Future Therapeutic Directions for Smac-Mimetics


Abstract
:1. The Relevance of Programmed Cell Death in Cancer
2. Inhibitor of Apoptosis Proteins
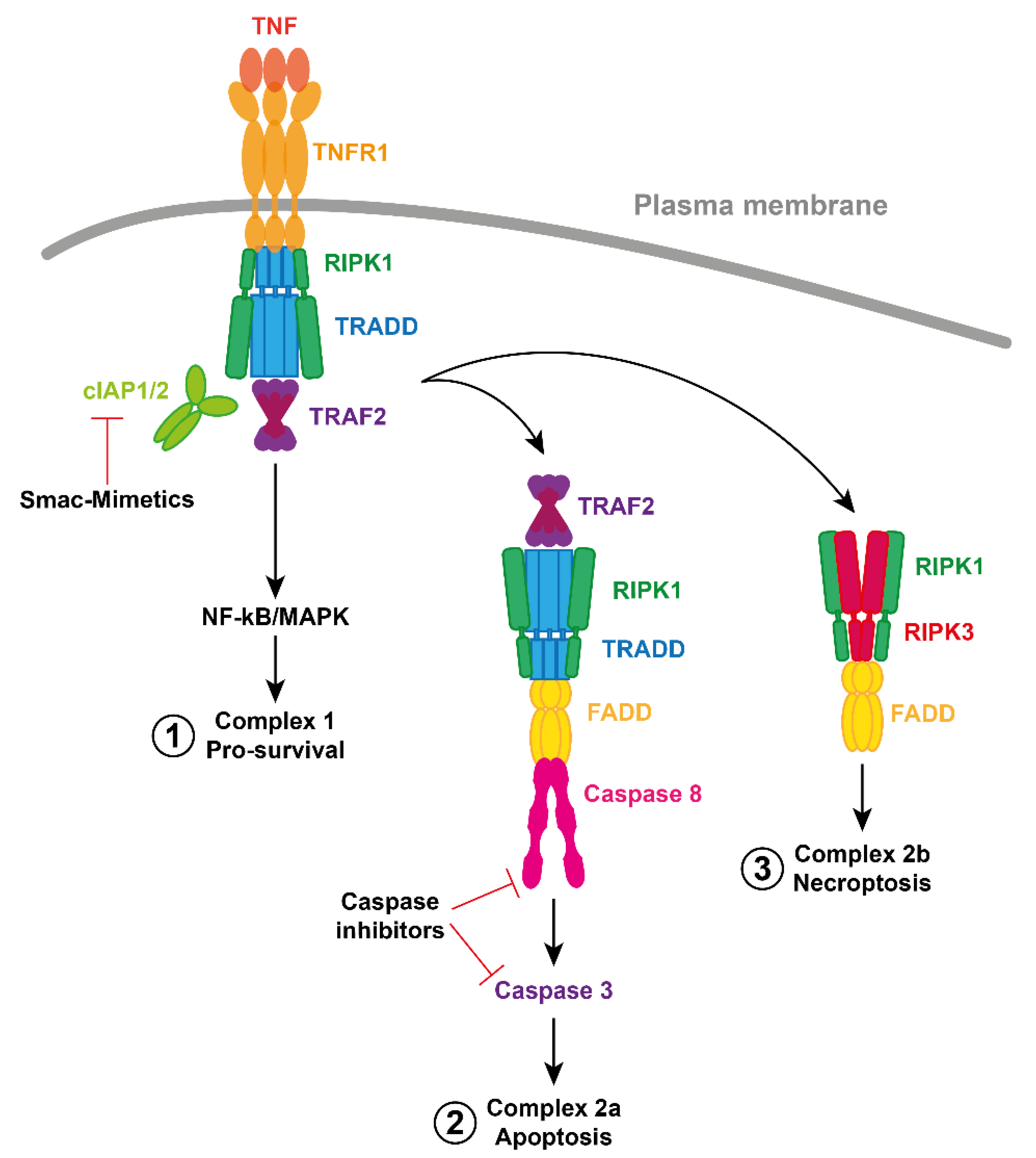
3. TNF Signaling Pathway
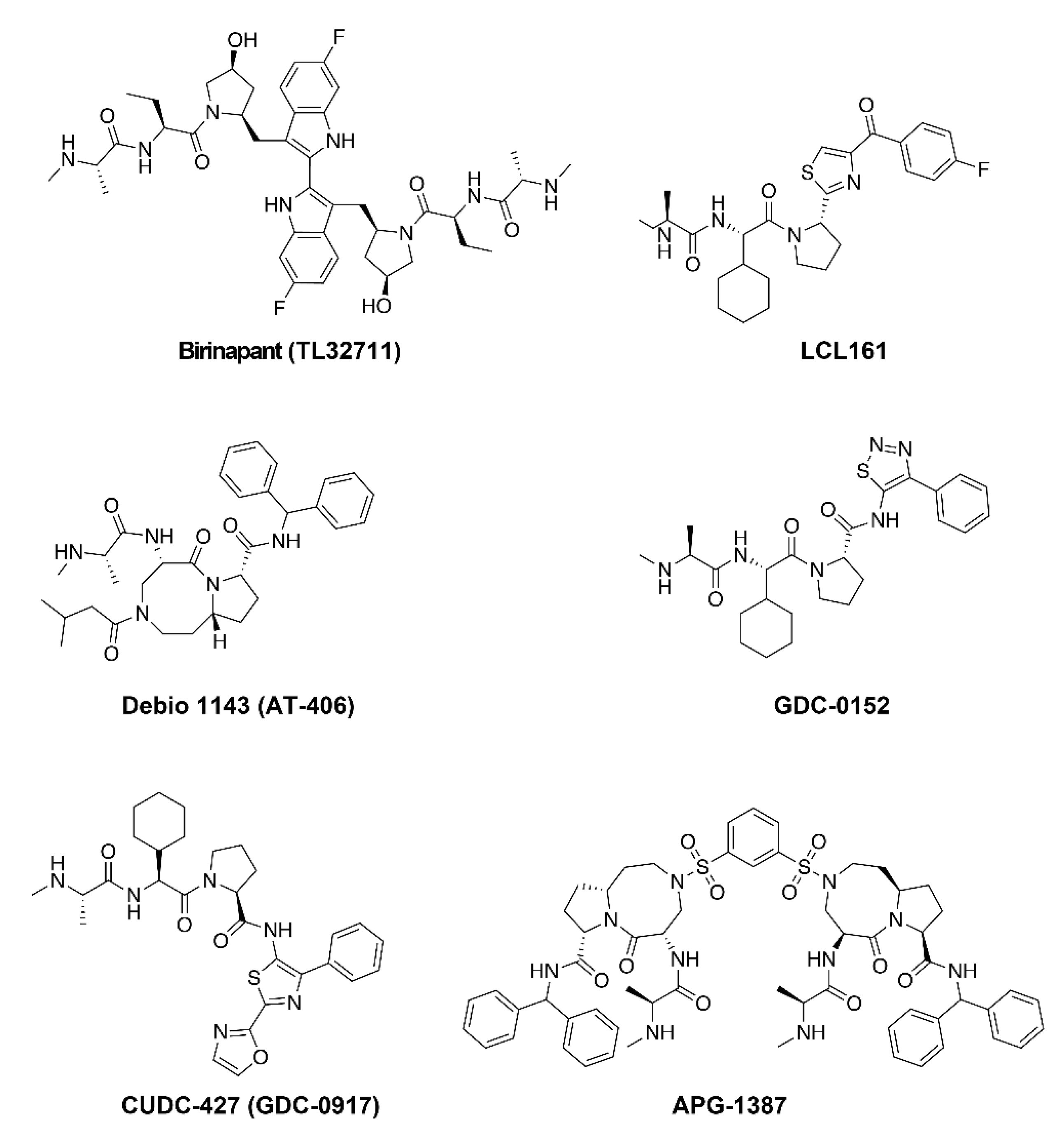
4. Development of Smac-Mimetics
5. Smac-Mimetics as Single Agents in Pre-Clinical and Clinical Studies
5.1. Birinapant
5.2. LCL161
5.3. Debio 1143
5.4. GDC Smac-Mimetics
5.5. Other Smac-Mimetic Drugs
6. Mechanisms of Resistance to Smac-Mimetics and Strategies to Overcome Them
6.1. Importance of TNF
6.2. Combination with Radiation
6.3. Combination with Chemotherapy
6.4. Combination with Bcl-2 Inhibitors
6.5. Combination with Immunotherapy
6.6. Inducing Necroptosis
6.7. Mechanical Resistance
7. Future Directions and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting autophagy in cancer: Recent advances and future directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvesen, G.S.; Duckett, C.S. Apoptosis: IAP proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar] [CrossRef]
- Brumatti, G.; Ma, C.; Lalaoui, N.; Nguyen, N.-Y.; Navarro, M.; Tanzer, M.C.; Richmond, J.; Ghisi, M.; Salmon, J.M.; Silke, N.; et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci. Transl. Med. 2016, 8, 339–369. [Google Scholar] [CrossRef] [Green Version]
- Schenk, B.; Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 2015, 34, 5796. [Google Scholar] [CrossRef]
- Silke, J.; Rickard, J.A.; Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 2015, 16, 689. [Google Scholar] [CrossRef]
- De Almagro, M.; Vucic, D. The inhibitor of apoptosis (IAP) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Exp. Oncol. 2012, 34, 200–211. [Google Scholar]
- Vaux, D.L.; Silke, J. IAPs, RINGs and ubiquitylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 287. [Google Scholar] [CrossRef] [PubMed]
- Obexer, P.; Ausserlechner, M.J. X-linked inhibitor of apoptosis protein–a critical death resistance regulator and therapeutic target for personalized cancer therapy. Front. Oncol. 2014, 4, 197. [Google Scholar] [CrossRef] [Green Version]
- Gyrd-Hansen, M.; Darding, M.; Miasari, M.; Santoro, M.M.; Zender, L.; Xue, W.; Tenev, T.; da Fonseca, P.C.A.; Zvelebil, M.; Bujnicki, J.M.; et al. IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-kB as well as cell survival and oncogenesis. Nat. Cell Biol. 2008, 10, 1309–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darding, M.; Feltham, R.; Tenev, T.; Bianchi, K.; Benetatos, C.; Silke, J.; Meier, P. Molecular determinants of Smac mimetic induced degradation of cIAP1 and cIAP2. Cell Death Differ. 2011, 18, 1376–1386. [Google Scholar] [CrossRef]
- Silke, J.; Ekert, P.G.; Day, C.L.; Hawkins, C.J.; Baca, M.; Chew, J.; Pakusch, M.; Verhagen, A.M.; Vaux, D.L. Direct inhibition of caspase 3 is dispensable for the anti-apoptotic activity of XIAP. EMBO J. 2001, 20, 3114–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galbán, S.; Hwang, C.; Rumble, J.M.; Oetjen, K.A.; Wright, C.W.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; et al. Cytoprotective effects of IAPs revealed by a small molecule antagonist. Biochem. J. 2009, 417, 765–771. [Google Scholar]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.A.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Cheung, H.H.; Mrad, R.L.; Plenchette, S.; Simard, C.; Enwere, E.; Arora, V.; Mak, T.W.; Lacasse, E.C.; Waring, J.; et al. Both cIAP1 and cIAP2 regulate TNFα-mediated NF-κB activation. Proc. Natl. Acad. Sci. USA 2008, 105, 11778–11783. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Wong, W.W.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M.; et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef] [Green Version]
- Dowling, J.P.; Alsabbagh, M.; Del Casale, C.; Liu, Z.-G.; Zhang, J. TRADD regulates perinatal development and adulthood survival in mice lacking RIPK1 and RIPK3. Nat. Commun. 2019, 10, 705. [Google Scholar] [CrossRef] [PubMed]
- Füllsack, S.; Rosenthal, A.; Wajant, H.; Siegmund, D. Redundant and receptor-specific activities of TRADD, RIPK1 and FADD in death receptor signaling. Cell Death Dis. 2019, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Anderton, H.; Bandala-Sanchez, E.; Simpson, D.S.; Rickard, J.A.; Ng, A.P.; Di Rago, L.; Hall, C.; Vince, J.E.; Silke, J.; Liccardi, G.; et al. RIPK1 prevents TRADD-driven, but TNFR1 independent, apoptosis during development. Cell Death Differ. 2019, 26, 877. [Google Scholar] [CrossRef] [PubMed]
- Mace, P.D.; Smits, C.; Vaux, D.L.; Silke, J.; Day, C.L. Asymmetric recruitment of cIAPs by TRAF2. J. Mol. Biol. 2010, 400, 8–15. [Google Scholar] [CrossRef]
- Zheng, C.; Kabaleeswaran, V.; Wang, Y.; Cheng, G.; Wu, H. Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: Affinity, specificity, and regulation. Mol. Cell 2010, 38, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Vanden Berghe, T.; Kaiser, W.J.; Bertrand, M.J.M.; Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell. Oncol. 2015, 2, 975093. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.L.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef]
- Jaco, I.; Annibaldi, A.; Lalaoui, N.; Wilson, R.; Tenev, T.; Laurien, L.; Kim, C.; Jamal, K.; John, S.W.; Liccardi, G.; et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol. Cell 2017, 66, 698–710. [Google Scholar] [CrossRef]
- Menon, M.B.; Gropengießer, J.; Fischer, J.; Novikova, L.; Deuretzbacher, A.; Lafera, J.; Schimmeck, H.; Czymmeck, N.; Ronkina, N.; Kotlyarov, A.; et al. p38MAPK/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 2017, 19, 1248. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Delanghe, T.; Rojas-Rivera, D.; Priem, D.; Delvaeye, T.; Bruggeman, I.; Van Herreweghe, F.; Vandenabeele, P.; Bertrand, M.J.M. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 2017, 19, 1237. [Google Scholar] [CrossRef] [PubMed]
- Annibaldi, A.; John, S.W.; Berghe, T.V.; Swatek, K.N.; Ruan, J.; Liccardi, G.; Bianchi, K.; Elliott, P.R.; Choi, S.M.; Van Coillie, S.; et al. Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Mol. Cell 2018, 69, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Silke, J.; Hawkins, C.J.; Ekert, P.G.; Chew, J.; Day, C.L.; Pakusch, M.; Verhagen, A.M.; Vaux, D.L. The anti-apoptotic activity of XIAP is retained upon mutation of both the caspase 3–and caspase 9–interacting sites. J. Cell Biol. 2002, 157, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Bratton, S.B.; Walker, G.; Srinivasula, S.M.; Sun, X.-M.; Butterworth, M.; Alnemri, E.S.; Cohen, G.M. Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 2001, 20, 998–1009. [Google Scholar] [CrossRef]
- Wu, G.; Chai, J.; Suber, T.L.; Wu, J.-W.; Du, C.; Wang, X.; Shi, Y. Structural basis of IAP recognition by Smac/DIABLO. Nature 2000, 408, 1008–1012. [Google Scholar] [CrossRef]
- Hegde, R.; Srinivasula, S.M.; Zhang, Z.; Wassell, R.; Mukattash, R.; Cilenti, L.; DuBois, G.; Lazebnik, Y.; Zervos, A.S.; Fernandes-Alnemri, T.; et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 2002, 277, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Ekert, P.G.; Silke, J.; Hawkins, C.J.; Verhagen, A.M.; Vaux, D.L. DIABLO promotes apoptosis by removing MIHA/XIAP from processed caspase 9. J. Cell Biol. 2001, 152, 483–490. [Google Scholar] [CrossRef]
- Feltham, R.; Bettjeman, B.; Budhidarmo, R.; Mace, P.D.; Shirley, S.; Condon, S.M.; Chunduru, S.K.; McKinlay, M.A.; Vaux, D.L.; Silke, J.; et al. Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J. Biol. Chem. 2011, 286, 17015–17028. [Google Scholar] [CrossRef] [Green Version]
- Dueber, E.C.; Schoeffler, A.J.; Lingel, A.; Elliott, J.M.; Fedorova, A.V.; Giannetti, A.M.; Zobel, K.; Maurer, B.; Varfolomeev, E.; Wu, P.; et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science 2011, 334, 376–380. [Google Scholar] [CrossRef]
- Hu, S.; Yang, X. Cellular inhibitor of apoptosis 1 and 2 are ubiquitin ligases for the apoptosis inducer Smac/DIABLO. J. Biol. Chem. 2003, 278, 10055–10060. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.-H.; Du, C. Smac/DIABLO Selectively Reduces the Levels of c-IAP1 and c-IAP2 but Not That of XIAP and Livin in HeLa Cells. J. Biol. Chem. 2004, 279, 16963–16970. [Google Scholar] [CrossRef] [Green Version]
- Creagh, E.M.; Murphy, B.M.; Duriez, P.J.; Duckett, C.S.; Martin, S.J. Smac/Diablo antagonizes ubiquitin ligase activity of inhibitor of apoptosis proteins. J. Biol. Chem. 2004, 279, 26906–26914. [Google Scholar] [CrossRef] [Green Version]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Kitson, J.; Raven, T.; Jiang, Y.-P.; Goeddel, D.V.; Giles, K.M.; Pun, K.-T.; Grinham, C.J.; Brown, R.; Farrow, S.N. A death-domain-containing receptor that mediates apoptosis. Nature 1996, 384, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Dickens, L.S.; Boyd, R.S.; Jukes-Jones, R.; Hughes, M.A.; Robinson, G.L.; Fairall, L.; Schwabe, J.W.; Cain, K.; MacFarlane, M. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 2012, 47, 291–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Yu, J.; Zhang, L. Necroptosis: An alternative cell death program defending against cancer. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2016, 1865, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.-G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.M.; Silke, J. Ars Moriendi; the art of dying well–new insights into the molecular pathways of necroptotic cell death. EMBO Rep. 2014, 15, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.-F.; Wang, F.-S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Condon, S.M.; Mitsuuchi, Y.; Deng, Y.; LaPorte, M.G.; Rippin, S.R.; Haimowitz, T.; Alexander, M.D.; Kumar, P.T.; Hendi, M.S.; Lee, Y.H.; et al. Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J. Med. Chem. 2014, 57, 3666–3677. [Google Scholar] [CrossRef]
- Zender, L.; Spector, M.S.; Xue, W.; Flemming, P.; Cordon-Cardo, C.; Silke, J.; Fan, S.-T.; Luk, J.M.; Wigler, M.; Hannon, G.J.; et al. Identification and Validation of Oncogenes in Liver Cancer Using an Integrative Oncogenomic Approach. Cell 2006, 125, 1253–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Wick, W.; Weller, M.; Debatin, K.-M. Smac agonists sensitize for Apo2L/TRAIL-or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat. Med. 2002, 8, 808. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Milella, M.; Tsao, T.; McQueen, T.; Schober, W.; Hu, W.; Dean, N.; Steelman, L.; McCubrey, J.; Andreeff, M. Regulation and targeting of antiapoptotic XIAP in acute myeloid leukemia. Leukemia 2003, 17, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Arnt, C.R.; Chiorean, M.V.; Heldebrant, M.P.; Gores, G.J.; Kaufmann, S.H. Synthetic Smac/DIABLO peptides enhance the effects of chemotherapeutic agents by binding XIAP and cIAP1 in situ. J. Biol. Chem. 2002, 277, 44236–44243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silke, J.; Verhagen, A.M.; Ekert, P.G.; Vaux, D.L. Sequence as well as functional similarity for DIABLO/Smac and Grim, Reaper and Hid? Cell Death Differ. 2000, 7, 1275. [Google Scholar] [CrossRef] [PubMed]
- Kipp, R.A.; Case, M.A.; Wist, A.D.; Cresson, C.M.; Carrell, M.; Griner, E.; Wiita, A.; Albiniak, P.A.; Chai, J.; Shi, Y.; et al. Molecular Targeting of Inhibitor of Apoptosis Proteins Based on Small Molecule Mimics of Natural Binding Partners. Biochemistry 2002, 41, 7344–7349. [Google Scholar] [CrossRef]
- Schimmer, A.D.; Welsh, K.; Pinilla, C.; Wang, Z.; Krajewska, M.; Bonneau, M.-J.; Pedersen, I.M.; Kitada, S.; Scott, F.L.; Bailly-Maitre, B.; et al. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell 2004, 5, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Oost, T.K.; Sun, C.; Armstrong, R.C.; Al-Assaad, A.-S.; Betz, S.F.; Deckwerth, T.L.; Ding, H.; Elmore, S.W.; Meadows, R.P.; Olejniczak, E.T.; et al. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J. Med. Chem. 2004, 47, 4417–4426. [Google Scholar] [CrossRef]
- Li, L.; Thomas, R.M.; Suzuki, H.; De Brabander, J.K.; Wang, X.; Harran, P.G. A small molecule Smac mimic potentiates TRAIL-and TNFα-mediated cell death. Science 2004, 305, 1471–1474. [Google Scholar] [CrossRef] [Green Version]
- Zobel, K.; Wang, L.; Varfolomeev, E.; Franklin, M.C.; Elliott, L.O.; Wallweber, H.J.; Okawa, D.C.; Flygare, J.A.; Vucic, D.; Fairbrother, W.J.; et al. Design, synthesis, and biological activity of a potent Smac mimetic that sensitizes cancer cells to apoptosis by antagonizing IAPs. ACS Chem. Biol. 2006, 1, 525–533. [Google Scholar] [CrossRef]
- Chai, J.; Du, C.; Wu, J.-W.; Kyin, S.; Wang, X.; Shi, Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 2000, 406, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Moulin, M.; Anderton, H.; Voss, A.K.; Thomas, T.; Wong, W.W.L.; Bankovacki, A.; Feltham, R.; Chau, D.; Cook, W.D.; Silke, J.; et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 2012, 31, 1679–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heard, K.N.; Bertrand, M.J.; Barker, P.A. cIAP2 supports viability of mice lacking cIAP1 and XIAP. EMBO J. 2015, 34, 2393–2395. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; Wong, W.W.-L.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef] [Green Version]
- Anderton, H.; Rickard, J.A.; Varigos, G.A.; Lalaoui, N.; Silke, J. Inhibitor of apoptosis proteins (IAPs) limit RIPK1-mediated skin inflammation. J. Investig. Dermatol. 2017, 137, 2371–2379. [Google Scholar] [CrossRef] [Green Version]
- Ndubaku, C.; Varfolomeev, E.; Wang, L.; Zobel, K.; Lau, K.; Elliott, L.O.; Maurer, B.; Fedorova, A.V.; Dynek, J.N.; Koehler, M.; et al. Antagonism of c-IAP and XIAP proteins is required for efficient induction of cell death by small-molecule IAP antagonists. ACS Chem. Biol. 2009, 4, 557–566. [Google Scholar] [CrossRef]
- Benetatos, C.A.; Mitsuuchi, Y.; Burns, J.M.; Neiman, E.M.; Condon, S.M.; Yu, G.; Seipel, M.E.; Kapoor, G.S.; LaPorte, M.G.; Rippin, S.R.; et al. Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-κB activation, and is active in patient-derived xenograft models. Mol. Cancer Ther. 2014, 13, 867–879. [Google Scholar] [CrossRef] [Green Version]
- Krepler, C.; Chunduru, S.K.; Halloran, M.B.; He, X.; Xiao, M.; Vultur, A.; Villanueva, J.; Mitsuuchi, Y.; Neiman, E.M.; Benetatos, C.; et al. The novel SMAC mimetic birinapant exhibits potent activity against human melanoma cells. Clin. Cancer Res. 2013, 19, 1784–1794. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860. [Google Scholar] [CrossRef]
- Katerinaki, E.; Evans, G.; Lorigan, P.; MacNeil, S. TNF-α increases human melanoma cell invasion and migration in vitro: The role of proteolytic enzymes. Br. J. Cancer 2003, 89, 1123. [Google Scholar] [CrossRef] [Green Version]
- Beug, S.T.; Tang, V.A.; LaCasse, E.C.; Cheung, H.H.; Beauregard, C.E.; Brun, J.; Nuyens, J.P.; Earl, N.; St-Jean, M.; Holbrook, J.; et al. Smac mimetics and innate immune stimuli synergize to promote tumor death. Nat. Biotechnol. 2014, 32, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesi, M.; Mirza, N.N.; Garbitt, V.M.; Sharik, M.E.; Dueck, A.C.; Asmann, Y.W.; Akhmetzyanova, I.; Kosiorek, H.E.; Calcinotto, A.; Riggs, D.L.; et al. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat. Med. 2016, 22, 1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michie, J.; Beavis, P.A.; Freeman, A.J.; Vervoort, S.J.; Ramsbottom, K.M.; Narasimhan, V.; Lelliott, E.J.; Lalaoui, N.; Ramsay, R.G.; Johnstone, R.W.; et al. Antagonism of IAPs enhances CAR T-cell efficacy. Cancer Immunol. Res. 2019, 7, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Eytan, D.F.; Snow, G.E.; Carlson, S.; Derakhshan, A.; Saleh, A.; Schiltz, S.; Cheng, H.; Mohan, S.; Cornelius, S.; Coupar, J.; et al. SMAC mimetic birinapant plus radiation eradicates human head and neck cancers with genomic amplifications of cell death genes FADD and BIRC2. Cancer Res. 2016, 76, 5442–5454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, C.G.A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eytan, D.F.; Snow, G.E.; Carlson, S.G.; Schiltz, S.; Chen, Z.; Van Waes, C. Combination effects of SMAC mimetic birinapant with TNF α, TRAIL, and docetaxel in preclinical models of HNSCC. Laryngoscope 2015, 125, E118–E124. [Google Scholar] [CrossRef] [Green Version]
- Allensworth, J.L.; Aird, K.M.; Aldrich, A.J.; Batinic-Haberle, I.; Devi, G.R. XIAP Inhibition and Generation of Reactive Oxygen Species Enhances TRAIL Sensitivity in Inflammatory Breast Cancer Cells. Mol. Cancer Ther. 2012, 11, 1518–1527. [Google Scholar] [CrossRef] [Green Version]
- Allensworth, J.L.; Sauer, S.J.; Lyerly, H.K.; Morse, M.A.; Devi, G.R. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-α-independent mechanism. Breast Cancer Res. Treat. 2013, 137, 359–371. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Schilder, R.J.; Martin, L.P.; Levin, M.; Graham, M.A.; Weng, D.E.; Adjei, A.A. A Phase I Study of the SMAC-Mimetic Birinapant in Adults with Refractory Solid Tumors or Lymphoma. Mol. Cancer Ther. 2015, 14, 2569–2575. [Google Scholar] [CrossRef] [Green Version]
- Frey, N.V.; Luger, S.; Mangan, J.; Zebrowski, A.; Loren, A.W.; Minderman, H.; Baird, J.; Porter, D.L.; Hexner, E.O.; Kumar, A.J.; et al. Abstract: A phase I study using single agent birinapant in patients with relapsed myelodysplastic syndrome and acute myelogenous leukemia. Blood 2014, 124, 3758. [Google Scholar] [CrossRef]
- Noonan, A.M.; Bunch, K.P.; Chen, J.Q.; Herrmann, M.A.; Lee, J.M.; Kohn, E.C.; O’sullivan, C.C.; Jordan, E.; Houston, N.; Takebe, N.; et al. Pharmacodynamic markers and clinical results from the phase 2 study of the SMAC mimetic birinapant in women with relapsed platinum-resistant or-refractory epithelial ovarian cancer. Cancer 2016, 122, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Ray, A.; Barrett, R.; Nelson, E.; Christie, A.L.; Porter, D.; Straub, C.; Zawel, L.; Daley, J.F.; Lazo-Kallanian, S.; et al. Smac mimetics: Implications for enhancement of targeted therapies in leukemia. Leukemia 2010, 24, 2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, E.; Kung, A.L.; Wright, R.D.; Moreno, D.; Catley, L.; Ray, A.; Zawel, L.; Tran, M.; Cools, J.; Gilliland, G.; et al. Potentiation of antileukemic therapies by Smac mimetic, LBW242: Effects on mutant FLT3-expressing cells. Mol. Cancer Ther. 2007, 6, 1951–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condon, S.M. The discovery and development of Smac mimetics—small-molecule antagonists of the inhibitor of apoptosis proteins. In Annual reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2011; Volume 46, pp. 211–226. [Google Scholar]
- Hird, A.W.; Aquila, B.M.; Hennessy, E.J.; Vasbinder, M.M.; Yang, B. Small molecule inhibitor of apoptosis proteins antagonists: A patent review. Expert Opin. Ther. Pat. 2015, 25, 755–774. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-F.; Lin, J.-P.; Shiau, C.-W.; Tai, W.-T.; Liu, C.-Y.; Yu, H.-C.; Chen, P.-J.; Cheng, A.-L. Inhibition of Bcl-2 improves effect of LCL161, a SMAC mimetic, in hepatocellular carcinoma cells. Biochem. Pharmacol. 2012, 84, 268–277. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; Painuly, U.; Kimlinger, T.; Haug, J.; Rajkumar, S.V.; Kumar, S. Inhibitor of apoptosis proteins as therapeutic targets in multiple myeloma. Leukemia 2014, 28, 1519. [Google Scholar] [CrossRef] [Green Version]
- Houghton, P.J.; Kang, M.H.; Reynolds, C.P.; Morton, C.L.; Kolb, E.A.; Gorlick, R.; Keir, S.T.; Carol, H.; Lock, R.; Maris, J.M.; et al. Initial testing (stage 1) of LCL161, a SMAC mimetic, by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2012, 58, 636–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neale, G.; Su, X.; Morton, C.L.; Phelps, D.; Gorlick, R.; Lock, R.B.; Reynolds, C.P.; Maris, J.M.; Friedman, H.S.; Dome, J.; et al. Molecular characterization of the pediatric preclinical testing panel. Clin. Cancer Res. 2008, 14, 4572–4583. [Google Scholar] [CrossRef] [Green Version]
- Faye, M.; Beug, S.; Graber, T.; Earl, N.; Xiang, X.; Wild, B.; Langlois, S.; Michaud, J.; Cowan, K.; Korneluk, R.; et al. IGF2BP1 controls cell death and drug resistance in rhabdomyosarcomas by regulating translation of cIAP1. Oncogene 2015, 34, 1532. [Google Scholar] [CrossRef]
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 3103–3110. [Google Scholar] [CrossRef]
- Cai, Q.; Sun, H.; Peng, Y.; Lu, J.; Nikolovska-Coleska, Z.; McEachern, D.; Liu, L.; Qiu, S.; Yang, C.-Y.; Miller, R.; et al. A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J. Med. Chem. 2011, 54, 2714–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunckhorst, M.K.; Lerner, D.; Wang, S.; Yu, Q. AT-406, an orally active antagonist of multiple inhibitor of apoptosis proteins, inhibits progression of human ovarian cancer. Cancer Biol. Ther. 2012, 13, 804–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Li, Y.; Zou, P.; Yu, J.y.; McEachern, D.; Wang, S.; Sun, D. Physiologically based pharmacokinetic and pharmacodynamic modeling of an antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in a mouse xenograft model of human breast cancer. Biopharm. Drug Dispos. 2013, 34, 348–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, H.I.; Smith, D.C.; Pitot, H.C.; Brill, J.M.; Chugh, R.; Rouits, E.; Rubin, J.; Strickler, J.; Vuagniaux, G.; Sorensen, J.M.; et al. Safety, pharmacokinetics, and pharmacodynamic properties of oral DEBIO1143 (AT-406) in patients with advanced cancer: Results of a first-in-man study. Cancer Chemother. Pharmacol. 2015, 75, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Flygare, J.A.; Beresini, M.; Budha, N.; Chan, H.; Chan, I.T.; Cheeti, S.; Cohen, F.; Deshayes, K.; Doerner, K.; Eckhardt, S.G.; et al. Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152). J. Med. Chem. 2012, 55, 4101–4113. [Google Scholar] [CrossRef]
- Wong, H.; Gould, S.E.; Budha, N.; Darbonne, W.C.; Kadel, E.E.; La, H.; Alicke, B.; Halladay, J.S.; Erickson, R.; Portera, C.; et al. Learning and confirming with preclinical studies: Modeling and simulation in the discovery of GDC-0917, an inhibitor of apoptosis proteins antagonist. Drug Metab. Dispos. 2013, 41, 2104–2113. [Google Scholar] [CrossRef] [Green Version]
- Tchoghandjian, A.; Soubéran, A.; Tabouret, E.; Colin, C.; Denicolaï, E.; Jiguet-Jiglaire, C.; El-Battari, A.; Villard, C.; Baeza-Kallee, N.; Figarella-Branger, D. Inhibitor of apoptosis protein expression in glioblastomas and their in vitro and in vivo targeting by SMAC mimetic GDC-0152. Cell Death Dis. 2016, 7, 2325. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Bendell, J.C.; Papadopoulos, K.P.; Burris, H.A.; Patnaik, A.; Fairbrother, W.J.; Wong, H.; Budha, N.; Darbonne, W.C.; Peale, F.; et al. A phase I dose-escalation study evaluating the safety tolerability and pharmacokinetics of CUDC-427, a potent, oral, monovalent IAP antagonist, in patients with refractory solid tumors. Clin. Cancer Res. 2016, 22, 4567–4573. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Yu, Y.; Li, Z.-L.; Chen, M.-Y.; Deng, R.; Huang, X.; Wang, G.-F.; Zhang, M.-X.; Yang, Q.; Ravichandran, S.; et al. XIAP Limits Autophagic Degradation of Sox2 and is a therapeutic target in nasopharyngeal carcinoma stem cells. Theranostics 2018, 8, 1494. [Google Scholar] [CrossRef] [Green Version]
- Li, B.-X.; Wang, H.-B.; Qiu, M.-Z.; Luo, Q.-Y.; Yi, H.-J.; Yan, X.-L.; Pan, W.-T.; Yuan, L.-P.; Zhang, Y.-X.; Xu, J.-H.; et al. Novel smac mimetic APG-1387 elicits ovarian cancer cell killing through TNF-alpha, Ripoptosome and autophagy mediated cell death pathway. J. Exp. Clin. Cancer Res. 2018, 37, 53. [Google Scholar] [CrossRef]
- Li, N.; Feng, L.; Han, H.-Q.; Yuan, J.; Qi, X.-K.; Lian, Y.-F.; Kuang, B.-H.; Zhang, Y.-C.; Deng, C.-C.; Zhang, H.-J.; et al. A novel Smac mimetic APG-1387 demonstrates potent antitumor activity in nasopharyngeal carcinoma cells by inducing apoptosis. Cancer Lett. 2016, 381, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Li, Y.; Ji, J.; Qiu, M.; Zhang, Y.; Liu, W.; Tian, X.; Li, S.; Wang, H.; Wang, F.; et al. A phase I study of a novel IAP inhibitor APG-1387 in patients with advanced solid tumors. Am. Soc. Clin. Oncol. 2018, 36, 2593. [Google Scholar] [CrossRef]
- Rasco, D.W.; Li, Y.; Tang, Y.; Men, L.; Wang, H.; Ji, J.; Liang, Z.; Sun, J.; Amaya, A.; Huang, Y.; et al. A phase I study of a novel IAP inhibitor APG-1387 as a monotherapy or in combination with pembrolizumab in treatments of patients with advanced solid tumors. Am. Soc. Clin. Oncol. 2019, 37, 3125. [Google Scholar] [CrossRef]
- Humphreys, R.C.; Poortman, C.; McCormick, K.; Carrell, J.; Shepard, L. Abstract C10: A novel combination of a small molecule IAP inhibitor and a TRAIL-R1 monoclonal antibody synergize to induce apoptosis in pancreatic tumor cell lines. Mol. Cancer Ther. 2009, 8, C10. [Google Scholar]
- Eckhardt, S.G.; Gallant, G.; Sikic, B.I.; Camidge, D.R.; Burris III, H.A.; Wakelee, H.A.; Messersmith, W.A.; Jones, S.F.; Colevas, A.D.; Infante, J.R. Phase I study evaluating the safety, tolerability, and pharmacokinetics (PK) of HGS1029, a small-molecule inhibitor of apoptosis protein (IAP), in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2010, 28, 2580. [Google Scholar] [CrossRef]
- Sikic, B.I.; Eckhardt, S.G.; Gallant, G.; Burris, H.A.; Camidge, D.R.; Colevas, A.D.; Jones, S.F.; Messersmith, W.A.; Wakelee, H.A.; Li, H.; et al. Safety, pharmacokinetics (PK), and pharmacodynamics (PD) of HGS1029, an inhibitor of apoptosis protein (IAP) inhibitor, in patients (Pts) with advanced solid tumors: Results of a phase I study. J. Clin. Oncol. 2011, 29, 3008. [Google Scholar] [CrossRef]
- Impagnatiello, M.A.; Reschke, M.; Reiser, U.; Scharn, D.; Spevak, W.; Savchenko, A.; Tirapu, I.; Wernitznig, A.; Sykora, M.; Langlois, R.; et al. BI5 (BI 891065): A Novel SMAC Mimetic that Triggers Tumor Cell Death and Potentiates PD-1 Mediated Cancer Therapy. In Proceedings of the AACR Annual Meeting, Washington, DC, USA, 1–5 April 2017. [Google Scholar]
- Petersen, S.L.; Wang, L.; Yalcin-Chin, A.; Li, L.; Peyton, M.; Minna, J.; Harran, P.; Wang, X. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 2007, 12, 445–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaither, A.; Porter, D.; Yao, Y.; Borawski, J.; Yang, G.; Donovan, J.; Sage, D.; Slisz, J.; Tran, M.; Straub, C.; et al. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-α signaling. Cancer Res. 2007, 67, 11493–11498. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.J.; Zhou, S.; Diaz Jr, L.A.; Holdhoff, M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget 2011, 2, 739. [Google Scholar] [CrossRef] [Green Version]
- Lejeune, F.J.; Liénard, D.; Matter, M.; Rüegg, C. Efficiency of recombinant human TNF in human cancer therapy. Cancer Immun. Arch. 2006, 6, 6. [Google Scholar]
- Yuan, Z.; Syrkin, G.; Adem, A.; Geha, R.; Pastoriza, J.; Vrikshajanani, C.; Smith, T.; Quinn, T.; Alemu, G.; Cho, H.; et al. Blockade of inhibitors of apoptosis (IAPs) in combination with tumor-targeted delivery of tumor necrosis factor-α leads to synergistic antitumor activity. Cancer Gene Ther. 2013, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Hajitou, A.; Trepel, M.; Lilley, C.E.; Soghomonyan, S.; Alauddin, M.M.; Marini, F.C.; Restel, B.H.; Ozawa, M.G.; Moya, C.A.; Rangel, R.; et al. A Hybrid Vector for Ligand-Directed Tumor Targeting and Molecular Imaging. Cell 2006, 125, 385–398. [Google Scholar] [CrossRef]
- Hajitou, A.; Rangel, R.; Trepel, M.; Soghomonyan, S.; Gelovani, J.G.; Alauddin, M.M.; Pasqualini, R.; Arap, W. Design and construction of targeted AAVP vectors for mammalian cell transduction. Nat. Protoc. 2007, 2, 523. [Google Scholar] [CrossRef]
- Beug, S.T.; Pichette, S.J.; St-Jean, M.; Holbrook, J.; Walker, D.E.; LaCasse, E.C.; Korneluk, R.G. Combination of IAP Antagonists and TNF-α-Armed Oncolytic Viruses Induce Tumor Vascular Shutdown and Tumor Regression. Mol. Ther.-Oncolytics 2018, 10, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Lalaoui, N.; Hänggi, K.; Brumatti, G.; Chau, D.; Nguyen, N.-Y.; Vasilikos, L.; Spilgies, L.M.; Heckmann, D.A.; Ma, C.; Ghisi, M.; et al. Targeting p38 or MK2 Enhances the Anti-Leukemic Activity of Smac-Mimetics. Cancer Cell 2016, 29, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallahan, D.E.; Spriggs, D.R.; Beckett, M.A.; Kufe, D.W.; Weichselbaum, R.R. Increased tumor necrosis factor alpha mRNA after cellular exposure to ionizing radiation. Proc. Natl. Acad. Sci. USA 1989, 86, 10104–10107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Q.; Zuo, Y.; Yang, X.; Lu, J.; Zhan, L.; Xu, L.; Zhang, C.; Zhu, H.; Liu, J.; Liu, Z.; et al. Smac mimetic compound LCL161 sensitizes esophageal carcinoma cells to radiotherapy by inhibiting the expression of inhibitor of apoptosis protein. Tumor Biol. 2014, 35, 2565–2574. [Google Scholar] [CrossRef]
- Liu, N.; Tao, Z.; Le Blanc, J.M.; Zaorsky, N.G.; Sun, Y.; Vuagniaux, G.; Dicker, A.P.; Lu, B. Debio 1143, an antagonist of multiple inhibitor-of-apoptosis proteins, activates apoptosis and enhances radiosensitization of non-small cell lung cancer cells in vitro. Am. J. Cancer Res. 2014, 4, 943. [Google Scholar]
- Matzinger, O.; Viertl, D.; Tsoutsou, P.; Kadi, L.; Rigotti, S.; Zanna, C.; Wiedemann, N.; Vozenin, M.-C.; Vuagniaux, G.; Bourhis, J. The radiosensitizing activity of the SMAC-mimetic, Debio 1143, is TNFα-mediated in head and neck squamous cell carcinoma. Radiother. Oncol. 2015, 116, 495–503. [Google Scholar] [CrossRef]
- Adrain, C.; Creagh, E.M.; Martin, S.J. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 2001, 20, 6627–6636. [Google Scholar] [CrossRef] [Green Version]
- Chu, Q.; Vincent, M.; Logan, D.; Mackay, J.A.; Evans, W.K.; Lung Cancer Disease Site Group of Cancer Care Ontario’s Program in Evidence-based Care. Taxanes as first-line therapy for advanced non-small cell lung cancer: A systematic review and practice guideline. Lung Cancer 2005, 50, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, H.; Zhang, B.; Chen, Y.; Zhang, Y.; Sun, X.; Xiao, G.; Nan, K.; Ren, H.; Qin, S. LCL161 increases paclitaxel-induced apoptosis by degrading cIAP1 and cIAP2 in NSCLC. J. Exp. Clin. Cancer Res. 2016, 35, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimoto, T.; Tsuda, H.; Imazeki, N.; Ohno, Y.; Imoto, I.; Inazawa, J.; Matsubara, O. Nuclear expression of cIAP-1, an apoptosis inhibiting protein, predicts lymph node metastasis and poor patient prognosis in head and neck squamous cell carcinomas. Cancer Lett. 2005, 224, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Esposito, I.; Kleeff, J.; Abiatari, I.; Shi, X.; Giese, N.; Bergmann, F.; Roth, W.; Friess, H.; Schirmacher, P. Overexpression of cellular inhibitor of apoptosis protein 2 is an early event in the progression of pancreatic cancer. J. Clin. Pathol. 2007, 60, 885–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.; Zhu, W.-G.; Morrison, C.D.; Brena, R.M.; Smiraglia, D.J.; Raval, A.; Wu, Y.-Z.; Rush, L.J.; Ross, P.; Molina, J.R.; et al. A comprehensive search for DNA amplification in lung cancer identifies inhibitors of apoptosis cIAP1 and cIAP2 as candidate oncogenes. Hum. Mol. Genet. 2003, 12, 791–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannistra, S.A. Cancer of the ovary. New Engl. J. Med. 2004, 351, 2519–2529. [Google Scholar] [CrossRef]
- Cannistra, S.A. Cancer of the ovary. New Engl. J. Med. 1993, 329, 1550–1559. [Google Scholar] [CrossRef]
- Janzen, D.; Tiourin, E.; Salehi, J.; Paik, D.; Lu, J.; Pellegrini, M.; Memarzadeh, S. An apoptosis-enhancing drug overcomes platinum resistance in a tumour-initiating subpopulation of ovarian cancer. Nat. Commun. 2015, 6, 7956. [Google Scholar] [CrossRef] [Green Version]
- Thibault, B.; Genre, L.; Le Naour, A.; Broca, C.; Mery, E.; Vuagniaux, G.; Delord, J.P.; Wiedemann, N.; Couderc, B. DEBIO 1143, an IAP inhibitor, reverses carboplatin resistance in ovarian cancer cells and triggers apoptotic or necroptotic cell death. Sci. Rep. 2018, 8, 17862. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Senzer, N.N.; Martin, L.P.; Schilder, R.J.; LoRusso, P.; Papadopoulos, K.P.; Weng, D.E.; Graham, M.; Adjei, A.A. A phase I study of birinapant (TL32711) combined with multiple chemotherapies evaluating tolerability and clinical activity for solid tumor patients. J. Clin. Oncol. 2013, 31, 2504. [Google Scholar]
- Senzer, N.N.; LoRusso, P.; Martin, L.P.; Schilder, R.J.; Amaravadi, R.K.; Papadopoulos, K.P.; Segota, Z.E.; Weng, D.E.; Graham, M.; Adjei, A.A. Phase II clinical activity and tolerability of the SMAC-mimetic birinapant (TL32711) plus irinotecan in irinotecan-relapsed/refractory metastatic colorectal cancer. J. Clin. Oncol. 2013, 31, 3621. [Google Scholar]
- Dienstmann, R.; Vidal, L.; Dees, E.; Chia, S.; Mayer, E.; Porter, D.; Baney, T.; Dhuria, S.; Sen, S.; Firestone, B.; et al. Abstract P6-11-06: A phase Ib Study of LCL161, an oral Inhibitor of Apoptosis (IAP) Antagonist, in Combination with Weekly Paclitaxel in Patients with Advanced Solid Tumors; AACR: Philadelphia, PA, USA, 2012. [Google Scholar]
- Bardia, A.; Cameron, J.S.; Venkatesan, K.; Robinson, D.; Porter, D.; Kim, S.B.; Parton, M.; Kümmel, S.; Estevez, L.G.; Huang, C.S.; et al. 1977 Synergy of LCL161, an antagonist of inhibitor of apoptosis proteins (IAPs), with paclitaxel in a gene expression signature-enriched cohort of triple-negative breast cancer (TNBC). Eur. J. Cancer 2015, 51, S326. [Google Scholar] [CrossRef]
- Firestone, B.; Conway, C.; Yang, G.; Gao, H.; Porter, D.; Slisz, J.; He, D.; Mosher, R.; Monahan, J.; Straub, C.; et al. Abstract B27: Correlation between TNFα and LCL161 Anti-Tumor Activity in Patient Derived Xenograft Models of Human Cancer; AACR: Philadelphia, PA, USA, 2009. [Google Scholar]
- Bardia, A.; Parton, M.; Kummel, S.; Estévez, L.G.; Huang, C.-S.; Cortés, J.; Ruiz-Borrego, M.; Telli, M.L.; Martin-Martorell, P.; López, R.; et al. Paclitaxel with inhibitor of apoptosis antagonist, LCL161, for localized triple-negative breast cancer, prospectively stratified by gene signature in a biomarker-driven neoadjuvant trial. J. Clin. Oncol. 2018, 36, 3126–3133. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.-I.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99. [Google Scholar] [CrossRef]
- Pettersson, M.; Jernberg-Wiklund, H.; Larsson, L.; Sundstrom, C.; Givol, I.; Tsujimoto, Y.; Nilsson, K. Expression of the bcl-2 gene in human multiple myeloma cell lines and normal plasma cells. Blood 1992, 79, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Wuilleme-Toumi, S.; Robillard, N.; Gomez, P.; Moreau, P.; Le Gouill, S.; Avet-Loiseau, H.; Harousseau, J.; Amiot, M.; Bataille, R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005, 19, 1248. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, Y.; Abe, S.; Kurata, M.; Hasegawa, M.; Yamamoto, K.; Inoue, M.; Takemura, T.; Suzuki, K.; Kitagawa, M. IAP family protein expression correlates with poor outcome of multiple myeloma patients in association with chemotherapy-induced overexpression of multidrug resistance genes. Am. J. Hematol. 2006, 81, 824–831. [Google Scholar] [CrossRef]
- Desplanques, G.; Giuliani, N.; Delsignore, R.; Rizzoli, V.; Bataille, R.; Barillé-Nion, S. Impact of XIAP protein levels on the survival of myeloma cells. Haematologica 2009, 94, 87–93. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; Gomez, M.; Prasad, V.; Kimlinger, T.; Painuly, U.; Mukhopadhyay, B.; Haug, J.; Bi, L.; Rajkumar, S.V.; Kumar, S. Smac mimetic LCL161 overcomes protective ER stress induced by obatoclax, synergistically causing cell death in multiple myeloma. Oncotarget 2016, 7, 56253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogler, M.; Weber, K.; Dinsdale, D.; Schmitz, I.; Schulze-Osthoff, K.; Dyer, M.; Cohen, G. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009, 16, 1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalobos-Ortiz, M.; Ryan, J.; Mashaka, T.N.; Opferman, J.T.; Letai, A. BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics. Cell Death Differ. 2019, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Perimenis, P.; Galaris, A.; Voulgari, A.; Prassa, M.; Pintzas, A. IAP antagonists Birinapant and AT-406 efficiently synergise with either TRAIL, BRAF, or BCL-2 inhibitors to sensitise BRAFV600E colorectal tumour cells to apoptosis. BMC Cancer 2016, 16, 624. [Google Scholar] [CrossRef] [Green Version]
- Kearney, C.J.; Lalaoui, N.; Freeman, A.J.; Ramsbottom, K.M.; Silke, J.; Oliaro, J. PD-L1 and IAPs co-operate to protect tumors from cytotoxic lymphocyte-derived TNF. Cell Death Differ. 2017, 24, 1705. [Google Scholar] [CrossRef] [Green Version]
- Beug, S.T.; Beauregard, C.E.; Healy, C.; Sanda, T.; St-Jean, M.; Chabot, J.; Walker, D.E.; Mohan, A.; Earl, N.; Lun, X.; et al. Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kearney, C.J.; Vervoort, S.J.; Hogg, S.J.; Ramsbottom, K.M.; Freeman, A.J.; Lalaoui, N.; Pijpers, L.; Michie, J.; Brown, K.K.; Knight, D.A.; et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci. Immunol. 2018, 3, 3451. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. New Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Vredevoogd, D.W.; Kuilman, T.; Ligtenberg, M.A.; Boshuizen, J.; Stecker, K.E.; de Bruijn, B.; Krijgsman, O.; Huang, X.; Kenski, J.C.; Lacroix, R.; et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell 2019, 178, 585–599. [Google Scholar] [CrossRef]
- Michie, J.; Kearney, C.J.; Hawkins, E.D.; Silke, J.; Oliaro, J. The Immuno-Modulatory Effects of Inhibitor of Apoptosis Protein Antagonists in Cancer Immunotherapy. Cells 2020, 9, 207. [Google Scholar] [CrossRef] [Green Version]
- Laukens, B.; Jennewein, C.; Schenk, B.; Vanlangenakker, N.; Schier, A.; Cristofanon, S.; Zobel, K.; Deshayes, K.; Vucic, D.; Jeremias, I.; et al. Smac mimetic bypasses apoptosis resistance in FADD-or caspase-8-deficient cells by priming for tumor necrosis factor α-induced necroptosis. Neoplasia (New York NY) 2011, 13, 971. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Exploiting inhibitor of apoptosis proteins as therapeutic targets in hematological malignancies. Leukemia 2012, 26, 1155–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinwascher, S.; Nugues, A.-L.; Schoeneberger, H.; Fulda, S. Identification of a novel synergistic induction of cell death by Smac mimetic and HDAC inhibitors in acute myeloid leukemia cells. Cancer Lett. 2015, 366, 32–43. [Google Scholar] [CrossRef] [PubMed]
- McComb, S.; Aguadé-Gorgorió, J.; Harder, L.; Marovca, B.; Cario, G.; Eckert, C.; Schrappe, M.; Stanulla, M.; von Stackelberg, A.; Bourquin, J.-P.; et al. Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci. Transl. Med. 2016, 8, 339–370. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9. [Google Scholar] [CrossRef]
- Han, D.; Ybanez, M.D.; Ahmadi, S.; Yeh, K.; Kaplowitz, N. Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal. 2009, 11, 2245–2263. [Google Scholar] [CrossRef] [Green Version]
- Seyfrid, M.; Marschall, V.; Fulda, S. Reactive oxygen species contribute toward Smac mimetic/temozolomide-induced cell death in glioblastoma cells. Anti-Cancer Drugs 2016, 27, 953–959. [Google Scholar] [CrossRef]
- Hugle, M.; Czaplinski, S.; Habermann, K.; Vogler, M.; Fulda, S. Identification of Smac mimetics as novel substrates for p-glycoprotein. Cancer Lett. 2019, 440, 126–134. [Google Scholar] [CrossRef]
- Talbott, R.L.; Borzilleri, R.M.; Chaudhry, C.; Fargnoli, J.; Shen, H.; Fairchild, C.; Barnhart, B.; Ortega, M.; McDonagh, T.E.; Vuppugalla, R.; et al. Pharmacology of smac mimetics; chemotype differentiation based on physical association with caspase regulators and cellular transport. Exp. Cell Res. 2015, 338, 251–260. [Google Scholar] [CrossRef]
- Adams, J. The proteasome: A suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349. [Google Scholar] [CrossRef]
- Silke, J.; Miasari, M.; Puthalakath, H. Ubiquitylation and Cancer Development. Curr. Cancer Drug Targets 2008, 8, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Hannan, R.D.; Drygin, D.; Pearson, R.B. Targeting RNA polymerase I transcription and the nucleolus for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Smac-Mimetic | cIAP1 | cIAP2 | XIAP | ML-IAP | Reference |
|---|---|---|---|---|---|
| Birinapant | ~1 | 36 | 50 ± 23 | ~1 | Condon et al., 2014 |
| LCL161 | − | − | − | − | Derakhshan et al., 2016 |
| Debio 1143 | 1.9 | 5.1 | 66.4 | − | Cai et al., 2011 |
| GDC-0152 | 17 | 43 | 28 | 14 | Flygare et al., 2013 |
| CUDC-427 | <60 | <60 | <60 | <60 | Wong et al., 2013 |
| Smac-Mimetic | Adjuvant Therapy | Cancer | Phase | Clinical Trial | Date |
|---|---|---|---|---|---|
| Birinapant | Pembrolizumab | Solid cancer | I/II | NCT02587962 | Aug-17 |
| Birinapant | Radiation | HNSCC | I | NCT03803774 | Jan-19 |
| LCL161 | None | Myelofibrosis | II | NCT02098161 | Dec-14 |
| LCL161 | Immunotherapy a | Solid tumors b | Ib | NCT02890069 | Oct-16 |
| LCL161 | Immunotherapy c | Multiple myeloma | I | NCT03111992 | Dec-17 |
| LCL161 | Topotecan | Solid tumors d | I/II | NCT02649673 | Mar-16 |
| Debio 1143 | Nivolumab | Solid cancer | I/II | NCT04122625 | Apr-19 |
| Debio 1143 | Pembrolizumab | Solid tumors e | I | NCT03871959 | Sep-19 |
| Debio 1143 | Avelumab | NSCLC | Ib | NCT03270176 | Oct-17 |
| Debio 1143 | Cisplatin/radiotherapy | HNSCC | I/II | NCT02022098 | Oct-13 |
| APG-1387 | None | Solid cancer/Hema | I/II | NCT03386526 | Nov-17 |
| BI 891065 | Immunotherapy f | Solid tumors g | I | NCT03166631 | Sep-17 |
| BI 891065 | Immunotherapy f | Neoplasm metastasis | II | NCT03697304 | Mar-19 |
| BI 891065 | Immunotherapy f | Neoplasm | I | NCT04138823 | Nov-19 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morrish, E.; Brumatti, G.; Silke, J. Future Therapeutic Directions for Smac-Mimetics. Cells 2020, 9, 406. https://doi.org/10.3390/cells9020406
Morrish E, Brumatti G, Silke J. Future Therapeutic Directions for Smac-Mimetics. Cells. 2020; 9(2):406. https://doi.org/10.3390/cells9020406
Chicago/Turabian StyleMorrish, Emma, Gabriela Brumatti, and John Silke. 2020. "Future Therapeutic Directions for Smac-Mimetics" Cells 9, no. 2: 406. https://doi.org/10.3390/cells9020406
APA StyleMorrish, E., Brumatti, G., & Silke, J. (2020). Future Therapeutic Directions for Smac-Mimetics. Cells, 9(2), 406. https://doi.org/10.3390/cells9020406