagner-Stippich, D.; Klubertz, D.; Kronke, M.; Abken, H.; Kashkar, H. Second mitochondria-derived activator of caspase (SMAC) mimetic potentiates tumor susceptibility toward natural killer cell-mediated killing. Leuk. Lymphoma 2014, 55, 645–651. [Google Scholar] [CrossRef]
- Sauer, M.; Reiners, K.S.; Hansen, H.P.; Engert, A.; Gasser, S.; von Strandmann, E.P. Induction of the DNA damage response by IAP inhibition triggers natural immunity via upregulation of NKG2D ligands in Hodgkin lymphoma in vitro. Biol. Chem. 2013, 394, 1325–1331. [Google Scholar] [CrossRef]
- Fischer, K.; Tognarelli, S.; Roesler, S.; Boedicker, C.; Schubert, R.; Steinle, A.; Klingebiel, T.; Bader, P.; Fulda, S.; Ullrich, E. The Smac Mimetic BV6 Improves NK Cell-Mediated Killing of Rhabdomyosarcoma Cells by Simultaneously Targeting Tumor and Effector Cells. Front. Immunol. 2017, 8, 202. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, J.; Liu, H.; Dong, W.; Huang, X.; Yang, D.; Hou, J.; Zhang, X. The SMAC Mimetic APG-1387 Sensitizes Immune-Mediated Cell Apoptosis in Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1298. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Invest. 2018, 128, 4654–4668. [Google Scholar] [CrossRef] [PubMed]
- Concha-Benavente, F.; Kansy, B.; Moskovitz, J.; Moy, J.; Chandran, U.; Ferris, R.L. PD-L1 Mediates Dysfunction in Activated PD-1(+) NK Cells in Head and Neck Cancer Patients. Cancer Immunol. Res. 2018, 6, 1548–1560. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017, 36, 6143–6153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigaud, S.; Fondaneche, M.C.; Lambert, N.; Pasquier, B.; Mateo, V.; Soulas, P.; Galicier, L.; Le Deist, F.; Rieux-Laucat, F.; Revy, P.; et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 2006, 444, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Clancy-Thompson, E.; Ali, L.; Bruck, P.T.; Exley, M.A.; Blumberg, R.S.; Dranoff, G.; Dougan, M.; Dougan, S.K. IAP Antagonists Enhance Cytokine Production from Mouse and Human iNKT Cells. Cancer Immunol. Res. 2018, 6, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruschinski, A.; Moosmann, A.; Poschke, I.; Norell, H.; Chmielewski, M.; Seliger, B.; Kiessling, R.; Blankenstein, T.; Abken, H.; Charo, J. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc. Natl. Acad. Sci. USA 2008, 105, 17481–17486. [Google Scholar] [CrossRef] [Green Version]
- Boissel, L.; Betancur, M.; Wels, W.S.; Tuncer, H.; Klingemann, H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk. Res. 2009, 33, 1255–1259. [Google Scholar] [CrossRef] [Green Version]
- Varela-Guruceaga, M.; Tejada-Solis, S.; Garcia-Moure, M.; Fueyo, J.; Gomez-Manzano, C.; Patino-Garcia, A.; Alonso, M.M. Oncolytic Viruses as Therapeutic Tools for Pediatric Brain Tumors. Cancers (Basel) 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Garber, K. China approves world’s first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Terawaki, S.; Chikuma, S.; Shibayama, S.; Hayashi, T.; Yoshida, T.; Okazaki, T.; Honjo, T. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J. Immunol. 2011, 186, 2772–2779. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Dastidar, H.; Zhang, C.; Zemp, F.J.; Lau, K.; Ernst, M.; Rakic, A.; Sikdar, S.; Rajwani, J.; Naumenko, V.; et al. Smac mimetics and oncolytic viruses synergize in driving anticancer T-cell responses through complementary mechanisms. Nat. Commun. 2017, 8, 344. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Lin, Y.; Zhang, H.; Liang, J.; Tan, Y.; Cavenee, W.K.; Yan, G. Selective replication of oncolytic virus M1 results in a bystander killing effect that is potentiated by Smac mimetics. Proc. Natl. Acad. Sci. USA 2017, 114, 6812–6817. [Google Scholar] [CrossRef] [Green Version]
- Beug, S.T.; Pichette, S.J.; St-Jean, M.; Holbrook, J.; Walker, D.E.; LaCasse, E.C.; Korneluk, R.G. Combination of IAP Antagonists and TNF-alpha-Armed Oncolytic Viruses Induce Tumor Vascular Shutdown and Tumor Regression. Mol. Ther. Oncolytics 2018, 10, 28–39. [Google Scholar] [CrossRef]
- Witt, A.; Seeger, J.M.; Coutelle, O.; Zigrino, P.; Broxtermann, P.; Andree, M.; Brinkmann, K.; Jungst, C.; Schauss, A.C.; Schull, S.; et al. IAP antagonization promotes inflammatory destruction of vascular endothelium. EMBO Rep. 2015, 16, 719–727. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Identifier | Smac- mimetic | Combination Therapy | Malignancy | Phase |
|---|---|---|---|---|
| NCT03270176 | Debio 1143 (AT- 406) | Avelumab (α-PDL-1) | Advances solid tumours and advanced/metastatic non-small cell lung carcinoma | 1 |
| NCT02587962 | Birinapant (TL32711) | Pembrolizumab (α-PD-1) | Various solid tumours | 1/2 |
| NCT03111992 | LCL161 | PDR001 (α-PD-1) CJM112 (anti-IL-17a) | Multiple myeloma | 1 |
| NCT02890069 | LCL161 | PDR001 (α-PD-1) Everolimus (mToR inhibitor) Panobinostat (HDAC inhibitor) | Colorectal cancer, non-small cell lung carcinoma, triple negative breast cancer, renal cell carcinoma | 1 |
| NCT03166631 | BI891065 | B I754091 (α-PD-1) | Advanced and/or metastatic malignancies | 1 |
| NCT03697304 | BI891065 | BI 754111 (anti-LAG-3) BI 754091 (α-PD-1) | Advanced and/or metastatic solid tumours (pre-treated) | 2 |
| NCT03871959 | Debio 1143 | Pembrolizumab (α-PD-1) | Non-MSI-high advanced or metastatic pancreatic ductal adenocarcinoma or colorectal cancer | 1 |
| NCT03386526 | APG-1387 | Pembrolizumab (α-PD-1) | Advanced solid tumours or haematological malignancies | 1/2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michie, J.; Kearney, C.J.; Hawkins, E.D.; Silke, J.; Oliaro, J. The Immuno-Modulatory Effects of Inhibitor of Apoptosis Protein Antagonists in Cancer Immunotherapy. Cells 2020, 9, 207. https://doi.org/10.3390/cells9010207
Michie J, Kearney CJ, Hawkins ED, Silke J, Oliaro J. The Immuno-Modulatory Effects of Inhibitor of Apoptosis Protein Antagonists in Cancer Immunotherapy. Cells. 2020; 9(1):207. https://doi.org/10.3390/cells9010207
Chicago/Turabian StyleMichie, Jessica, Conor J. Kearney, Edwin D. Hawkins, John Silke, and Jane Oliaro. 2020. "The Immuno-Modulatory Effects of Inhibitor of Apoptosis Protein Antagonists in Cancer Immunotherapy" Cells 9, no. 1: 207. https://doi.org/10.3390/cells9010207
APA StyleMichie, J., Kearney, C. J., Hawkins, E. D., Silke, J., & Oliaro, J. (2020). The Immuno-Modulatory Effects of Inhibitor of Apoptosis Protein Antagonists in Cancer Immunotherapy. Cells, 9(1), 207. https://doi.org/10.3390/cells9010207