Interaction between Polycomb and SSX Proteins in Pericentromeric Heterochromatin Function and Its Implication in Cancer


{kind=link}
{kind=link}
Abstract
:1. Structure and Function of Polycomb Protein Complexes
2. Genomic Targeting of PcG Complexes
3. The Role of PcG Complexes in Repression of Pericentromeric Heterochromatin
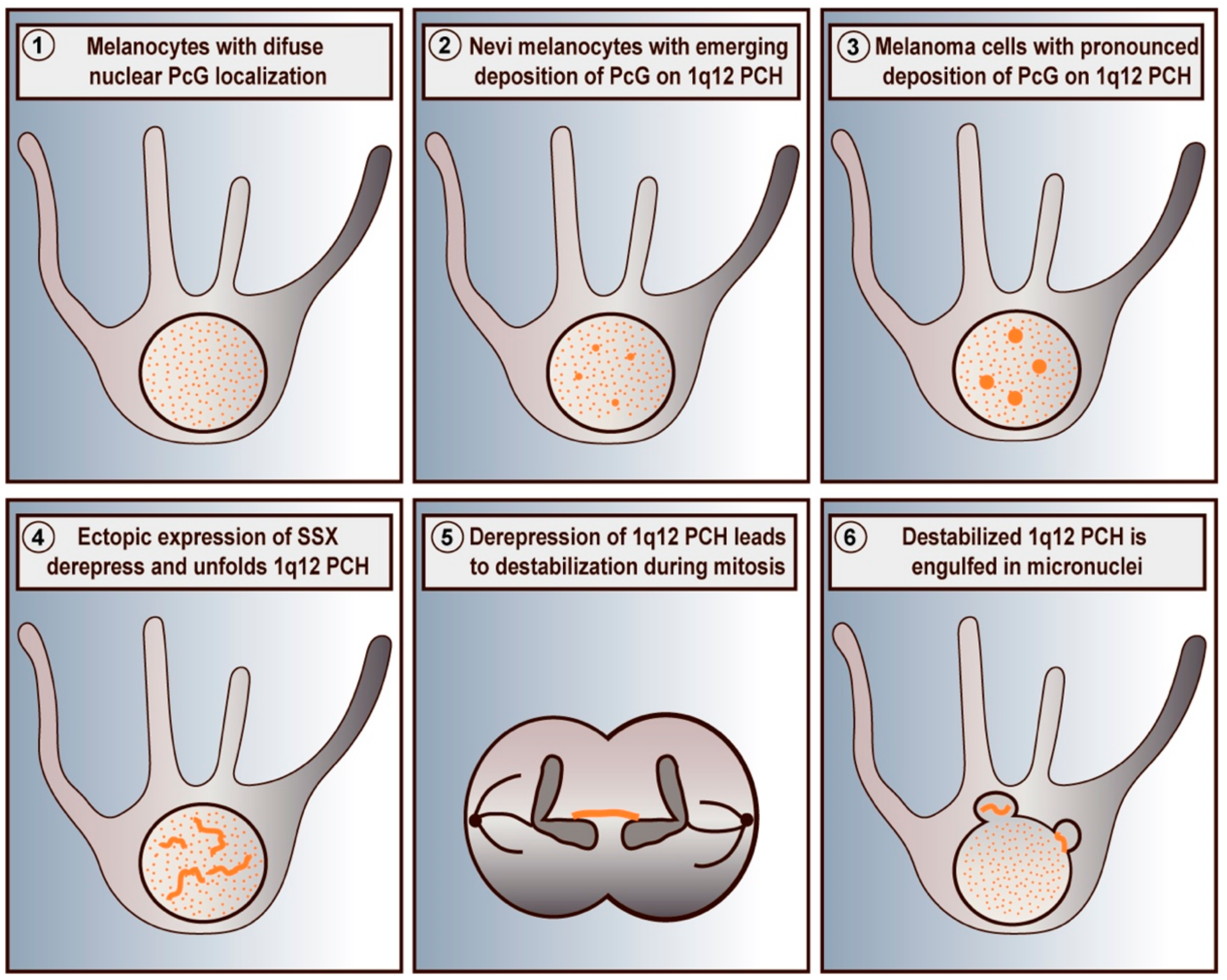
4. PcG Bodies in Premalignant and Malignant Cells
5. The SSX Family of Chromatin-Modulating Proteins
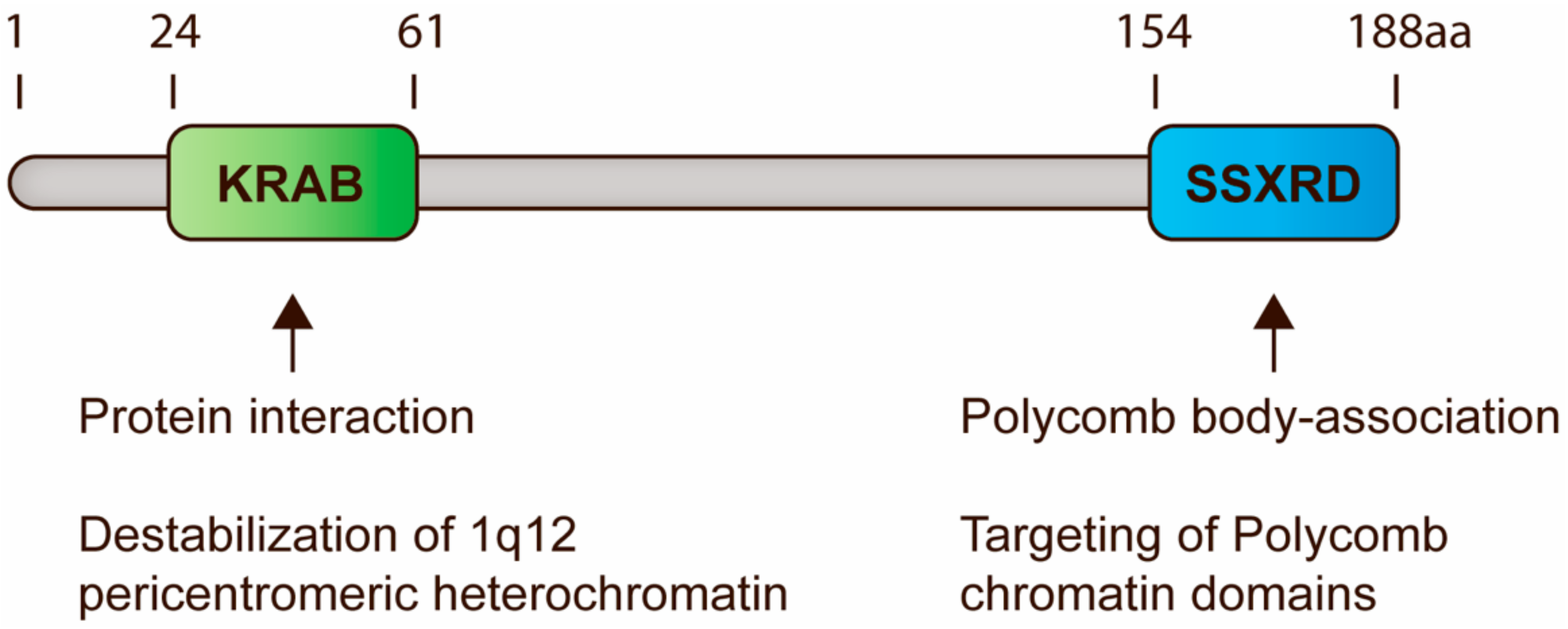
6. Interaction between SSX and PcG Factors
7. SSX-Mediated Derepression of PcG-Silenced Heterochromatin
8. Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Lewis, E.B. A gene complex controlling segmentation in Drosophila. Nature 1978, 276, 565–570. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, Y.B.; Kahn, T.G.; Nix, D.A.; Li, X.Y.; Bourgon, R.; Biggin, M.; Pirrotta, V. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat. Genet. 2006, 38, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.I.; et al. Control of Developmental Regulators by Polycomb in Human Embryonic Stem Cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef]
- Sparmann, A.; Lohuizen, M.V. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006, 6, 846–856. [Google Scholar] [CrossRef]
- Mills, A.A. Throwing the cancer switch: Reciprocal roles of polycomb and trithorax proteins. Nat. Rev. Cancer 2010, 10, 669–682. [Google Scholar] [CrossRef]
- Chittock, E.C.; Latwiel, S.; Miller, T.C.R.; Müller, C.W. Molecular architecture of polycomb repressive complexes. Biochem. Soc. Trans. 2017, 45, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF Homologs, CBX Proteins, and RYBP Define Functionally Distinct PRC1 Family Complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Tavares, L.; Dimitrova, E.; Oxley, D.; Webster, J.; Poot, R.; Demmers, J.; Bezstarosti, K.; Taylor, S.; Ura, H.; Koide, H.; et al. RYBP-PRC1 Complexes Mediate H2A Ubiquitylation at Polycomb Target Sites Independently of PRC2 and H3K27me3. Cell 2012, 148, 664–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.T.C.; Müller, C.W.; Vermeulen, M.; Müller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 Complex-Dependent H2A Ubiquitylation Drives PRC2 Recruitment and Polycomb Domain Formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S.; Dienstbier, M.; Hassan, R.; Schermelleh, L.; Sharif, J.; Blackledge, N.P.; De Marco, V.; Elderkin, S.; Koseki, H.; Klose, R.; et al. Targeting Polycomb to Pericentric Heterochromatin in Embryonic Stem Cells Reveals a Role for H2AK119u1 in PRC2 Recruitment. Cell Rep. 2014, 7, 1456–1470. [Google Scholar] [CrossRef] [Green Version]
- Fursova, N.A.; Blackledge, N.P.; Nakayama, M.; Ito, S.; Koseki, Y.; Farcas, A.M.; King, H.W.; Koseki, H.; Klose, R.J. Synergy between Variant PRC1 Complexes Defines Polycomb-Mediated Gene Repression. Mol. Cell 2019, 74, 1020–1036. e1028. [Google Scholar] [CrossRef] [Green Version]
- Kahn, T.G.; Stenberg, P.; Pirrotta, V.; Schwartz, Y.B. Combinatorial Interactions Are Required for the Efficient Recruitment of Pho Repressive Complex (PhoRC) to Polycomb Response Elements. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef] [Green Version]
- Schuettengruber, B.; Oded Elkayam, N.; Sexton, T.; Entrevan, M.; Stern, S.; Thomas, A.; Yaffe, E.; Parrinello, H.; Tanay, A.; Cavalli, G. Cooperativity, Specificity, and Evolutionary Stability of Polycomb Targeting in Drosophila. Cell Rep. 2014, 9, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide Analysis of PRC1 and PRC2 Occupancy Identifies Two Classes of Bivalent Domains. PLoS Genet. 2008, 4. [Google Scholar] [CrossRef]
- Mendenhall, E.M.; Koche, R.P.; Truong, T.; Zhou, V.W.; Issac, B.; Chi, A.S.; Ku, M.; Bernstein, B.E. GC-Rich Sequence Elements Recruit PRC2 in Mammalian ES Cells. PLoS Genet. 2010, 6, e1001244. [Google Scholar] [CrossRef] [Green Version]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.K.; McGouran, J.F.; Rose, N.R.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.W.; et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. ELife 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Blackledge, N.P.; Rose, N.R.; Klose, R.J. Targeting polycomb systems to regulate gene expression: Modifications to a complex story. Nat. Reviews. Mol. Cell Biol. 2015, 16, 643–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Rocha, S.T.; Boeva, V.; Escamilla-Del-Arenal, M.; Ancelin, K.; Granier, C.; Matias, N.R.; Sanulli, S.; Chow, J.; Schulz, E.; Picard, C.; et al. Jarid2 Is Implicated in the Initial Xist-Induced Targeting of PRC2 to the Inactive X Chromosome. Mol. Cell 2014, 53, 301–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarma, K.; Cifuentes-Rojas, C.; Ergun, A.; Del Rosario, A.; Jeon, Y.; White, F.; Sadreyev, R.; Lee, J.T. ATRX Directs Binding of PRC2 to Xist RNA and Polycomb Targets. Cell 2014, 159, 869–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidovich, C.; Cech, T.R. The recruitment of chromatin modifiers by long noncoding RNAs: Lessons from PRC2. RNA 2015, 21, 2007–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanhere, A.; Viiri, K.; Araújo, C.C.; Rasaiyaah, J.; Bouwman, R.D.; Whyte, W.A.; Pereira, C.F.; Brookes, E.; Walker, K.; Bell, G.W.; et al. Short RNAs are transcribed from repressed Polycomb target genes and interact with Polycomb Repressive Complex-2. Mol. Cell 2010, 38, 675–688. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, S.; Son, J.; Bonasio, R.; Shen, S.S.; Reinberg, D. Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev. 2014, 28, 1983–1988. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes-Rojas, C.; Hernandez, A.J.; Sarma, K.; Lee, J.T. Regulatory interactions between RNA and polycomb repressive complex 2. Mol. Cell 2014, 55, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Simon, J.A.; Kingston, R.E. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 2013, 49, 808–824. [Google Scholar] [CrossRef] [Green Version]
- Stock, J.K.; Giadrossi, S.; Casanova, M.; Brookes, E.; Vidal, M.; Koseki, H.; Brockdorff, N.; Fisher, A.G.; Pombo, A. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat. Cell Biol. 2007, 9, 1428–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Zhu, P.; Wang, J.; Pascual, G.; Ohgi, K.A.; Lozach, J.; Glass, C.K.; Rosenfeld, M.G. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol. Cell 2008, 29, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Kajitani, T.; Togo, S.; Masuko, N.; Ohdan, H.; Hishikawa, Y.; Koji, T.; Matsuyama, T.; Ikura, T.; Muramatsu, M.; et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 2008, 22, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, A.H.; Boettiger, A.N.; Schorderet, P.; Ergun, A.; Münger, C.; Sadreyev, R.I.; Zhuang, X.; Kingston, R.E.; Francis, N.J. Chromatin topology is coupled to Polycomb group protein subnuclear organization. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Isono, K.; Endo, T.A.; Ku, M.; Yamada, D.; Suzuki, R.; Sharif, J.; Ishikura, T.; Toyoda, T.; Bernstein, B.E.; Koseki, H. SAM Domain Polymerization Links Subnuclear Clustering of PRC1 to Gene Silencing. Dev. Cell 2013, 26, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, M.S.; Schwartz, M.G.; Kundu, S.; Savol, A.J.; Wang, P.I.; Marr, S.K.; Grau, D.J.; Schorderet, P.; Sadreyev, R.I.; Tabin, C.J.; et al. Mutation of a nucleosome compaction region disrupts Polycomb-mediated axial patterning. Science 2017, 355, 1081–1084. [Google Scholar] [CrossRef] [Green Version]
- Francis, N.J.; Kingston, R.E.; Woodcock, C.L. Chromatin Compaction by a Polycomb Group Protein Complex. Science 2004, 306, 1574–1577. [Google Scholar] [CrossRef] [Green Version]
- Entrevan, M.; Schuettengruber, B.; Cavalli, G. Regulation of Genome Architecture and Function by Polycomb Proteins. Trends Cell Biol. 2016, 26, 511–525. [Google Scholar] [CrossRef]
- Pasini, D.; Hansen, K.H.; Christensen, J.; Agger, K.; Cloos, P.A.C.; Helin, K. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Developmen. 2008, 22, 1345–1355. [Google Scholar] [CrossRef] [Green Version]
- Tie, F.; Banerjee, R.; Fu, C.; Stratton, C.A.; Fang, M.; Harte, P.J. Polycomb inhibits histone acetylation by CBP by binding directly to its catalytic domain. Proc. Natl. Acad. Sci. USA 2016, 113, E744–E753. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stützer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-Dependent H3K27me1 and H3K27me2 Regulate Active Transcription and Enhancer Fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Saksouk, N.; Barth, T.K.; Ziegler-Birling, C.; Olova, N.; Nowak, A.; Rey, E.; Mateos-Langerak, J.; Urbach, S.; Reik, W.; Torres-Padilla, M.E.; et al. Redundant Mechanisms to Form Silent Chromatin at Pericentromeric Regions Rely on BEND3 and DNA Methylation. Mol. Cell 2014, 56, 580–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puschendorf, M.; Terranova, R.; Boutsma, E.; Mao, X.; Isono, K.I.; Brykczynska, U.; Kolb, C.; Otte, A.P.; Koseki, H.; Orkin, S.H.; et al. PRC1 and Suv39h specify parental asymmetry at constitutive heterochromatin in early mouse embryos. Nat. Genet. 2008, 40, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Abdouh, M.; Hanna, R.; El Hajjar, J.; Flamier, A.; Bernier, G. The Polycomb Repressive Complex 1 Protein BMI1 Is Required for Constitutive Heterochromatin Formation and Silencing in Mammalian Somatic Cells. J. Biol. Chem. 2016, 291, 182–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saurin, A.J.; Shiels, C.; Williamson, J.; Satijn, D.P.; Otte, A.P.; Sheer, D.; Freemont, P.S. The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J. Cell Biol. 1998, 142, 887–898. [Google Scholar] [CrossRef] [Green Version]
- Déjardin, J. Switching between Epigenetic States at Pericentromeric Heterochromatin. Trends Genet. 2015, 31, 661–672. [Google Scholar] [CrossRef]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.H.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; Li, E.; Jenuwein, T.; Peters, A.H.F.M. Suv39h-Mediated Histone H3 Lysine 9 Methylation Directs DNA Methylation to Major Satellite Repeats at Pericentric Heterochromatin. Curr. Biol. 2003, 13, 1192–1200. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.H.F.M.; O’Carroll, D.; Scherthan, H.; Mechtler, K.; Sauer, S.; Schöfer, C.; Weipoltshammer, K.; Pagani, M.; Lachner, M.; Kohlmaier, A.; et al. Loss of the Suv39h Histone Methyltransferases Impairs Mammalian Heterochromatin and Genome Stability. Cell 2001, 107, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Jagannathan, M.; Cummings, R.; Yamashita, Y.M. A conserved function for pericentromeric satellite DNA. Elife 2018, 7. [Google Scholar] [CrossRef]
- Hall, L.L.; Byron, M.; Carone, D.M.; Whitfield, T.W.; Pouliot, G.P.; Fischer, A.; Jones, P.; Lawrence, J.B. Demethylated HSATII DNA and HSATII RNA Foci Sequester PRC1 and MeCP2 into Cancer-Specific Nuclear Bodies. Cell Rep. 2017, 18, 2943–2956. [Google Scholar] [CrossRef]
- Pirrotta, V.; Li, H.B. A view of nuclear Polycomb bodies. Curr. Opin. Genet. Dev. 2012, 22, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruckmann, N.H.; Pedersen, C.B.; Ditzel, H.J.; Gjerstorff, M.F. Epigenetic Reprogramming of Pericentromeric Satellite DNA in Premalignant and Malignant Lesions. Mol. Cancer Res. 2018, 16, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, F.; Peters, A.H.; Otte, A.P.; Reik, W.; Dean, W. Dynamic chromatin modifications characterise the first cell cycle in mouse embryos. Dev. Biol. 2005, 280, 225–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardat, M.; Albert, M.; Kunzmann, R.; Liu, Z.; Kaustov, L.; Thierry, R.; Duan, S.; Brykczynska, U.; Arrowsmith, C.H.; Peters, A.H. Cbx2 targets PRC1 to constitutive heterochromatin in mouse zygotes in a parent-of-origin-dependent manner. Mol. Cell. 2015, 58, 157–171. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, N.; Salmon-Divon, M.; Dvinge, H.; Hynes-Allen, A.; Balasooriya, G.; Leaford, D.; Behrens, A.; Bertone, P.; Hendrich, B. NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. EMBO J. 2012, 31, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Narayan, A.; Ji, W.; Zhang, X.Y.; Marrogi, A.; Graff, J.R.; Baylin, S.B.; Ehrlich, M. Hypomethylation of pericentromeric DNA in breast adenocarcinomas. Int. J. Cancer 1998, 77, 833–838. [Google Scholar] [CrossRef]
- Fanelli, M.; Caprodossi, S.; Ricci-Vitiani, L.; Porcellini, A.; Tomassoni-Ardori, F.; Amatori, S.; Andreoni, F.; Magnani, M.; De Maria, R.; Santoni, A.; et al. Loss of pericentromeric DNA methylation pattern in human glioblastoma is associated with altered DNA methyltransferases expression and involves the stem cell compartment. Oncogene 2008, 27, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Kanai, Y.; Ushijima, S.; Kitamura, T.; Kakizoe, T.; Hirohashi, S. Dna hypomethylation on pericentromeric satellite regions significantly correlates with loss of heterozygosity on chromosome 9 in urothelial carcinomas. J. Urol. 2005, 173, 243–246. [Google Scholar] [CrossRef]
- Ribera, J.; Zamora, L.; Morgades, M.; Mallo, M.; Solanes, N.; Batlle, M.; Vives, S.; Granada, I.; Junca, J.; Malinverni, R.; et al. Copy number profiling of adult relapsed B-cell precursor acute lymphoblastic leukemia reveals potential leukemia progression mechanisms. Genes Chromosomes Cancer 2017, 56, 810–820. [Google Scholar] [CrossRef]
- Schubert, S.A.; Ruano, D.; Elsayed, F.A.; Boot, A.; Crobach, S.; Sarasqueta, A.F.; Wolffenbuttel, B.; van der Klauw, M.M.; Oosting, J.; Tops, C.M.; et al. Evidence for genetic association between chromosome 1q loci and predisposition to colorectal neoplasia. Br. J. Cancer 2017, 117, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, J.R.; Tricot, G.; Mattox, S.; Jagannath, S.; Barlogie, B. Jumping translocations of chromosome 1q in multiple myeloma: Evidence for a mechanism involving decondensation of pericentromeric heterochromatin. Blood 1998, 91, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Le Baccon, P.; Leroux, D.; Dascalescu, C.; Duley, S.; Marais, D.; Esmenjaud, E.; Sotto, J.J.; Callanan, M. Novel evidence of a role for chromosome 1 pericentric heterochromatin in the pathogenesis of B-cell lymphoma and multiple myeloma. Genes Chromosomes Cancer 2001, 32, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Millington, K.; Hudnall, S.D.; Northup, J.; Panova, N.; Velagaleti, G. Role of chromosome 1 pericentric heterochromatin (1q) in pathogenesis of myelodysplastic syndromes: Report of 2 new cases. Exp. Mol. Pathol. 2008, 84, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Tatavosian, R.; Kent, S.; Brown, K.; Yao, T.; Duc, H.N.; Huynh, T.N.; Zhen, C.Y.; Ma, B.; Wang, H.; Ren, X. Nuclear condensates of the Polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem. 2019, 294, 1451–1463. [Google Scholar] [CrossRef] [Green Version]
- Plys, A.J.; Davis, C.P.; Kim, J.; Rizki, G.; Keenen, M.M.; Marr, S.K.; Kingston, R.E. Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes. Dev. 2019, 33, 799–813. [Google Scholar] [CrossRef] [Green Version]
- Mertens, F.; Johansson, B.; Hoglund, M.; Mitelman, F. Chromosomal imbalance maps of malignant solid tumors: A cytogenetic survey of 3185 neoplasms. Cancer Res. 1997, 57, 2765–2780. [Google Scholar]
- Sawyer, J.R.; Swanson, C.M.; Wheeler, G.; Cunniff, C. Chromosome instability in ICF syndrome: Formation of micronuclei from multibranched chromosomes 1 demonstrated by fluorescence in situ hybridization. Am. J. Med. Genet. 1995, 56, 203–209. [Google Scholar] [CrossRef]
- Tuck-Muller, C.M.; Narayan, A.; Tsien, F.; Smeets, D.F.; Sawyer, J.; Fiala, E.S.; Sohn, O.S.; Ehrlich, M. DNA hypomethylation and unusual chromosome instability in cell lines from ICF syndrome patients. Cytogenet. Cell Genet. 2000, 89, 121–128. [Google Scholar] [CrossRef]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Pequignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Hassan, K.M.; Norwood, T.; Gimelli, G.; Gartler, S.M.; Hansen, R.S. Satellite 2 methylation patterns in normal and ICF syndrome cells and association of hypomethylation with advanced replication. Hum. Genet. 2001, 109, 452–462. [Google Scholar] [CrossRef]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Poynter, J.N.; Elder, J.T.; Fullen, D.R.; Nair, R.P.; Soengas, M.S.; Johnson, T.M.; Redman, B.; Thomas, N.E.; Gruber, S.B. BRAF and NRAS mutations in melanoma and melanocytic nevi. Melanoma Res. 2006, 16, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Swanson, E.C.; Rapkin, L.M.; Bazett-Jones, D.P.; Lawrence, J.B. Unfolding the story of chromatin organization in senescent cells. Nucleus 2015, 6, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traynor, S.; Mollegaard, N.E.; Jorgensen, M.G.; Bruckmann, N.H.; Pedersen, C.B.; Terp, M.G.; Johansen, S.; Dejardin, J.; Ditzel, H.J.; Gjerstorff, M.F. Remodeling and destabilization of chromosome 1 pericentromeric heterochromatin by SSX proteins. Nucleic Acids Res. 2019, 47, 6668–6684. [Google Scholar] [CrossRef] [Green Version]
- Gure, A.O.; Wei, I.J.; Old, L.J.; Chen, Y.T. The SSX gene family: Characterization of 9 complete genes. Int. J. Cancer. 2002, 101, 448–453. [Google Scholar] [CrossRef]
- Smith, H.A.; McNeel, D.G. The SSX family of cancer-testis antigens as target proteins for tumor therapy. Clin. Dev. Immunol. 2010, 2010, 150591. [Google Scholar] [CrossRef] [Green Version]
- Greve, K.B.; Pohl, M.; Olsen, K.E.; Nielsen, O.; Ditzel, H.J.; Gjerstorff, M.F. SSX2-4 expression in early-stage non-small cell lung cancer. Tissue Antigens 2014, 83, 344–349. [Google Scholar] [CrossRef]
- dos Santos, N.R.; Torensma, R.; de Vries, T.J.; Schreurs, M.W.; de Bruijn, D.R.; Kater-Baats, E.; Ruiter, D.J.; Adema, G.J.; van Muijen, G.N.; van Kessel, A.G. Heterogeneous expression of the SSX cancer/testis antigens in human melanoma lesions and cell lines. Cancer Res. 2000, 60, 1654–1662. [Google Scholar]
- Gjerstorff, M.F.; Andersen, M.H.; Ditzel, H.J. Oncogenic cancer/testis antigens: Prime candidates for immunotherapy. Oncotarget 2015, 6, 15772–15787. [Google Scholar] [CrossRef] [Green Version]
- Brett, D.; Whitehouse, S.; Antonson, P.; Shipley, J.; Cooper, C.; Goodwin, G. The SYT protein involved in the t(X;18) synovial sarcoma translocation is a transcriptional activator localised in nuclear bodies. Hum. Mol. Genet. 1997, 6, 1559–1564. [Google Scholar] [CrossRef]
- Lim, F.L.; Soulez, M.; Koczan, D.; Thiesen, H.J.; Knight, J.C. A KRAB-related domain and a novel transcription repression domain in proteins encoded by SSX genes that are disrupted in human sarcomas. Oncogene 1998, 17, 2013–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urrutia, R. KRAB-containing zinc-finger repressor proteins. Genome Biol. 2003, 4, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bruijn, D.R.; dos Santos, N.R.; Kater-Baats, E.; Thijssen, J.; van den Berk, L.; Stap, J.; Balemans, M.; Schepens, M.; Merkx, G.; van Kessel, A.G. The cancer-related protein SSX2 interacts with the human homologue of a Ras-like GTPase interactor, RAB3IP, and a novel nuclear protein, SSX2IP. Genes Chromosomes Cancer 2002, 34, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, S.A.; Nekludova, L.; Pabo, C.O. DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 183–212. [Google Scholar] [CrossRef] [PubMed]
- Gjerstorff, M.F.; Relster, M.M.; Greve, K.B.; Moeller, J.B.; Elias, D.; Lindgreen, J.N.; Schmidt, S.; Mollenhauer, J.; Voldborg, B.; Pedersen, C.B.; et al. SSX2 is a novel DNA-binding protein that antagonizes polycomb group body formation and gene repression. Nucleic Acids Res. 2014, 42, 11433–11446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntley, S.; Baggott, D.M.; Hamilton, A.T.; Tran-Gyamfi, M.; Yang, S.; Kim, J.; Gordon, L.; Branscomb, E.; Stubbs, L. A comprehensive catalog of human KRAB-associated zinc finger genes: Insights into the evolutionary history of a large family of transcriptional repressors. Genome Res. 2006, 16, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.; Rocques, P.J.; Crew, A.J.; Gill, S.; Shipley, J.; Chan, A.M.; Gusterson, B.A.; Cooper, C.S. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat. Genet. 1994, 7, 502–508. [Google Scholar] [CrossRef]
- McBride, M.J.; Pulice, J.L.; Beird, H.C.; Ingram, D.R.; D’Avino, A.R.; Shern, J.F.; Charville, G.W.; Hornick, J.L.; Nakayama, R.T.; Garcia-Rivera, E.M.; et al. The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 2018, 33, 1128–1141 e1127. [Google Scholar] [CrossRef] [Green Version]
- Banito, A.; Li, X.; Laporte, A.N.; Roe, J.S.; Sanchez-Vega, F.; Huang, C.H.; Dancsok, A.R.; Hatzi, K.; Chen, C.C.; Tschaharganeh, D.F.; et al. The SS18-SSX Oncoprotein Hijacks KDM2B-PRC1.1 to Drive Synovial Sarcoma. Cancer Cell 2018, 34, 346–348. [Google Scholar] [CrossRef] [Green Version]
- Kadoch, C.; Crabtree, G.R. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 2013, 153, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Greve, K.B.; Lindgreen, J.N.; Terp, M.G.; Pedersen, C.B.; Schmidt, S.; Mollenhauer, J.; Kristensen, S.B.; Andersen, R.S.; Relster, M.M.; Ditzel, H.J.; et al. Ectopic expression of cancer/testis antigen SSX2 induces DNA damage and promotes genomic instability. Mol. Oncol. 2015, 9, 437–449. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, P.; Maruwge, W.; Wolahan, B.; Ma, L.; Brodin, B. Oncogenic functions of the cancer-testis antigen SSX on the proliferation, survival, and signaling pathways of cancer cells. PLoS ONE 2014, 9, e95136. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Bartkova, J.; Lukas, J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007, 26, 7773–7779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Land, H.; Parada, L.F.; Weinberg, R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983, 304, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, N.R.; de Bruijn, D.R.; Kater-Baats, E.; Otte, A.P.; van Kessel, A.G. Delineation of the protein domains responsible for SYT, SSX, and SYT-SSX nuclear localization. Exp. Cell Res. 2000, 256, 192–202. [Google Scholar] [CrossRef]
- Soulez, M.; Saurin, A.J.; Freemont, P.S.; Knight, J.C. SSX and the synovial-sarcoma-specific chimaeric protein SYT-SSX co-localize with the human Polycomb group complex. Oncogene 1999, 18, 2739–2746. [Google Scholar] [CrossRef] [Green Version]
- Garcia, C.B.; Shaffer, C.M.; Eid, J.E. Genome-wide recruitment to Polycomb-modified chromatin and activity regulation of the synovial sarcoma oncogene SYT-SSX2. BMC Genomics 2012, 13, 189. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Sampaio, A.V.; Jones, K.B.; Pacheco, M.; Goytain, A.; Lin, S.; Poulin, N.; Yi, L.; Rossi, F.M.; Kast, J.; et al. Deconstruction of the SS18-SSX fusion oncoprotein complex: Insights into disease etiology and therapeutics. Cancer Cell 2012, 21, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Barco, R.; Garcia, C.B.; Eid, J.E. The synovial sarcoma-associated SYT-SSX2 oncogene antagonizes the polycomb complex protein Bmi1. PLoS ONE 2009, 4, e5060. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johansen, S.; Gjerstorff, M.F. Interaction between Polycomb and SSX Proteins in Pericentromeric Heterochromatin Function and Its Implication in Cancer. Cells 2020, 9, 226. https://doi.org/10.3390/cells9010226
Johansen S, Gjerstorff MF. Interaction between Polycomb and SSX Proteins in Pericentromeric Heterochromatin Function and Its Implication in Cancer. Cells. 2020; 9(1):226. https://doi.org/10.3390/cells9010226
Chicago/Turabian StyleJohansen, Simone, and Morten Frier Gjerstorff. 2020. "Interaction between Polycomb and SSX Proteins in Pericentromeric Heterochromatin Function and Its Implication in Cancer" Cells 9, no. 1: 226. https://doi.org/10.3390/cells9010226
APA StyleJohansen, S., & Gjerstorff, M. F. (2020). Interaction between Polycomb and SSX Proteins in Pericentromeric Heterochromatin Function and Its Implication in Cancer. Cells, 9(1), 226. https://doi.org/10.3390/cells9010226