Aspirin Enhances the Protection of Hsp90 from Heat-Stressed Injury in Cardiac Microvascular Endothelial Cells Through PI3K-Akt and PKM2 Pathways

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Treatments and Cell Viability Assay
2.2. SiRNA Transfection
- S1:
- sense sequence 5’-GGAAGGAGCUGCACAUUAAdTdT-3’anti-sense sequence 5’-UUAAUGUGCAGCUCCUUCCdTdT-3’
- S2:
- sense sequence 5’-GGUGUCAGUUACCAAAGAAdTdT-3’anti-sense sequence 5’-UUCUUUGGUAACUGACACCdTdT-3’
- S3:
- sense sequence 5’-GACUUAAGUUUCAUCUUAAdTdT-3’anti-sense sequence 5’-UUAAGAUGAAACUUAAGUCdTdT-3’
2.3. ASA Treatment
2.4. GA/TR Treatment
2.5. Cell Viability
2.6. Detection of Lactate Dehydrogenase (LDH), Lipid Peroxide (LPO), Malondialdehyde (MDA) and Nitric Oxide (NO)
2.7. Observation of Cell Morphology
2.8. Flow Cytometric Analysis of Cell Apoptosis
2.9. Cell Protein Extraction and Western Blot
2.10. Immunofluorescence Analysis
2.11. Enzyme Linked Immunosorbent Assay (ELISA) Analysis for Hsp70 in Culture Medium
2.12. Statistical Analysis
3. Results
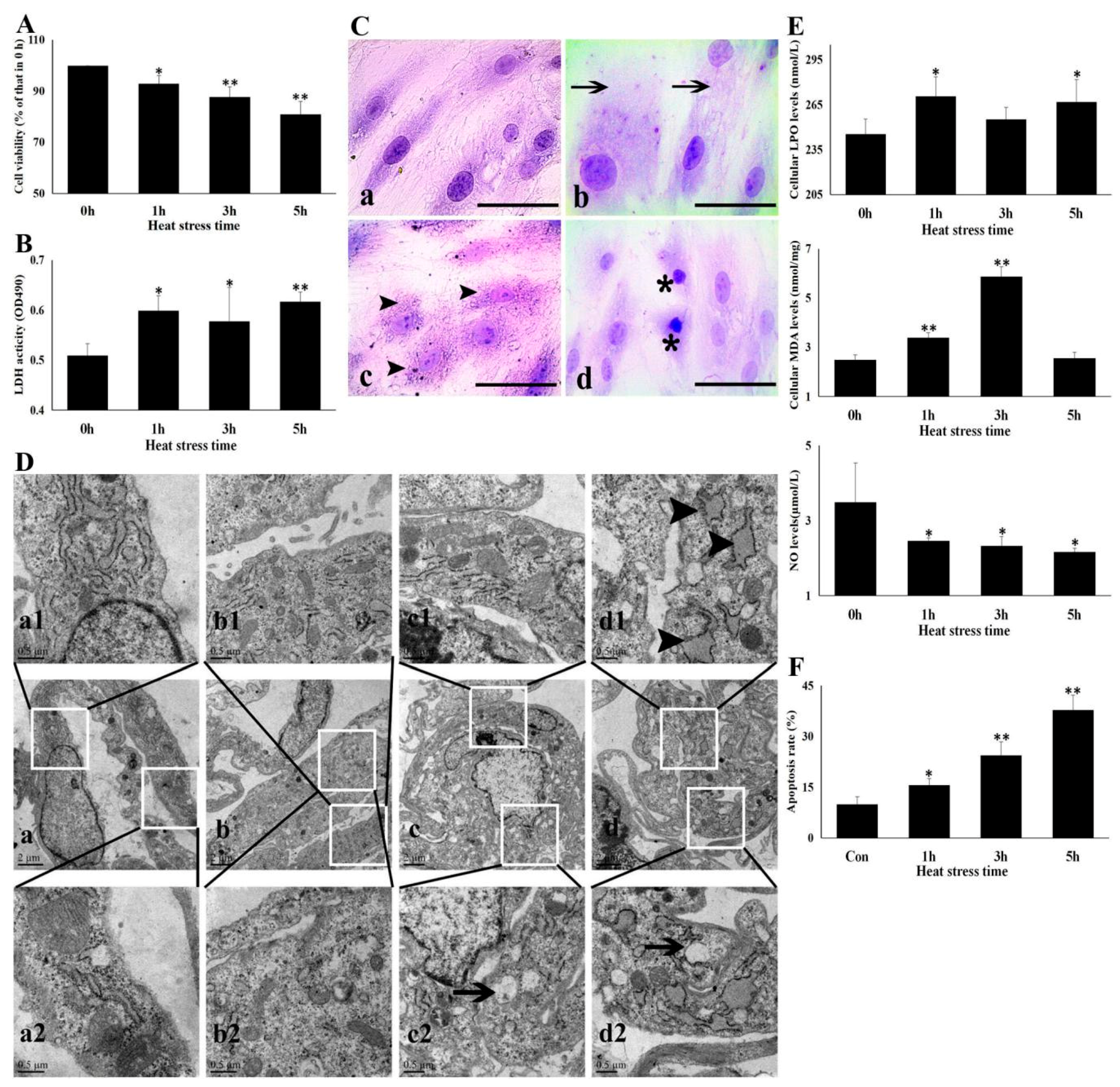
3.1. Heat Stress Damaged CMVECs
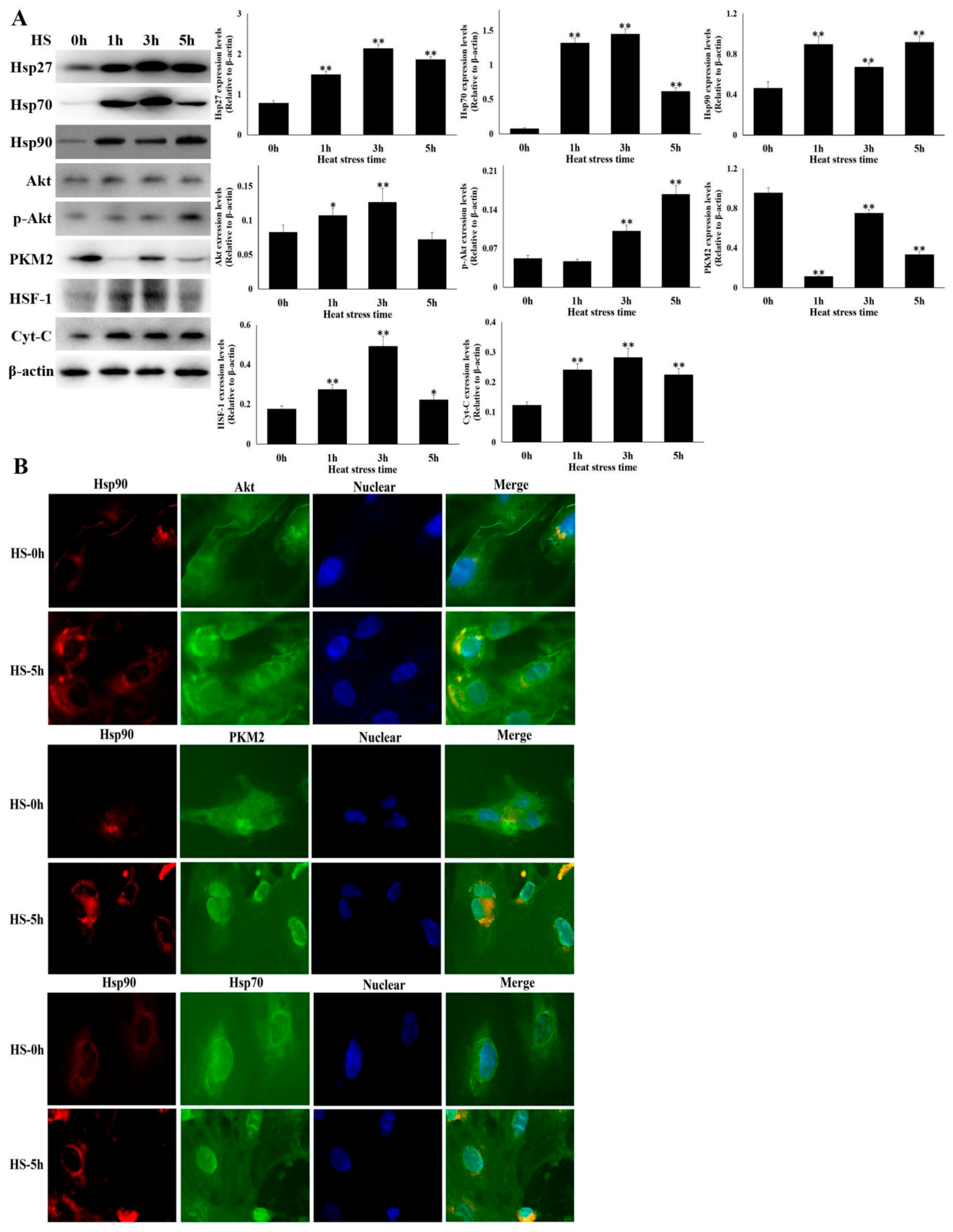
3.2. Hsp90 Expression were Up-Regulated in Heat Stressed CMVECs
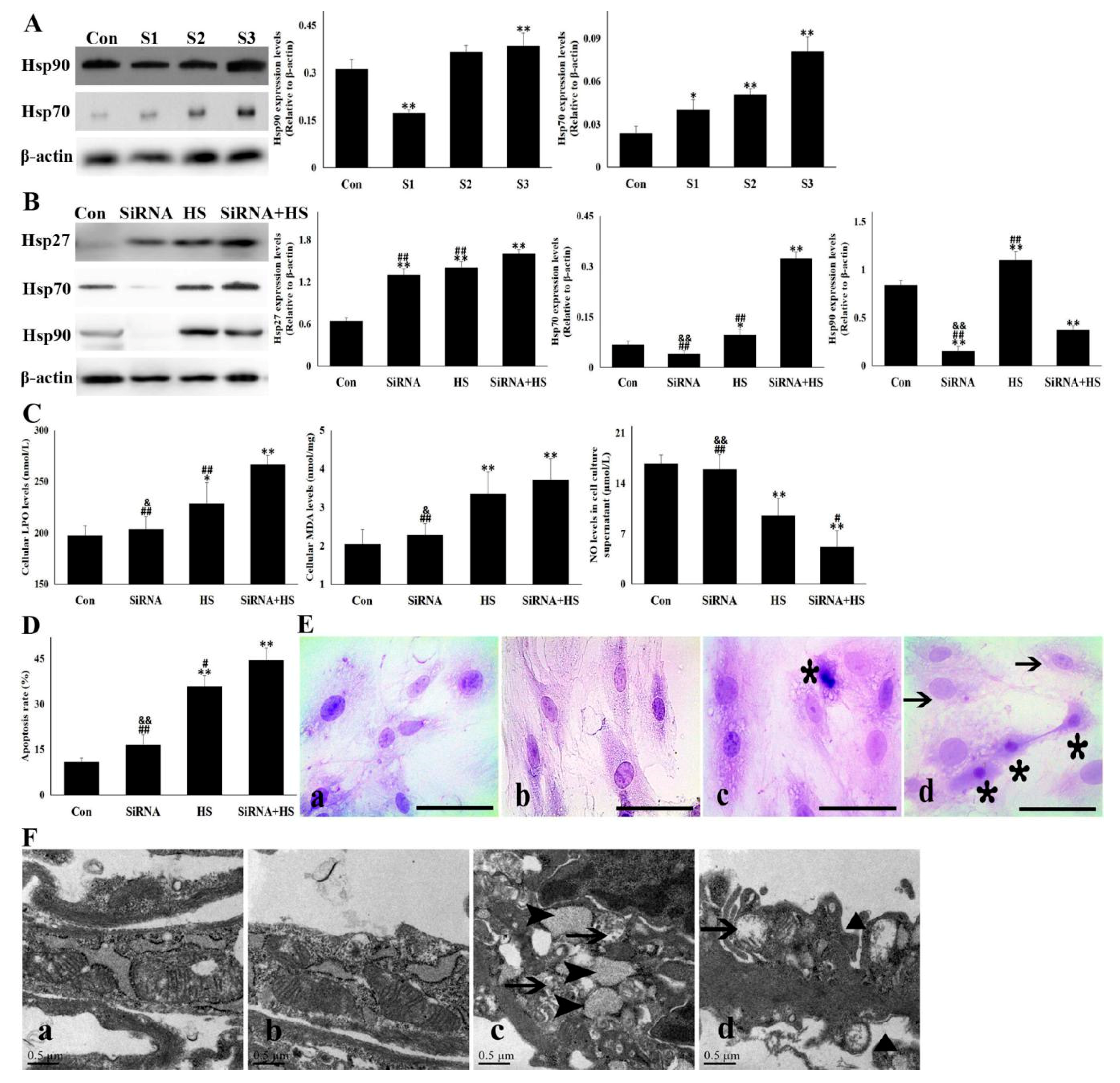
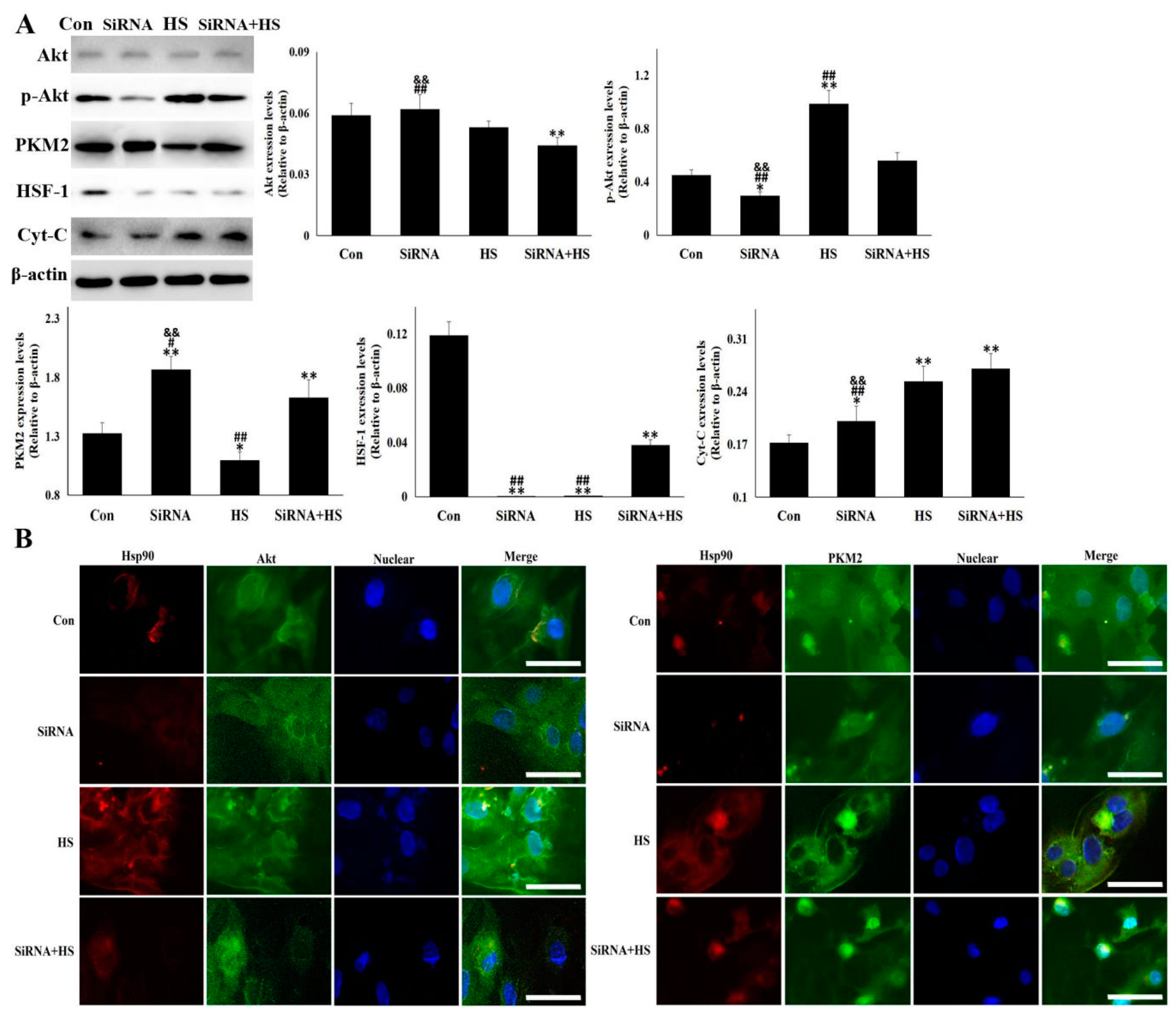
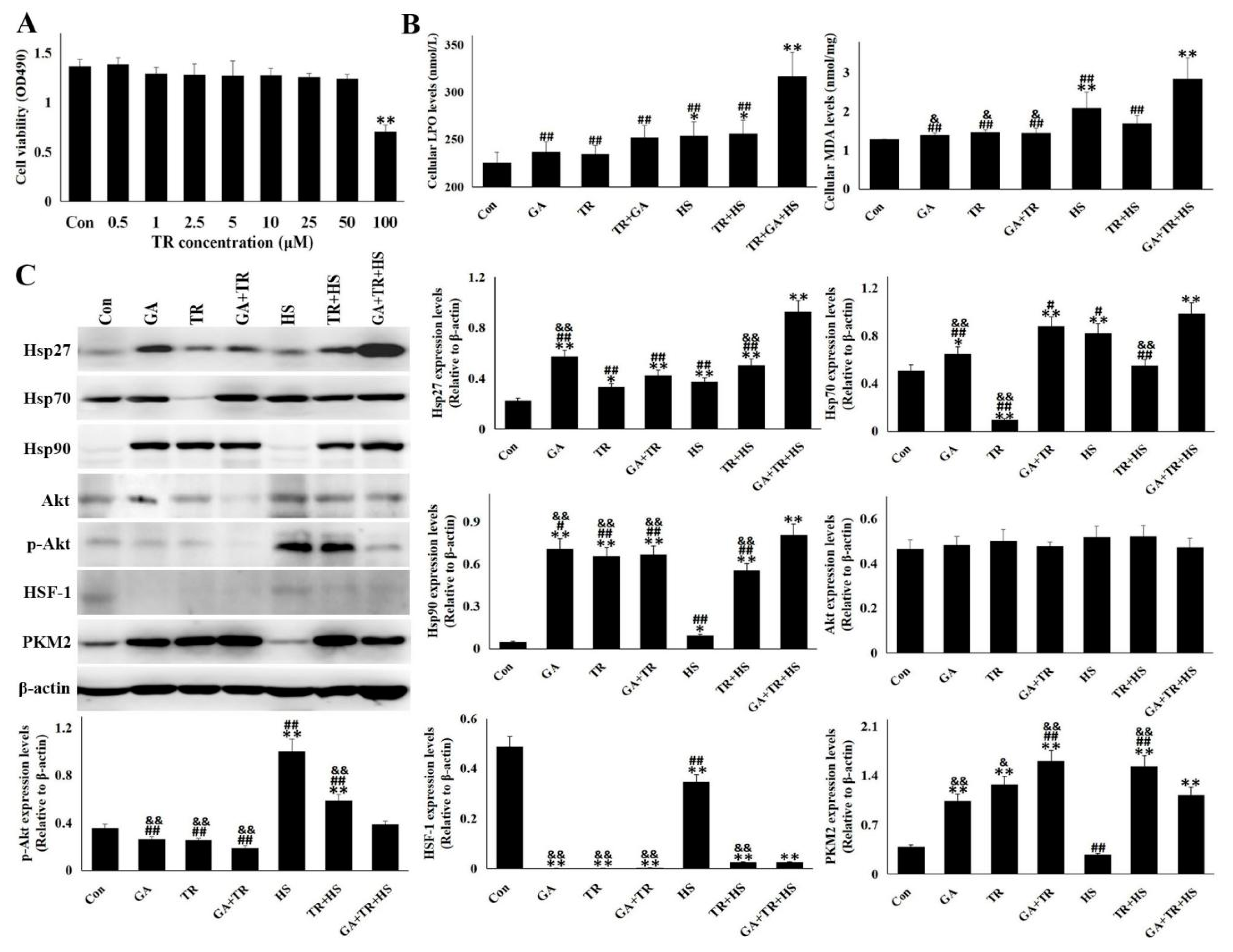
3.3. Knockdown of Hsp90 Aggravated the Cellular Damage Induced by Heat Stress
3.4. Akt and PKM2 were Regulated Differentially by Hsp90
3.5. PKM2 is a Alternative Pathway Under the Deficiency of Hsp90 Function and Akt Phosphorylation
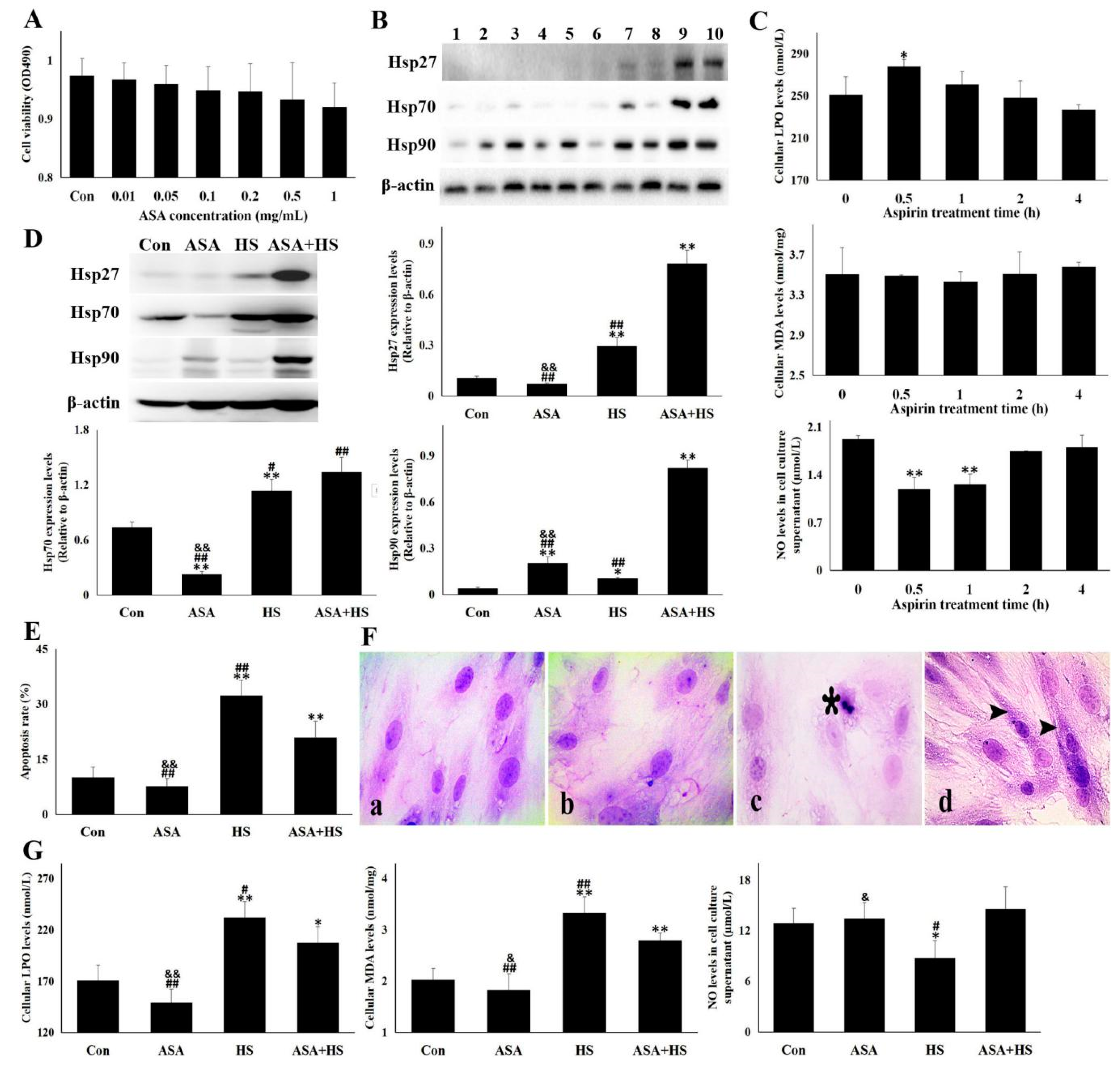
3.6. Hsp90 Induction by Aspirin Relieved the Heat-Stressed Damage
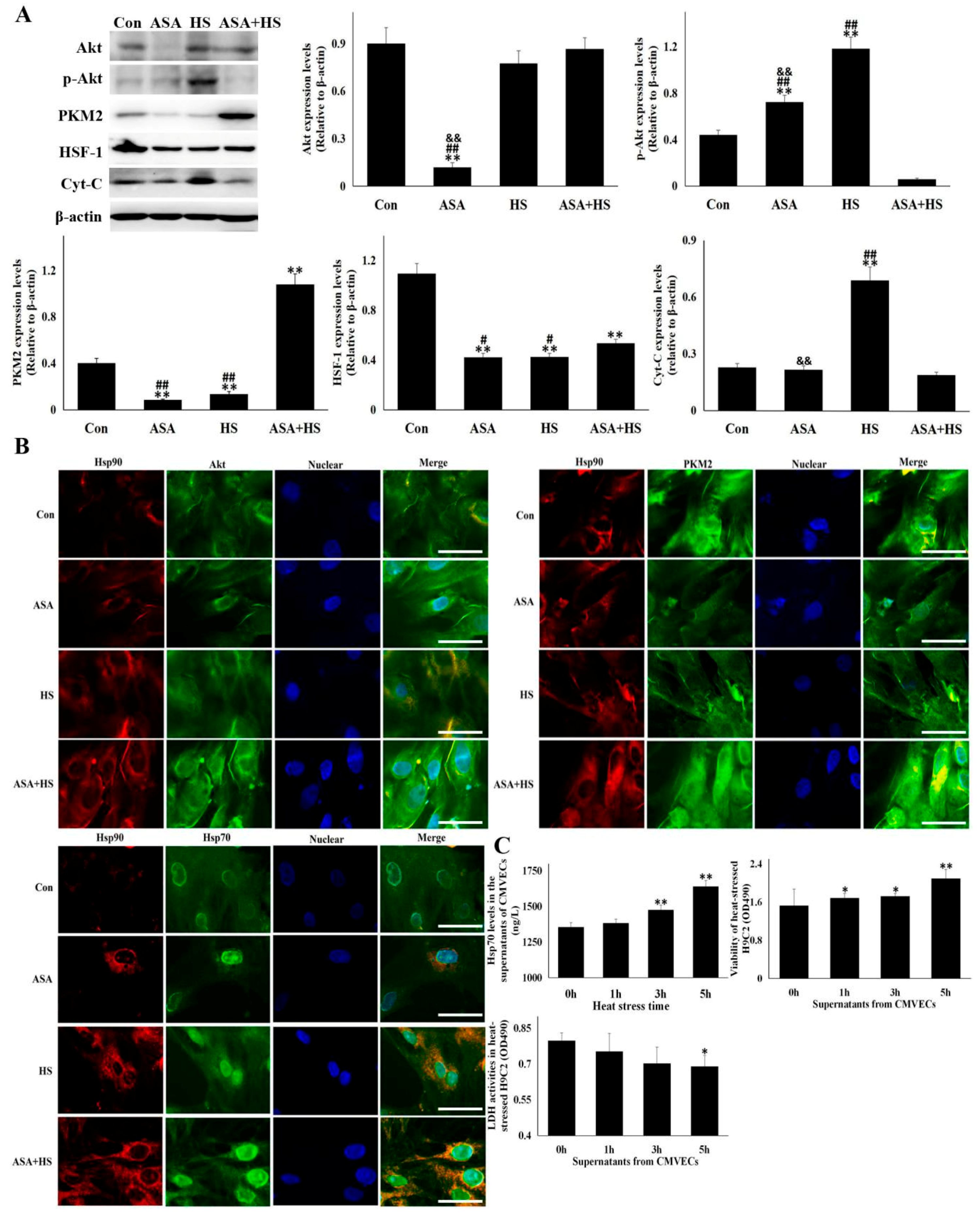
3.7. Both Akt and PKM2 Signals were Strengthened by Hsp90 Induction Caused by ASA
3.8. Extracellular Hsp70 Release from CMVECs Inhibited Heat-Stressed Damage of Myocardial Cell Line, H9C2
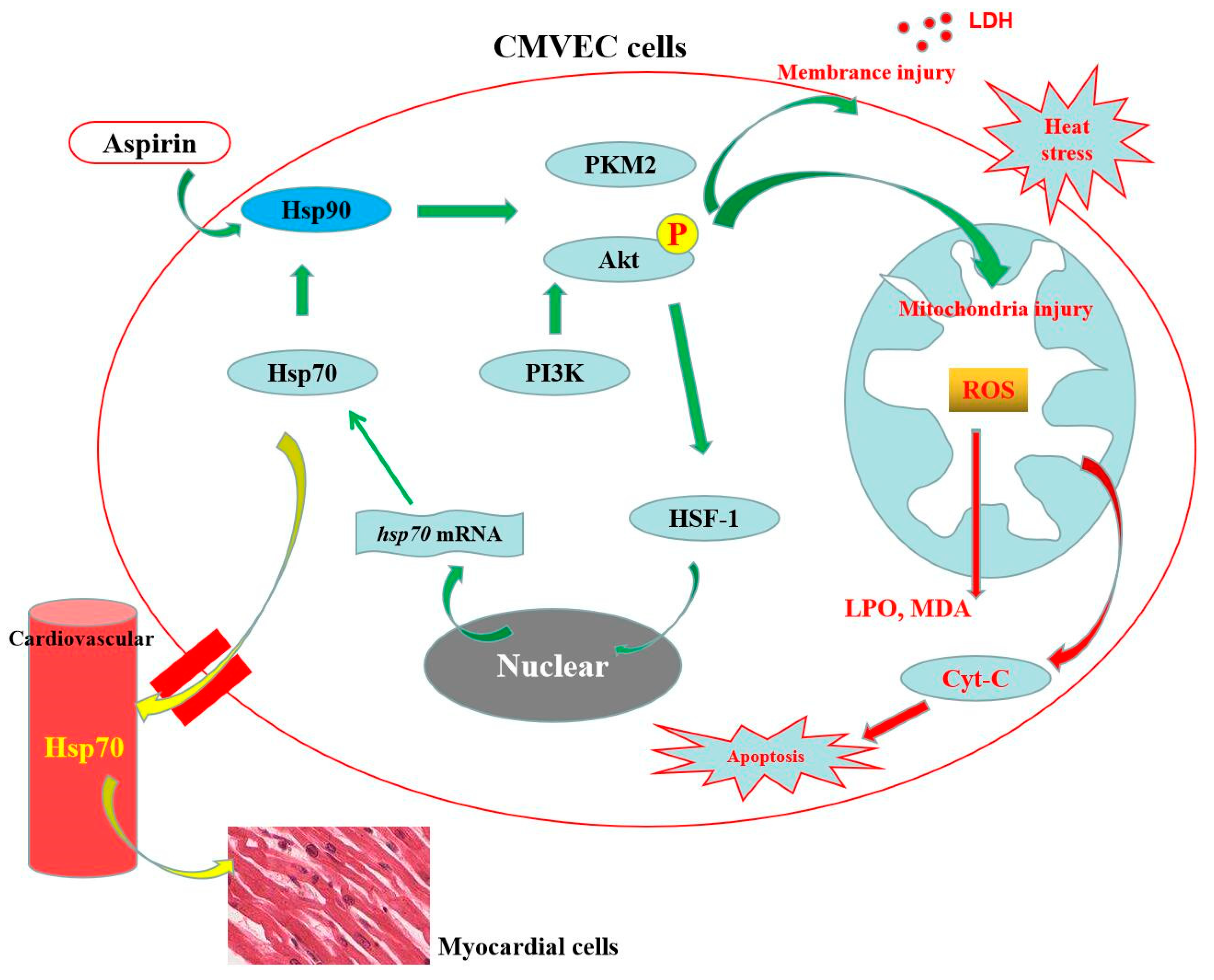
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bouchama, A.; Knochel, J.P. Heat stroke. N. Engl. J. Med. 2002, 346, 1978–1988. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Amador, N.M.; Rothenhaus, T.; Moyer, P. Heat-related illness. Emerg. Med. Clin. N. Am. 2004, 22, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Patz, J.A.; Campbell-Lendrum, D.; Holloway, T.; Foley, J.A. Impact of regional climate change on human health. Nature. 2005, 438, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Christen, F.; Desrosiers, V.; Dupont-Cyr, B.A.; Vandenberg, G.W.; Le François, N.R.; Tardif, J.C.; Dufresne, F.; Lamarre, S.G.; Blier, P.U. Thermal tolerance and thermal sensitivity of heart mitochondria: Mitochondrial integrity and ROS production. Free Radic. Biol. Med. 2018, 116, 11–18. [Google Scholar] [CrossRef]
- Tang., S.; Lv, Y.; Chen, H.; Adam, A.; Cheng, Y.; Hartung, J.; Bao, E. Comparative analysis of αB-crystallin expression in heat-stressed myocardial cells in vivo and in vitro. PLoS ONE 2014, 9, e86937. [Google Scholar] [CrossRef]
- Bao, E.; Sultan, K.R.; Nowak, B.; Hartung, J. Expression and distribution of heat shock proteins in the heart of transported pigs. Cell Stress Chaperones 2008, 13, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X. The types, mechanism and harm of animal stress. Mod. Anim. Husb. Sci. Technol. 2012, 4, 20. [Google Scholar]
- Liu, Y.R.; Chen, J.J.; Dai, M. Paeonol protects rat vascular endothelial cells from ox-LDL induced injury in vitro via downregulating micro RNA21 expression and TNF-α release. Acta Pharmacol. Sin. 2014, 35, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Michiels, C. Endothelial Cell Functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef]
- Roberts, G.T.; Ghebeh, H.; Chishti, M.A.; Al-Mohanna, F.; El-Sayed, R.; Al-Mohanna, F.; Bouchama, A. Microvascular injury, thrombosis, inflammation, and apoptosis in the pathogenesis of heatstroke: A study in baboon model. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1130–1136. [Google Scholar] [CrossRef] [Green Version]
- Lan, S.; Yi, F.; Shuang, L.; Chenjie, W.; Zheng, X.W. Chemical constituents from the fibrous root of Ophiopogon japonicus, and their effect on tube formation in human myocardial microvascular endothelial cells. Fitoterapia 2013, 85, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.L.; Li, Y. Endothelial cell senescence and agerelated vascular diseases. J. Genet. Genom. 2014, 41, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Nolan, D.J.; Ginsberg, M.; Israely, E.; Palikuqi, B.; Poulos, M.G.; James, D.; Ding, B.S.; Schachterle, W.; Liu, Y.; Rosenwaks, Z.; et al. Molecular signatures of tissue specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 2013, 26, 204–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Tan, H.; Yang, H.; Li, F.; He, X.; Gu, Z.; Zhao, M.; Su, L. Reactive oxygen species mediate heat stress-induced apoptosis via ERK dephosphorylation and Bcl-2 ubiquitination in human umbilical vein endothelial cells. Oncotarget 2017, 8, 12902–12916. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tan, H.; Gu, Z.; Liu, Z.; Geng, Y.; Liu, Y.; Tong, H.; Tang, Y.; Qiu, J.; Su, L. Heat stress induces apoptosis through a Ca2+-mediated mitochondrial apoptotic pathway in human umbilical vein endothelial cells. PLoS ONE 2014, 9, e111083. [Google Scholar] [CrossRef] [PubMed]
- Kishore, A.; Sodhi, M.; Kumari, P.; Mohanty, A.K.; Sadana, D.K.; Kapila, N.; Khate, K.; Shandilya, U.; Kataria, R.S.; Mukesh, M. Peripheral blood mononuclear cells: A potential cellular system to understand differential heat shock response across native cattle (Bos indicus), exotic cattle (Bos taurus), and riverine buffaloes (Bubalus bubalis) of India. Cell Stress Chaperones 2014, 19, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottile, M.L.; Nadin, S.B. Heat shock proteins and DNA repair mechanisms: An updated overview. Cell Stress Chaperones. Cell Stress Chaperones 2018, 23, 303–315. [Google Scholar] [CrossRef]
- Langer, T.; Käser, M.; Klanner, C.; Leonhard, K. AAA proteases of mitochondria: Quality control of membrane proteins and regulatory function during mitochondria biogenesis. Biochem. Soc. Trans. 2001, 29, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Zhu, H.S.; Qian, Z.; Tang, S.; Wu, D.; Kemper, N.; Hartung, J.; Bao, E.D. The association of Hsp90 expression induced by aspirin with anti-stress damage in chicken myocardial cells. J. Vet. Sci. 2016, 17, 35–44. [Google Scholar] [CrossRef]
- Yu, J.; Bao, E.; Yan, J.; Lei, L. Expression and localization of Hsps in the heart and blood vessel of heat-stressed broilers. Cell Stress Chaperones 2008, 13, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Wu, H.; Tang, S.; Li, Q.N.; Xu, J.; Zhang, M.; Su, Y.N.; Yin, B.; Zhao, Q.L.; Kemper, N.; et al. Apoptosis in response to heat stress is positively associated with heat-shock protein 90 expression in chicken myocardial cells in vitro. J. Vet. Sci. 2017, 18, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qian, Z.; Zhu, H.; Tang, S.; Wu, D.; Zhang, M.; Kemper, N.; Hartung, J.; Bao, E. HSP90 gene expression induced by aspirin is associated with damage remission in a chicken myocardial cell culture exposed to heat stress. Br. Poult. Sci. 2016, 57, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Andrulis, M.; Stühmer, T.; Müller, E.; Hofmann, C.; Steinbrunn, T.; Heimberger, T.; Schraud, H.; Kressmann, S.; Einsele, H.; et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013, 98, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Cao, R.; Wang, X.; Zhang, Y.; Wang, P.; Gao, H.; Li, C.; Yang, F.; Zeng, R.; Wei, P.; et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res. 2017, 27, 329–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, N.; Jin, J.; Liu, D.; Yan, R.; Zhang, S.; Yu, X.; Chen, X. Antiproliferative effect of HSP90 inhibitor Y306zh against pancreatic cancer is mediated by interruption of AKT and MAPK signaling pathways. Curr. Cancer Drug Targets 2014, 14, 671–683. [Google Scholar] [CrossRef]
- Xu, Q.; Tu, J.; Dou, C.; Zhang, J.; Yang, L.; Liu, X.; Lei, K.; Liu, Z.; Wang, Y.; Li, L.; et al. HSP90 promotes cell glycolysis, proliferation and inhibits apoptosis by regulating PKM2 abundance via Thr-328 phosphorylation in hepatocellular carcinoma. Mol. Cancer 2017, 16, 178. [Google Scholar] [CrossRef]
- Zhou, P.; Xie, W.; Luo, Y.; Lu, S.; Dai, Z.; Wang, R.; Sun, G.; Sun, X. Protective Effects of Total Saponins of Aralia elata (Miq.) on Endothelial Cell Injury Induced by TNF-α via Modulation of the PI3K/Akt and NF-κB Signalling Pathways. Int. J. Mol. Sci. 2018, 20, 36. [Google Scholar] [CrossRef] [Green Version]
- Dybdahl, B.; Wahba, A.; Lien, E.; Flo, T.H.; Waage, A.; Qureshi, N.; Sellevold, O.F.; Espevik, T.; Sundan, A. Inflammatory response after open heart surgery: Release of heart-shock protein 70 and signaling through toll-like receptor-4. Circulation 2002, 105, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Pucciariello, C.; Banti, V.; Perata, P. ROS signaling as common element in low oxygen and heat stresses. Plant Physiol. Bioch. 2012, 59, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Galeano, E.; Nieto, E.; Delgado, M.D.; García-Pérez, A.I. Dequalinium induces cell death in human leukemia cells by early mitochondrial alterations which enhance ROS production. Leukemia Res. 2007, 31, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jin, Q.; Li, Y.; Ma, Q.; Wang, J.; Li, D.; Zhou, H.; Chen, Y. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca2+]c/VDAC-[Ca2+]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones 2018, 23, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Shimp, S.K., 3rd; Parson, C.D.; Regna, N.L.; Thomas, A.N.; Chafin, C.B.; Reilly, C.M.; Nichole Rylander, M. HSP90 inhibition by 17-DMAG reduces inflammation in J774 macrophages through suppression of Akt and nuclear factor-κB pathways. Inflamm. Res. 2012, 61, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Mardones, J.; Gallardo-Escárate, C. Immune response of apoptosis-related cysteine peptidases from the red abalone Haliotis rufescens (HrCas8 and HrCas3): Molecular characterization and transcription expression. Fish. Shellfish Immunol. 2014, 39, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Nakai, A. The Heat Shock Factor Family and Adaptation to Proteotoxic Stress. FEBS J. 2010, 277, 4112–4125. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Salamó, I.; Papdi, C.; Rigó, G.; Zsigmond, L.; Vilela, B.; Lumbreras, V.; Nagy, I.; Horváth, B.; Domoki, M.; Darula, Z. The heat shock factor A4A confers salt tolerance and is regulated by oxidative stress and the mitogen-activated protein kinases MPK3 and MPK6. Plant Physiol. 2014, 165, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, M.; Su, Y.; Wang, Z.; Zhao, Q.; Zhu, H.; Qian, Z.; Xu, J.; Tang, S.; Wu, D.; et al. Inhibition of heat stress-related apoptosis of chicken myocardial cells through inducing Hsp90 expression by aspirin administration in vivo. Br. Poult. Sci. 2018, 59, 308–317. [Google Scholar] [CrossRef]
- Mosser, D.D.; Kotzbauer, P.T.; Sarge, K.D.; Morimoto, R.I. In vitro activation of heat shock transcription factor DNA-binding by calcium and biochemical conditions that affect protein conformation. Proc. Natl. Acad. Sci. USA 1990, 87, 3748–3752. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Orosz, A.; Wu, C. Direct sensing of heat and oxidation by Drosophila heat shock transcription factor. Mol. Cell 1998, 2, 101–108. [Google Scholar] [CrossRef]
- Viswanathan, M.; Rivera, O.; Short, B.L. Heat-shock protein 90 involved in pulsatile flow induced dilatation of the rat middle cerebral artery. J. Vasc. Res. 1999, 36, 524–527. [Google Scholar] [CrossRef]
- Shi, P.; Cao, Y.; Gao, J.; Fu, B.; Ren, J.; Ba, L.; Song, C.; Qi, H.; Huang, W.; Guan, X.; et al. Allicin improves the function of cardiac microvascular endothelial cells by increasing PECAM-1 in rats with cardiac hypertrophy. Phytomedicine 2018, 51, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Q.; Zhang, Y.H.; Jiang, S.D.; Li, H.; Jiang, L.S.; Dai, L.Y. ADAMTS-5 and intervertebral disc degeneration: The results of tissue immunohistochemistry and in vitro cell culture. J. Orthop. Res. 2011, 29, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.K.; Wang, G.; Chen, Z.; Teruya-Feldstein, J.; Liu, Y.; Chan, C.H.; Yang, W.L.; Erdjument-Bromage, H.; Nakayama, K.I.; Nimer, S. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of skp2 by Akt/PKB. Nat. Cell Biol. 2009, 11, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Cantley, L.C. Akt/pkb signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Lu, Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle 2013, 12, 3154–3158. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.D.; Campisi, J.; Sharkey, C.M.; Kennedy, S.L.; Nickerson, M.; Fleshner, M. Adrenergic receptors mediate stress-induced elevations in extracellular Hsp72. J. Appl. Physiol. 2005, 99, 1789–1795. [Google Scholar] [CrossRef] [Green Version]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. Hsp70 stimulates cytokine production through a CD14-dependent pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Chen, B.; Wu, J.; Sha, J.; Yang, B.; Zhu, J.; Sun, J.; Hartung, J.; Bao, E. Aspirin Enhances the Protection of Hsp90 from Heat-Stressed Injury in Cardiac Microvascular Endothelial Cells Through PI3K-Akt and PKM2 Pathways. Cells 2020, 9, 243. https://doi.org/10.3390/cells9010243
Zhang X, Chen B, Wu J, Sha J, Yang B, Zhu J, Sun J, Hartung J, Bao E. Aspirin Enhances the Protection of Hsp90 from Heat-Stressed Injury in Cardiac Microvascular Endothelial Cells Through PI3K-Akt and PKM2 Pathways. Cells. 2020; 9(1):243. https://doi.org/10.3390/cells9010243
Chicago/Turabian StyleZhang, Xiaohui, Bixia Chen, Jiaxin Wu, Junzhou Sha, Bo Yang, Jie Zhu, Jiarui Sun, Jörg Hartung, and Endong Bao. 2020. "Aspirin Enhances the Protection of Hsp90 from Heat-Stressed Injury in Cardiac Microvascular Endothelial Cells Through PI3K-Akt and PKM2 Pathways" Cells 9, no. 1: 243. https://doi.org/10.3390/cells9010243
APA StyleZhang, X., Chen, B., Wu, J., Sha, J., Yang, B., Zhu, J., Sun, J., Hartung, J., & Bao, E. (2020). Aspirin Enhances the Protection of Hsp90 from Heat-Stressed Injury in Cardiac Microvascular Endothelial Cells Through PI3K-Akt and PKM2 Pathways. Cells, 9(1), 243. https://doi.org/10.3390/cells9010243