Establishment and Characterization of Paired Primary Cultures of Human Pancreatic Cancer Cells and Stellate Cells Derived from the Same Tumor

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Reagents
2.3. Cell Culture
2.4. Morphology and Immunocytochemistry
2.5. Western Blot Analysis
2.6. Mutational Analysis
2.7. Cell Growth and Proliferation
2.8. Preparation of Conditioned Media
2.9. Secretome Analysis
2.10. Effect of PSC-CM on Proliferation and Migration of PCCs
2.11. Statistical Analysis
3. Results
3.1. Clinical Summary of Patients
3.2. Outgrowth Efficiency
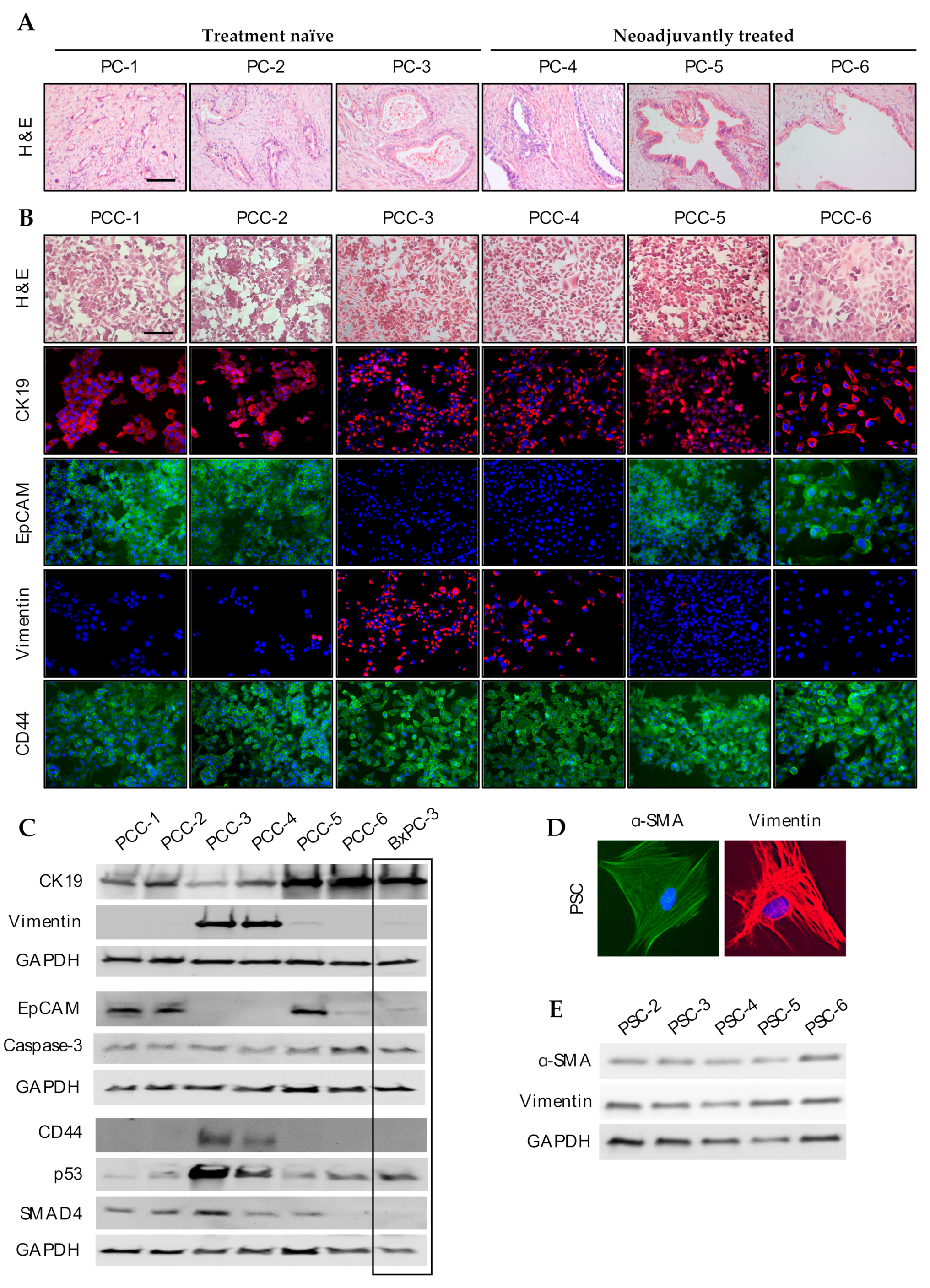
3.3. Phenotypic Characterization of PCCs and PSCs
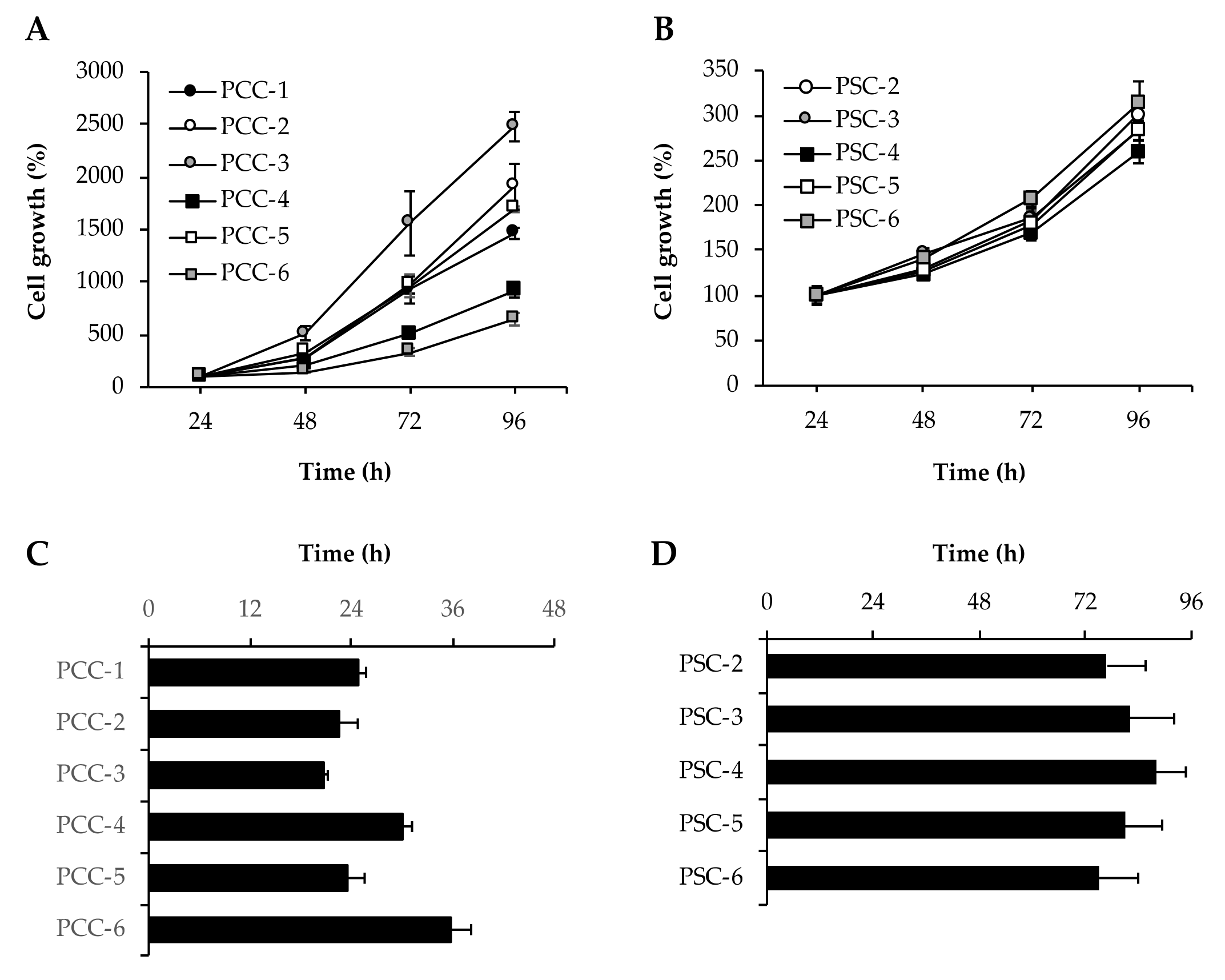
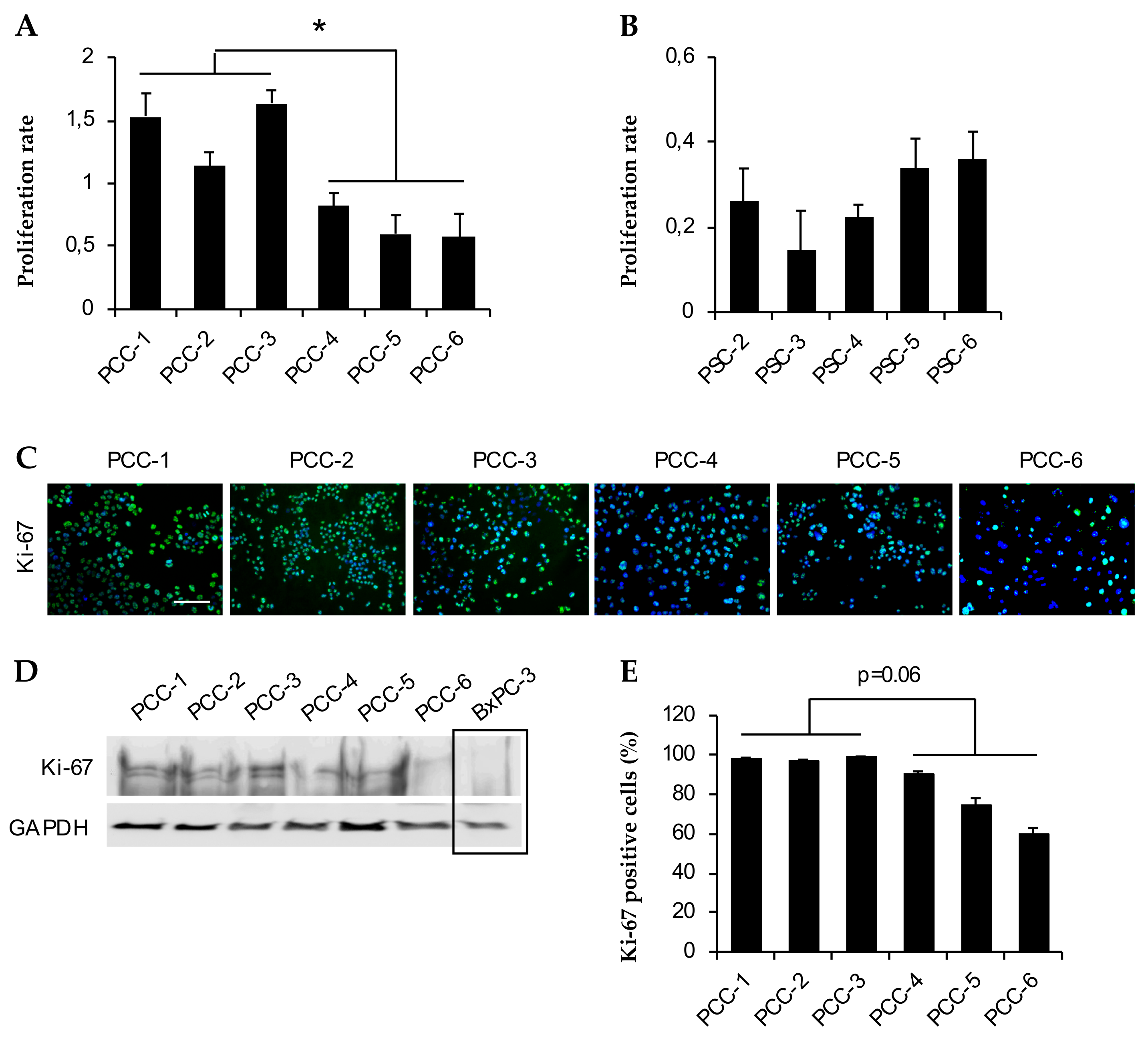
3.4. Assessment of Cell Growth and Proliferation
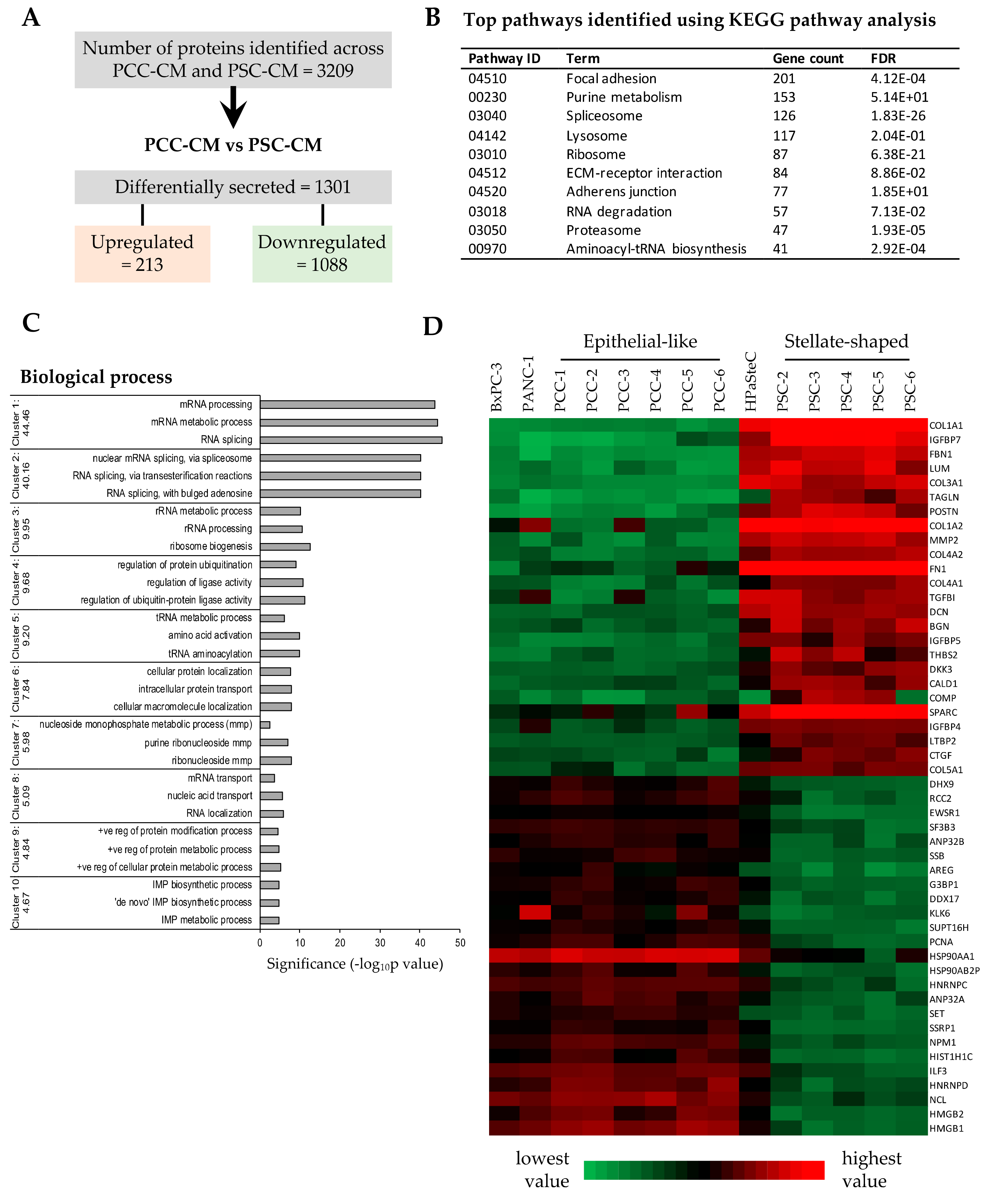
3.5. Proteomics Profiling of PCC- and PSC-Conditioned Medium
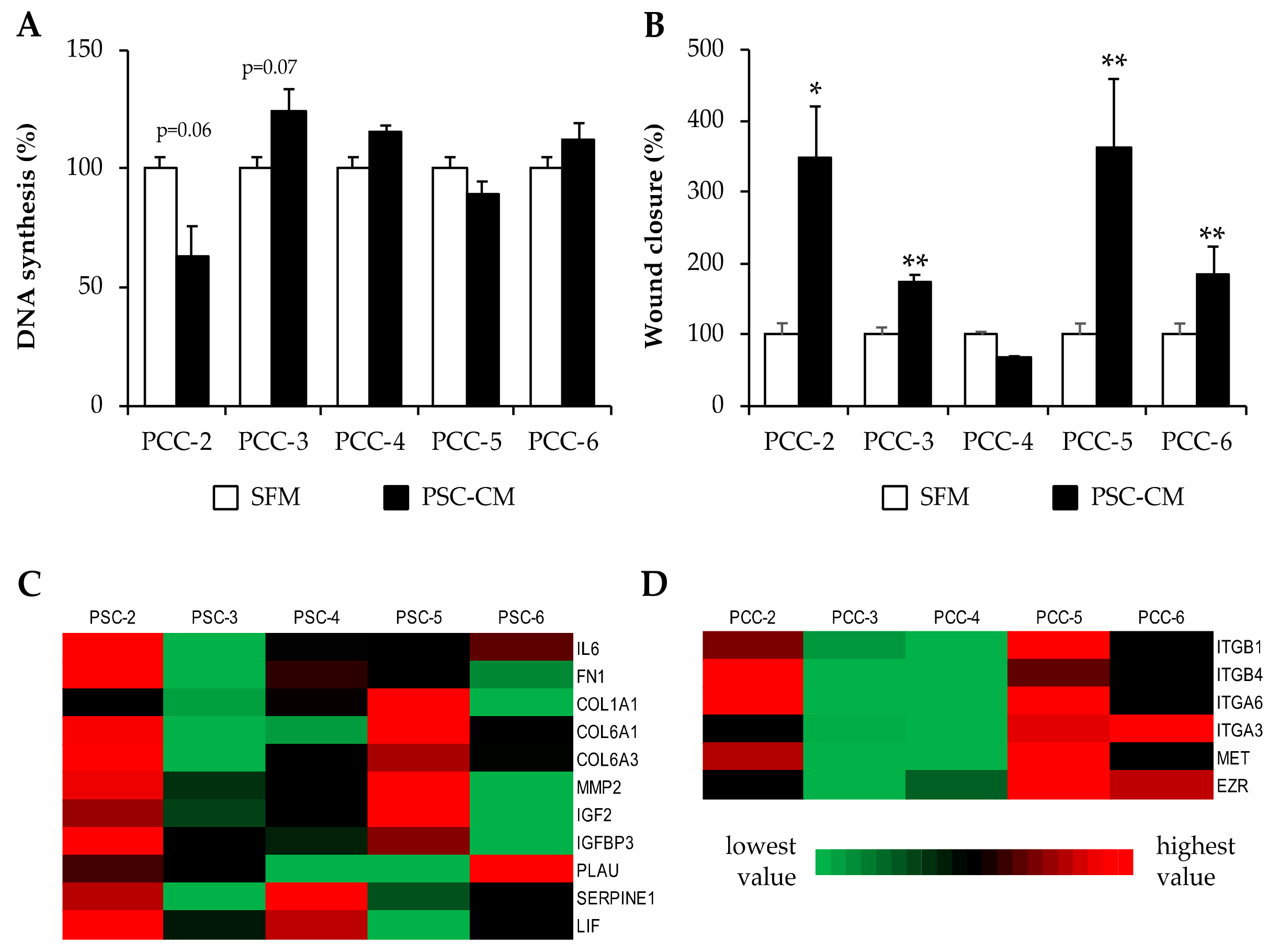
3.6. Effects of PSC-Conditioned Medium on DNA Synthesis and Migration of Paired PCCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Chandana, S.; Babiker, H.M.; Mahadevan, D. Therapeutic trends in pancreatic ductal adenocarcinoma (PDAC). Expert Opin. Investig. Drugs 2019, 28, 161–177. [Google Scholar] [CrossRef]
- Dreyer, S.B.; Chang, D.K.; Bailey, P.; Biankin, A.V. Pancreatic Cancer Genomes: Implications for Clinical Management and Therapeutic Development. Clin. Cancer Res. 2017, 23, 1638–1646. [Google Scholar] [CrossRef] [Green Version]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Labori, K.J.; Katz, M.H.; Tzeng, C.W.; Bjørnbeth, B.A.; Cvancarova, M.; Edwin, B.; Kure, E.H.; Eide, T.J.; Dueland, S.; Buanes, T.; et al. Impact of early disease progression and surgical complications on adjuvant chemotherapy completion rates and survival in patients undergoing the surgery first approach for resectable pancreatic ductal adenocarcinoma—A population-based cohort study. Acta Oncol. 2016, 55, 265–277. [Google Scholar] [CrossRef]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Kadaba, R.; Birke, H.; Wang, J.; Hooper, S.; Andl, C.D.; Di Maggio, F.; Soylu, E.; Ghallab, M.; Bor, D.; Froeling, F.E.; et al. Imbalance of desmoplastic stromal cell numbers drives aggressive cancer processes. J. Pathol. 2013, 230, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016, 381, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Bachem, M.G.; Schneider, E.; Gross, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grünert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef] [Green Version]
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141. [Google Scholar] [CrossRef] [Green Version]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef] [Green Version]
- Tjomsland, V.; Sandnes, D.; Pomianowska, E.; Cizmovic, S.T.; Aasrum, M.; Brusevold, I.J.; Christoffersen, T.; Gladhaug, I.P. The TGFbeta-SMAD3 pathway inhibits IL-1alpha induced interactions between human pancreatic stellate cells and pancreatic carcinoma cells and restricts cancer cell migration. J. Exp. Clin. Cancer Res. 2016, 35, 122. [Google Scholar] [CrossRef] [Green Version]
- Amrutkar, M.; Aasrum, M.; Verbeke, C.S.; Gladhaug, I.P. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer 2019, 19, 596. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.S.; Pirola, R.C.; Apte, M.V. Stars and stripes in pancreatic cancer: Role of stellate cells and stroma in cancer progression. Front. Physiol. 2014, 5, 52. [Google Scholar] [CrossRef] [Green Version]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, J.L.; Yoon, K.A.; Kim, W.H.; Jang, Y.; Suh, K.S.; Kim, S.W.; Park, Y.H.; Park, J.G. Establishment and characterization of four human pancreatic carcinoma cell lines. Genetic alterations in the TGFBR2 gene but not in the MADH4 gene. Cell Tissue Res. 2002, 308, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Rückert, F.; Aust, D.; Böhme, I.; Werner, K.; Brandt, A.; Diamandis, E.P.; Krautz, C.; Hering, S.; Saeger, H.D.; Grützmann, R.; et al. Five primary human pancreatic adenocarcinoma cell lines established by the outgrowth method. J. Surg. Res. 2012, 172, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, M.S.; Kim, S.J.; An, S.; Park, J.; Park, H.; Lee, J.H.; Song, K.B.; Hwang, D.W.; Chang, S.; et al. Establishment and characterization of 6 novel patient-derived primary pancreatic ductal adenocarcinoma cell lines from Korean pancreatic cancer patients. Cancer Cell Int. 2017, 17, 47. [Google Scholar] [CrossRef]
- Lenggenhager, D.; Amrutkar, M.; Sántha, P.; Aasrum, M.; Löhr, J.M.; Gladhaug, I.P.; Verbeke, C.S. Commonly Used Pancreatic Stellate Cell Cultures Differ Phenotypically and in Their Interactions with Pancreatic Cancer Cells. Cells 2019, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Neumann, C.C.M.; von Hörschelmann, E.; Reutzel-Selke, A.; Seidel, E.; Sauer, I.M.; Pratschke, J.; Bahra, M.; Schmuck, R.B. Tumor-stromal cross-talk modulating the therapeutic response in pancreatic cancer. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 461–472. [Google Scholar] [CrossRef]
- Ware, M.J.; Keshishian, V.; Law, J.J.; Ho, J.C.; Favela, C.A.; Rees, P.; Smith, B.; Mohammad, S.; Hwang, R.F.; Rajapakshe, K.; et al. Generation of an in vitro 3D PDAC stroma rich spheroid model. Biomaterials 2016, 108, 129–142. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, S.K.; Khawar, I.A.; Jeong, S.Y.; Chung, S.; Kuh, H.J. Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 4. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef]
- Hwang, H.J.; Oh, M.S.; Lee, D.W.; Kuh, H.J. Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix remodeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells. J. Exp. Clin. Cancer Res. 2019, 38, 258. [Google Scholar] [CrossRef] [Green Version]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell. 2017, 32, 169–184.e167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Santarius, T.; Shipley, J.; Brewer, D.; Stratton, M.R.; Cooper, C.S. A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer 2010, 10, 59–64. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef] [Green Version]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Pomianowska, E.; Sandnes, D.; Grzyb, K.; Schjolberg, A.R.; Aasrum, M.; Tveteraas, I.H.; Tjomsland, V.; Christoffersen, T.; Gladhaug, I.P. Inhibitory effects of prostaglandin E2 on collagen synthesis and cell proliferation in human stellate cells from pancreatic head adenocarcinoma. BMC Cancer 2014, 14, 413. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Tjomsland, V.; Aasrum, M.; Christoffersen, T.; Gladhaug, I.P. Functional heterogeneity in tumor-derived human pancreatic stellate cells: Differential expression of HGF and implications for mitogenic signaling and migration in pancreatic cancer cells. Oncotarget 2017, 8, 71672–71684. [Google Scholar] [CrossRef]
- Wehr, A.Y.; Furth, E.E.; Sangar, V.; Blair, I.A.; Yu, K.H. Analysis of the human pancreatic stellate cell secreted proteome. Pancreas 2011, 40, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzoq, A.J.; Mustafa, S.A.; Heidrich, L.; Hoheisel, J.D.; Alhamdani, M.S.S. Impact of the secretome of activated pancreatic stellate cells on growth and differentiation of pancreatic tumour cells. Sci. Rep. 2019, 9, 5303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafaro, K.J.; Melstrom, L.G. The Paradoxical Web of Pancreatic Cancer Tumor Microenvironment. Am. J. Pathol. 2019, 189, 44–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Tian, Y.; Zhang, J.; Zhang, H.; Gu, F.; Lu, Y.; Zou, S.; Chen, Y.; Sun, P.; Xu, M.; et al. Functions of pancreatic stellate cell-derived soluble factors in the microenvironment of pancreatic ductal carcinoma. Oncotarget 2017, 8, 102721–102738. [Google Scholar] [CrossRef] [Green Version]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Farran, B.; Nagaraju, G.P. The dynamic interactions between the stroma, pancreatic stellate cells and pancreatic tumor development: Novel therapeutic targets. Cytokine Growth Factor Rev. 2019, 48, 11–23. [Google Scholar] [CrossRef]
- Schüssler, M.H.; Skoudy, A.; Ramaekers, F.; Real, F.X. Intermediate filaments as differentiation markers of normal pancreas and pancreas cancer. Am. J. Pathol. 1992, 140, 559–568. [Google Scholar]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. CMLS 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [Green Version]
- Spizzo, G.; Fong, D.; Wurm, M.; Ensinger, C.; Obrist, P.; Hofer, C.; Mazzoleni, G.; Gastl, G.; Went, P. EpCAM expression in primary tumour tissues and metastases: An immunohistochemical analysis. J. Clin. Pathol. 2011, 64, 415–420. [Google Scholar] [CrossRef] [Green Version]
- Quint, K.; Tonigold, M.; Di Fazio, P.; Montalbano, R.; Lingelbach, S.; Ruckert, F.; Alinger, B.; Ocker, M.; Neureiter, D. Pancreatic cancer cells surviving gemcitabine treatment express markers of stem cell differentiation and epithelial-mesenchymal transition. Int. J. Oncol. 2012, 41, 2093–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.P.; Zhang, X.W.; Zheng, L.Z.; Guo, W.J. Expression of CD44 in pancreatic cancer and its significance. Int. J. Clin. Exp. Pathol. 2015, 8, 6724–6731. [Google Scholar] [PubMed]
- Kiuchi, S.; Ikeshita, S.; Miyatake, Y.; Kasahara, M. Pancreatic cancer cells express CD44 variant 9 and multidrug resistance protein 1 during mitosis. Exp. Mol. Pathol. 2015, 98, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression Model for Pancreatic Cancer. Clin. Cancer Res. 2000, 6, 2969. [Google Scholar]
- Wood, L.D.; Hruban, R.H. Pathology and molecular genetics of pancreatic neoplasms. Cancer J. 2012, 18, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Wu, W.; Huang, C.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Qiu, Z. SMAD4 and its role in pancreatic cancer. Tumor Biol. 2015, 36, 111–119. [Google Scholar] [CrossRef]
- Becker, A.E.; Hernandez, Y.G.; Frucht, H.; Lucas, A.L. Pancreatic ductal adenocarcinoma: Risk factors, screening, and early detection. World J. Gastroenterol. 2014, 20, 11182–11198. [Google Scholar] [CrossRef]
- Sipos, B.; Möser, S.; Kalthoff, H.; Török, V.; Löhr, M.; Klöppel, G. A comprehensive characterization of pancreatic ductal carcinoma cell lines: Towards the establishment of an in vitro research platform. Virchows Arch. 2003, 442, 444–452. [Google Scholar] [CrossRef]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimczak, M.; Biecek, P.; Zylicz, A.; Zylicz, M. Heat shock proteins create a signature to predict the clinical outcome in breast cancer. Sci. Rep. 2019, 9, 7507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, C.; Sun, J.; Lin, Z.; Jin, T.; Dong, B.; Meng, Z.; Piao, J. Ezrin promotes pancreatic cancer cell proliferation and invasion through activating the Akt/mTOR pathway and inducing YAP translocation. Cancer Manag. Res. 2019, 11, 6553–6566. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDAC ID | Tumor Site | TN-Stage | Tumor Size (mm) | Grade | Treatment | TRG | L | V | Pn | R | Survival (Months) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PC-1 | HOP | pT2(3)N0 | 35 × 28 × 25 | 2 | NA | NA | 0 | 0 | 0 | 0 | 19.3 |
| PC-2 | HOP | pT2(3) N2(1) | 39 × 35 × 30 | 2 | NA | NA | 1 | 1 | 1 | 1 | 10.3 |
| PC-3 | HOP | pT2(3) N1 | 26 × 24 × 21 | 3 | NA | NA | 1 | 1 | 1 | 0 | 11.9 |
| PC-4 | HOP | ypT3 N1 | 42 × 35 × 31 | NA | 4× Folfirinox | 3 | 1 | 0 | 1 | 1 | 33.0+ |
| PC-5 | HOP | ypT3 N1 | 55 × 21 × 10 | NA | 1× Folfirinox, 5× Gemzar -Abraxane | 2–3 | 1 | 1 | 1 | 1 | 11.5 |
| PC-6 | HOP | ypT3N2(1) | 52 × 50 × 32 | NA | 7× Gemzar | 3 | 1 | 0 | 1 | 1 | 12.8 |
| Target | PCC-1 | PCC-2 | PCC-3 | PCC-4 | PCC-5 | PCC-6 | BxPC-3 |
|---|---|---|---|---|---|---|---|
| CK19 | ++ | ++ | ++ | ++ | ++ | ++ | ++ |
| EpCAM | ++ | ++ | - | - | ++ | ++ | - |
| Vimentin | - | - | ++ | ++ | - | - | - |
| Ki-67 | ++ | ++ | ++ | + | + | + | - |
| p53 | + | + | ++ | ++ | + | + | + |
| SMAD4 | ++ | ++ | ++ | + | + | - | - |
| CD44 (ic) | + | + | + | + | + | + | - |
| Caspase-3 | ++ | ++ | + | + | + | + | + |
| p16 | ++ | ++ | + | + | + | ++ | - |
| Target (RefSeq) | PCC-1 | PCC-2 | PCC-3 | PCC-4 | PCC-5 | PCC-6 | AsPC-1 | Panc-1 |
|---|---|---|---|---|---|---|---|---|
| KRAS (nm_004985) | p.G13D (0.47) | p.G13D (0.46) | p.G12C (1.0) | p.G12C (1.0) | p.G12C (0.78) | p.G12V (0.67) | p.G12D (0.99) | p.G12C (0.49) |
| TP53 (nm-000546) | p.K373fs (0.33) | - | - | p.D21E (0.21) | - | p.A138V (1.0) | p.C135fs (0.86) | - |
| CDKN2A (nm_000077) | p.G23fs (0.37) p.E33fs (0.45) | p.G23fs (0.35) p.E33fs (0.40) | - | - | p.G23fs (0.35) p.E33fs (0.40) | - | p.L78fs (0.77) | p.G23fs (0.35) p.E33fs (0.40) |
| SMAD4 (nm_005359) | - | - | - | - | - | - | p.R100T (1.0) | - |
| BRCA1 (nm_007294) | - | - | - | - | - | - | p.D693N (1.0) | - |
| BRCA2 (nm_000059) | p.I2672fs (0.32) | p.I2672fs (0.37) | - | - | p.I2672fs (0.20) | p.N372H (0.54) | p.I2672fs (0.32) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amrutkar, M.; Larsen, E.K.; Aasrum, M.; Finstadsveen, A.V.; Andresen, P.A.; Verbeke, C.S.; Gladhaug, I.P. Establishment and Characterization of Paired Primary Cultures of Human Pancreatic Cancer Cells and Stellate Cells Derived from the Same Tumor. Cells 2020, 9, 227. https://doi.org/10.3390/cells9010227
Amrutkar M, Larsen EK, Aasrum M, Finstadsveen AV, Andresen PA, Verbeke CS, Gladhaug IP. Establishment and Characterization of Paired Primary Cultures of Human Pancreatic Cancer Cells and Stellate Cells Derived from the Same Tumor. Cells. 2020; 9(1):227. https://doi.org/10.3390/cells9010227
Chicago/Turabian StyleAmrutkar, Manoj, Emma Kristine Larsen, Monica Aasrum, Anette Vefferstad Finstadsveen, Per Arne Andresen, Caroline S. Verbeke, and Ivar P. Gladhaug. 2020. "Establishment and Characterization of Paired Primary Cultures of Human Pancreatic Cancer Cells and Stellate Cells Derived from the Same Tumor" Cells 9, no. 1: 227. https://doi.org/10.3390/cells9010227
APA StyleAmrutkar, M., Larsen, E. K., Aasrum, M., Finstadsveen, A. V., Andresen, P. A., Verbeke, C. S., & Gladhaug, I. P. (2020). Establishment and Characterization of Paired Primary Cultures of Human Pancreatic Cancer Cells and Stellate Cells Derived from the Same Tumor. Cells, 9(1), 227. https://doi.org/10.3390/cells9010227