Identification and Characterization of a New Platinum-Induced TP53 Mutation in MDAH Ovarian Cancer Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Compounds and Drugs Treatment
2.3. Vectors and Transfections
2.4. Preparation of Cell Lysates, Immunoblotting, and Immunoprecipitation
2.5. Protein Stability
2.6. RNA Isolation and Real-Time Polymerase Chain Reaction
2.7. In Vitro Cell Proliferation Analyses
2.8. Double Thymidine Block
2.9. Immunofluorescence Analyses
2.10. Reactive Oxygen Species (ROS) Analyses
2.11. Cell Cycle Analyses
2.12. Next Generation Sequencing (NGS)
2.13. Statistical Analysis
3. Results
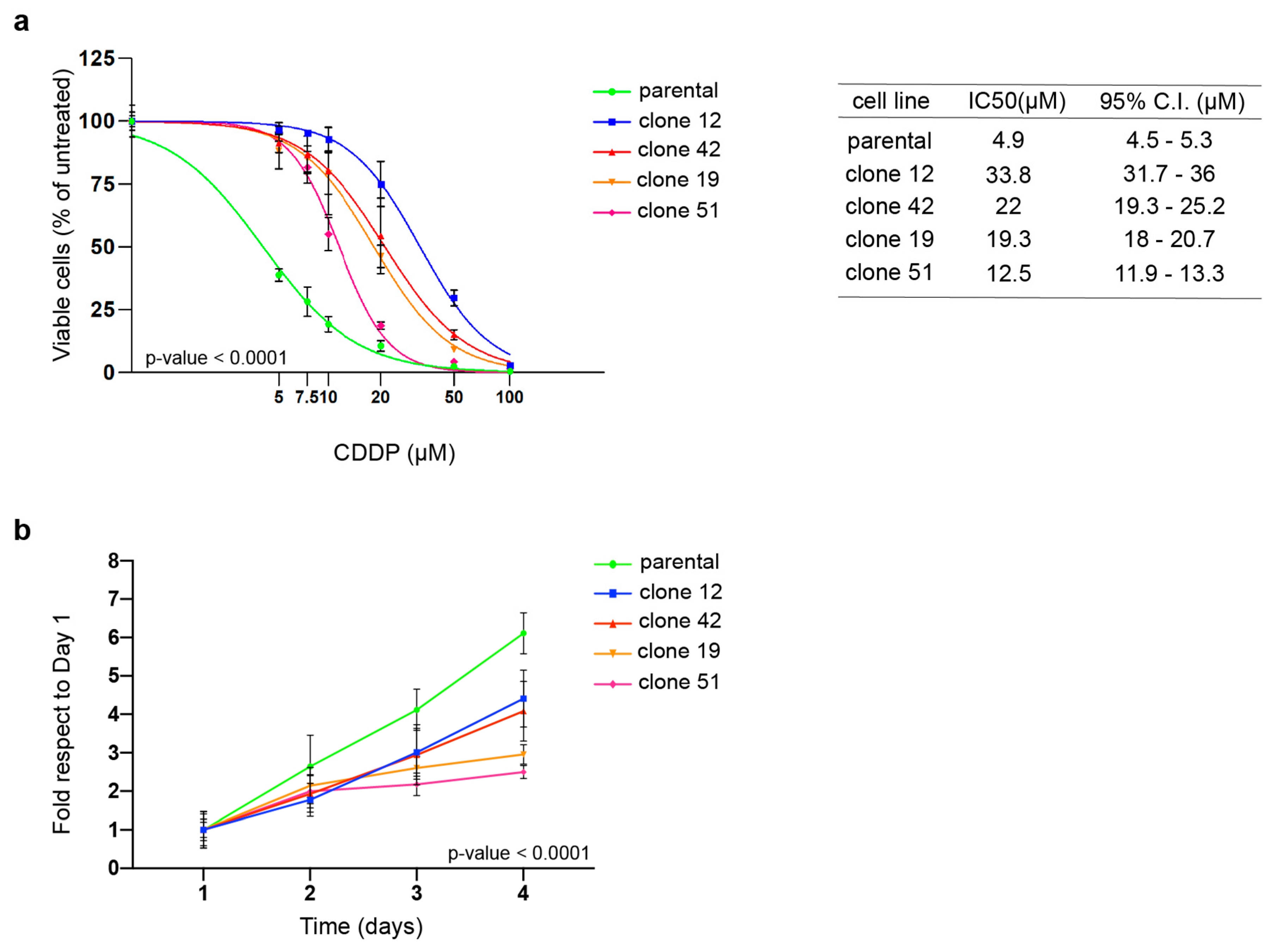
3.1. PT-res Clones Show an Impaired Growth Rate
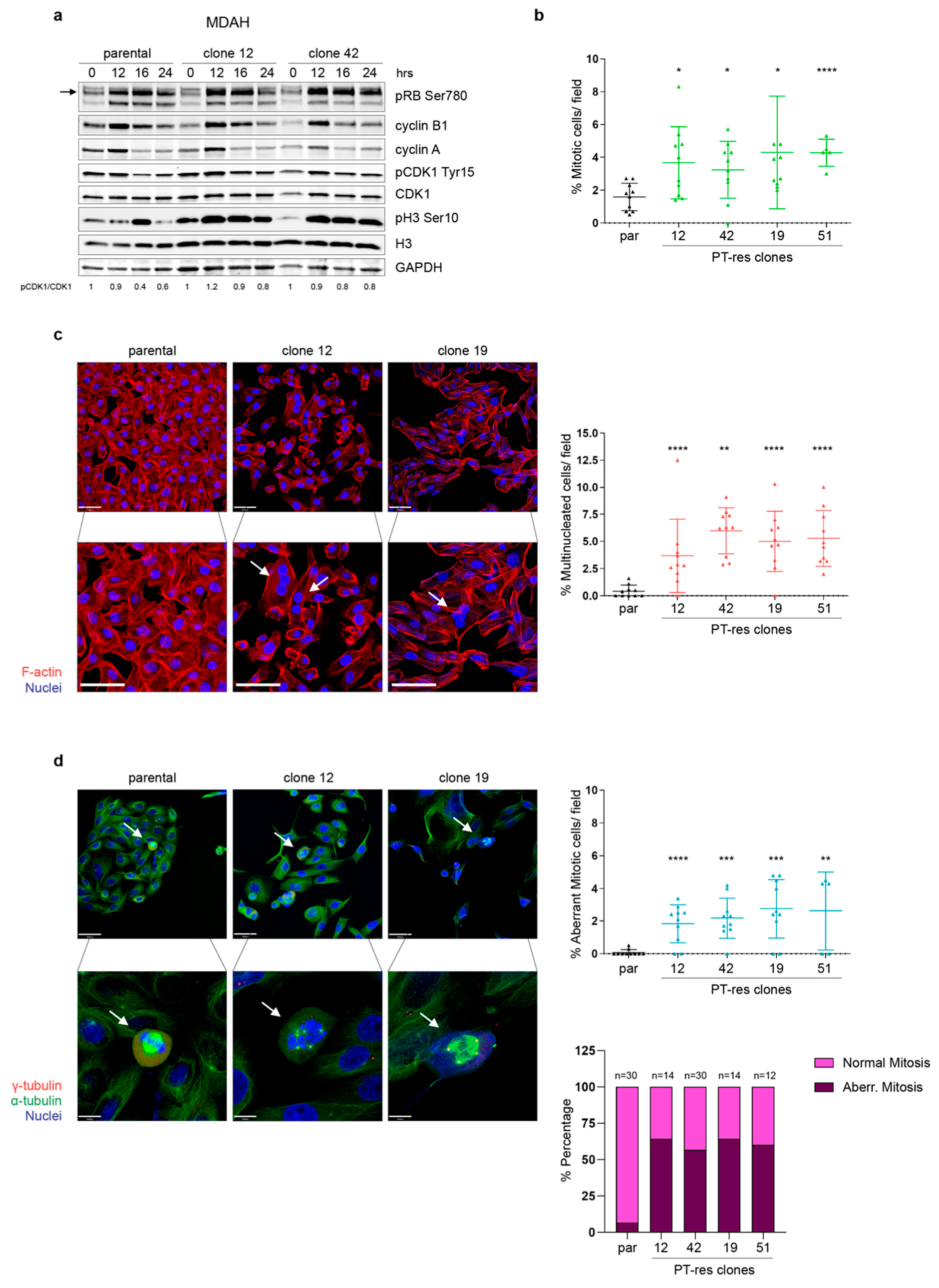
3.2. PT-res Clones Present an Increased Number of Aberrant Mitotic Figures
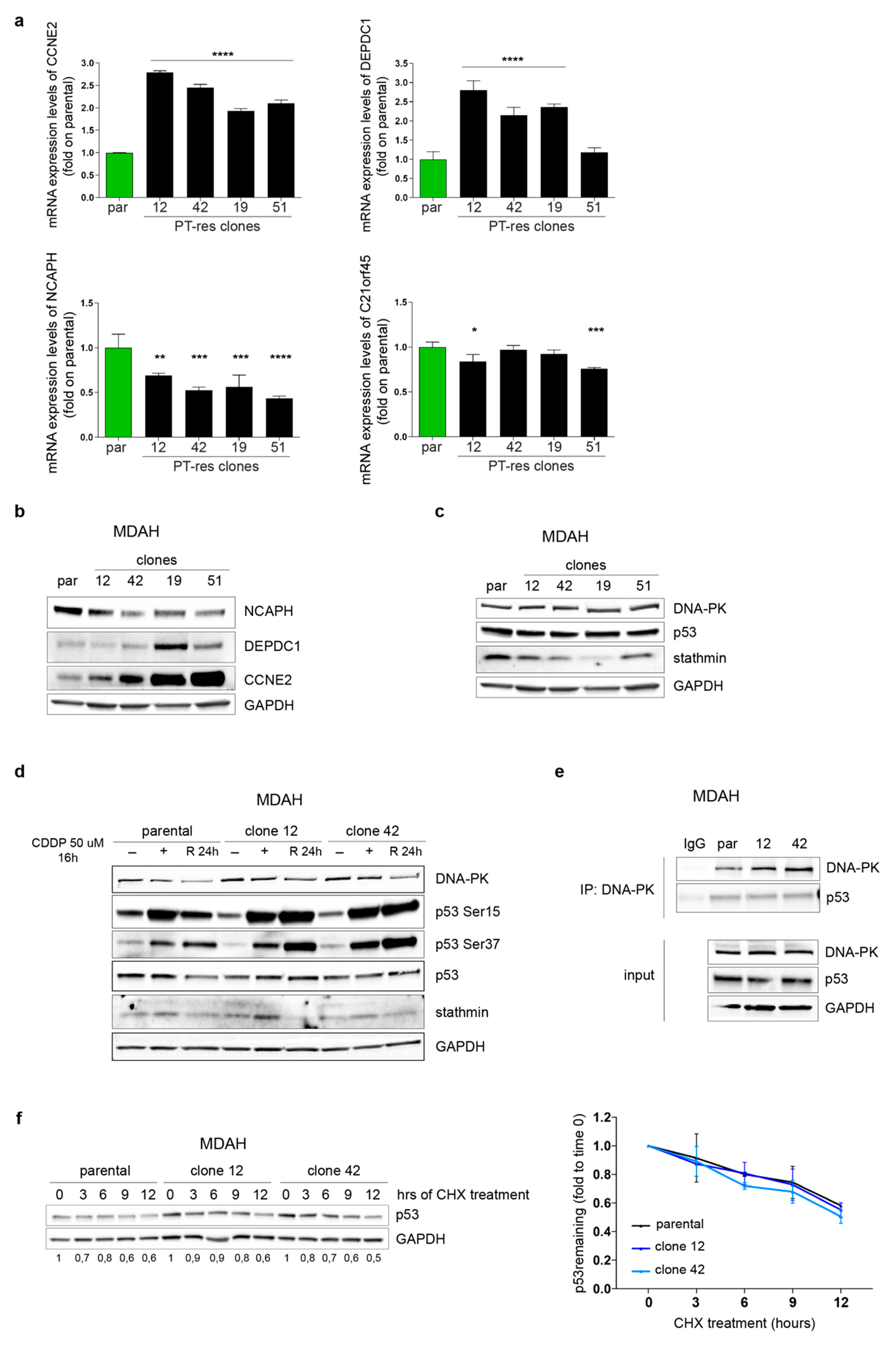
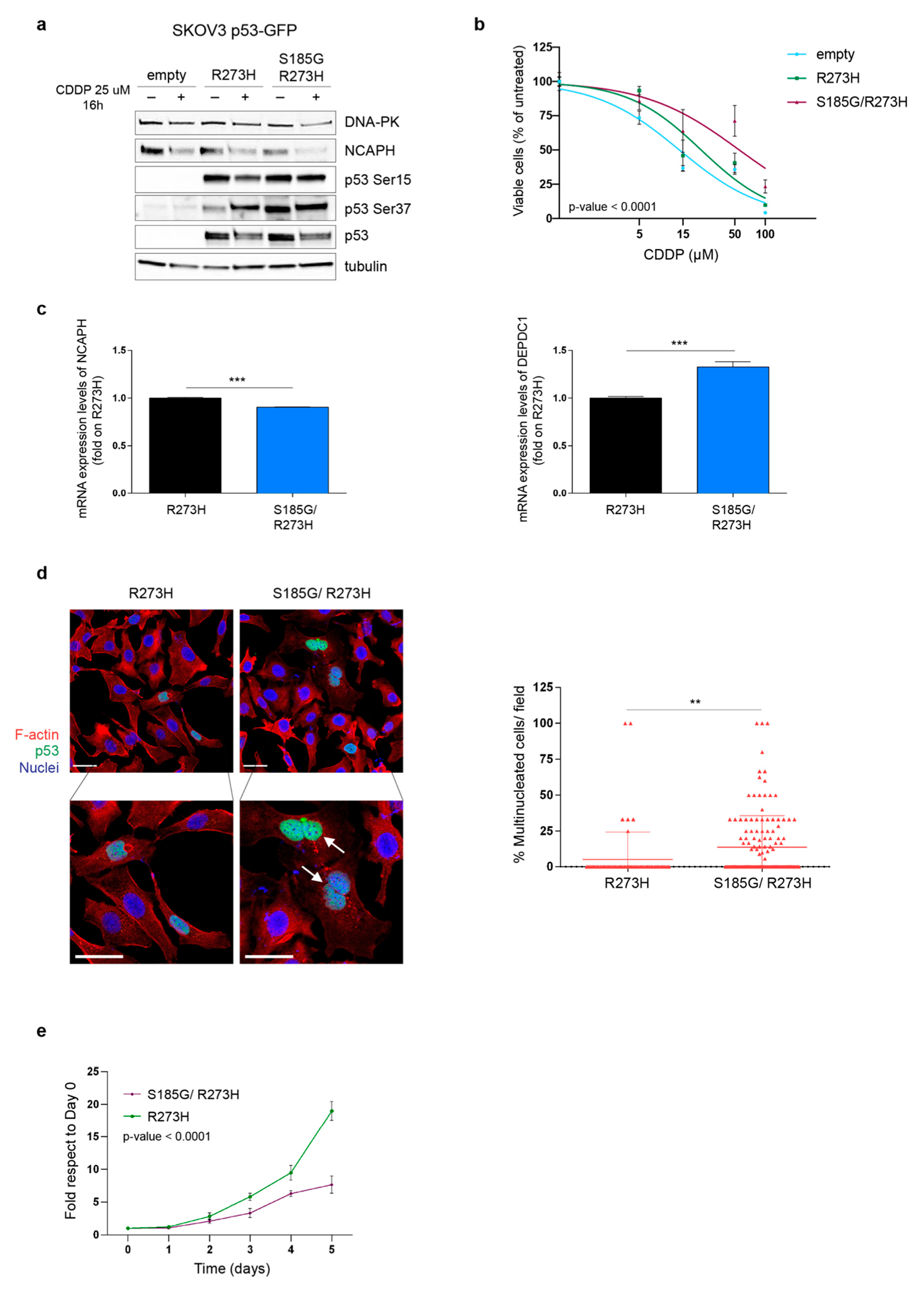
3.3. p53MUT Downstream Targets Are Differently Modulated in PT-res Clones
3.4. DNA-PKCS Is Not Involved in the Regulation of p53MUT Activity in PT-res Clones
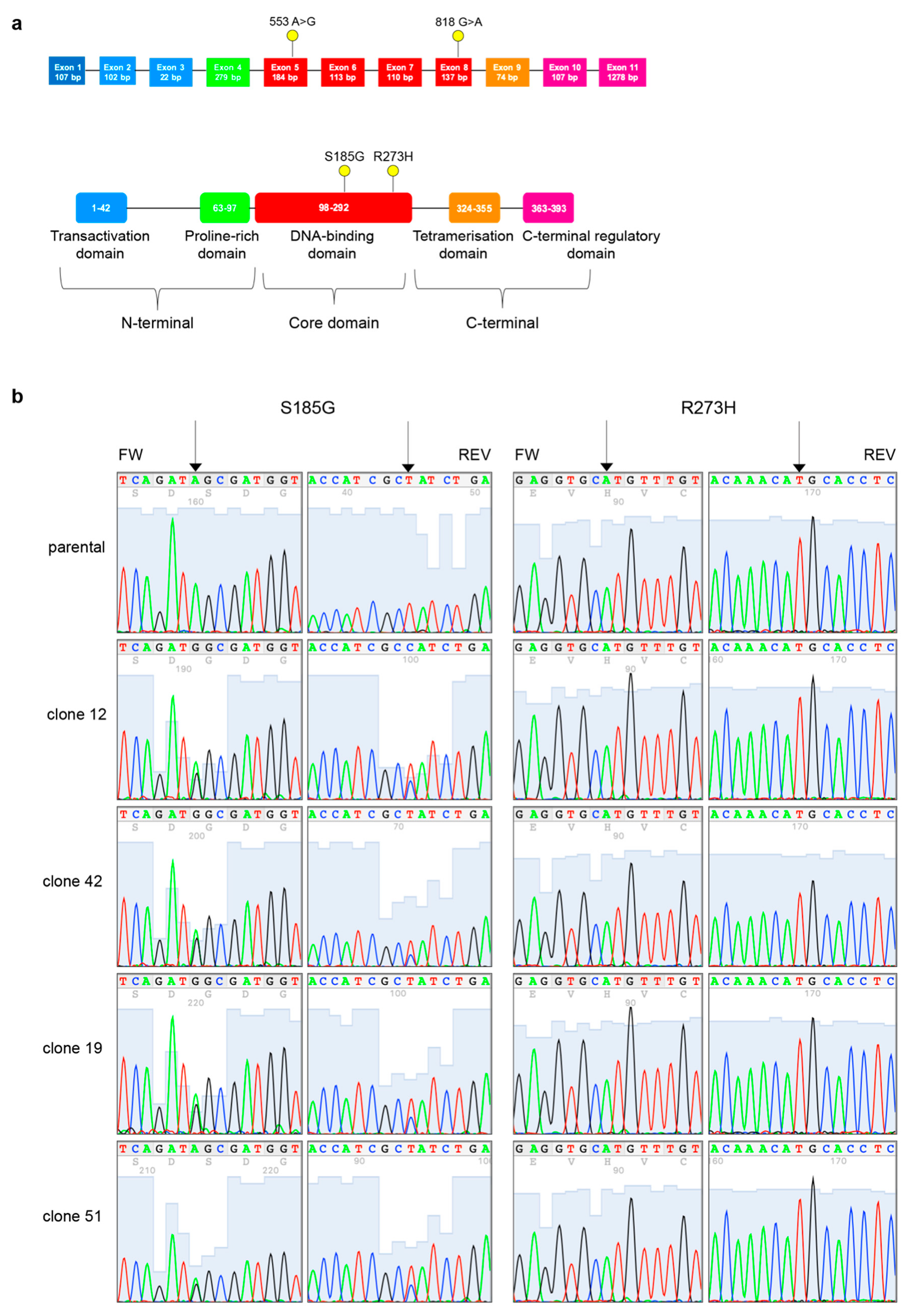
3.5. PT-res Clones Gain a New TP53 Missense Mutation after Selection Process
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Holmes, D. The problem with platinum. Nature 2015, 527, S218–S219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, D. Ovarian cancer: Beyond resistance. Nature 2015, 527, S217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, P.T.; Drapkin, R. Pathogenesis and heterogeneity of ovarian cancer. Curr. Opin. Obstet. Gynecol. 2017, 29, 26–34. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; Roth, A.; Laks, E.; Masud, T.; Bashashati, A.; Zhang, A.W.; Ha, G.; Biele, J.; Yap, D.; Wan, A.; et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016, 48, 758–767. [Google Scholar] [CrossRef]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [Green Version]
- Gatti, L.; Cassinelli, G.; Zaffaroni, N.; Lanzi, C.; Perego, P. New mechanisms for old drugs: Insights into DNA-unrelated effects of platinum compounds and drug resistance determinants. Drug Resist. Update 2015, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sonego, M.; Pellizzari, I.; Dall’Acqua, A.; Pivetta, E.; Lorenzon, I.; Benevol, S.; Bomben, R.; Spessotto, P.; Sorio, R.; Gattei, V.; et al. Common biological phenotypes characterize the acquisition of platinum-resistance in epithelial ovarian cancer cells. Sci. Rep. 2017, 7, 7104. [Google Scholar] [CrossRef] [PubMed]
- Dall’Acqua, A.; Sonego, M.; Pellizzari, I.; Pellarin, I.; Canzonieri, V.; D’Andrea, S.; Benevol, S.; Sorio, R.; Giorda, G.; Califano, D.; et al. CDK6 protects epithelial ovarian cancer from platinum-induced death via FOXO3 regulation. EMBO Mol. Med. 2017, 10, 1415–1433. [Google Scholar] [CrossRef] [PubMed]
- Sonego, M.; Pellarin, I.; Costa, A.; Vinciguerra, G.L.R.; Coan, M.; Kraut, A.; D’Andrea, S.; Dall’Acqua, A.; Castillo-Tong, D.C.; Califano, D.; et al. USP1 links platinum resistance to cancer cell dissemination by regulating Snail stability. Sci. Adv. 2019, 5, eaav3235. [Google Scholar] [CrossRef] [Green Version]
- Sonego, M.; Schiappacassi, M.; Lovisa, S.; Dall’Acqua, A.; Bagnoli, M.; Lovat, F.; Libra, M.; D’Andrea, S.; Canzonieri, V.; Militello, L.; et al. Stathmin regulates mutant p53 stability and transcriptional activity in ovarian cancer. EMBO Mol. Med. 2013, 5, 707–722. [Google Scholar] [CrossRef]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef]
- Diederichs, S.; Bartsch, L.; Berkmann, J.C.; Fröse, K.; Heitmann, J.; Hoppe, C.; Iggena, D.; Jazmati, D.; Karschnia, P.; Linsenmeier, M.; et al. The dark matter of the cancer genome: Aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol. Med. 2016, 8, 442–457. [Google Scholar] [CrossRef]
- Parekh, A.; Das, S.; Parida, S.; Das, C.K.; Dutta, D.; Mallick, S.K.; Wu, P.-H.; Kumar, B.N.P.; Bharti, R.; Dey, G.; et al. Multi-nucleated cells use ROS to induce breast cancer chemo-resistance in vitro and in vivo. Oncogene 2018, 37, 4546–4561. [Google Scholar] [CrossRef]
- Lukin, D.J.; Carvajal, L.A.; Liu, W.; Resnick-Silverman, L.; Manfredi, J.J. p53 Promotes cell survival due to the reversibility of its cell-cycle checkpoints. Mol. Cancer Res. MCR 2015, 13, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintás-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Allison, S.J.; Milner, J. Loss of p53 has site-specific effects on histone H3 modification, including serine 10 phosphorylation important for maintenance of ploidy. Cancer Res. 2003, 63, 6674–6679. [Google Scholar] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Kumar, P.; Murray, D. Multinucleated Giant Cancer Cells Produced in Response to Ionizing Radiation Retain Viability and Replicate Their Genome. Int. J. Mol. Sci. 2017, 18, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Caldon, C.E.; Sergio, C.M.; Burgess, A.; Deans, A.J.; Sutherland, R.L.; Musgrove, E.A. Cyclin E2 induces genomic instability by mechanisms distinct from cyclin E1. Cell Cycle 2013, 12, 606–617. [Google Scholar] [CrossRef] [Green Version]
- Motwani, M.; Li, X.; Schwartz, G.K. Flavopiridol, a cyclin-dependent kinase inhibitor, prevents spindle inhibitor-induced endoreduplication in human cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 924–932. [Google Scholar]
- Pan, D.; Klare, K.; Petrovic, A.; Take, A.; Walstein, K.; Singh, P.; Rondelet, A.; Bird, A.W.; Musacchio, A. CDK-regulated dimerization of M18BP1 on a Mis18 hexamer is necessary for CENP-A loading. Elife 2017, 6, e23352. [Google Scholar] [CrossRef]
- Spiller, F.; Medina-Pritchard, B.; Abad, M.A.; Wear, M.A.; Molina, O.; Earnshaw, W.C.; Jeyaprakash, A.A. Molecular basis for Cdk1-regulated timing of Mis18 complex assembly and CENP-A deposition. EMBO Rep. 2017, 18, 894–905. [Google Scholar] [CrossRef]
- Chen, D.; Ito, S.; Hyodo, T.; Asano-Inami, E.; Yuan, H.; Senga, T. Phosphorylation of DEPDC1 at Ser110 is required to maintain centrosome organization during mitosis. Exp. Cell Res. 2017, 358, 101–110. [Google Scholar] [CrossRef]
- Mi, Y.; Zhang, C.; Bu, Y.; Zhang, Y.; He, L.; Li, H.; Zhu, H.; Li, Y.; Lei, Y.; Zhu, J. DEPDC1 is a novel cell cycle related gene that regulates mitotic progression. BMB Rep. 2015, 48, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Du, Y.; Xu, S.; Jiang, Y.; Yuan, C.; Zhou, L.; Ma, X.; Bai, Y.; Lu, J.; Ma, J. DEPDC1, negatively regulated by miR-26b, facilitates cell proliferation via the up-regulation of FOXM1 expression in TNBC. Cancer Lett. 2019, 442, 242–251. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, P.; Tu, M.; Huang, Y.; Xiong, F.; Wu, Y. DEPDC1 promotes cell proliferation and suppresses sensitivity to chemotherapy in human hepatocellular carcinoma. Biosci. Rep. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batty, P.; Gerlich, D.W. Mitotic Chromosome Mechanics: How Cells Segregate Their Genome. Trends Cell Biol. 2019, 29, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C.-A.; Murray, J.E.; Carroll, P.; Leitch, A.; Mackenzie, K.J.; Halachev, M.; Fetit, A.E.; Keith, C.; Bicknell, L.S.; Fluteau, A.; et al. Mutations in genes encoding condensin complex proteins cause microcephaly through decatenation failure at mitosis. Genes Dev. 2016, 30, 2158–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Jiang, L.-P.; Shen, Q.-S.; Xiong, Q.-X.; Zhuo, X.; Zhang, L.-L.; Yu, H.-J.; Guo, X.; Luo, Y.; Dong, J.; et al. NCAPH plays important roles in human colon cancer. Cell Death Dis. 2017, 8, e2680. [Google Scholar] [CrossRef] [Green Version]
- Ralhan, R.; Arora, S.; Chattopadhyay, T.K.; Shukla, N.K.; Mathur, M. Circulating p53 antibodies, p53 gene mutational profile and product accumulation in esophageal squamous-cell carcinoma in India. Int. J. Cancer 2000, 85, 791–795. [Google Scholar] [CrossRef]
- Neungton, N.; Neungton, S.; Leelaphatanadit, C.; Dangrat, C.; Soiampornkul, R. p53 tumor suppressor gene mutation in ovarian cancer in Thai patients. J. Med. Assoc. Thail. Chotmaihet Thangphaet 2002, 85, 658–667. [Google Scholar]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Mittal, K.; Donthamsetty, S.; Kaur, R.; Yang, C.; Gupta, M.V.; Reid, M.D.; Choi, D.H.; Rida, P.C.G.; Aneja, R. Multinucleated polyploidy drives resistance to Docetaxel chemotherapy in prostate cancer. Br. J. Cancer 2017, 116, 1186–1194. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. The Growing Complexity of Cancer Cell Response to DNA-Damaging Agents: Caspase 3 Mediates Cell Death or Survival? Int. J. Mol. Sci. 2016, 17, 708. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.-F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.-C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Boland, K.; Flanagan, L.; Prehn, J.H.M. Paracrine control of tissue regeneration and cell proliferation by Caspase-3. Cell Death Dis. 2013, 4, e725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; He, Y.; Li, F.; Huang, Q.; Kato, T.A.; Hall, R.P.; Li, C.-Y. Caspase-3 promotes genetic instability and carcinogenesis. Mol. Cell 2015, 58, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Shi, Y.; Zhang, L.; Zhang, D.; Liu, G.; Yang, Z.; Li, Y.; Fei, F.; Zhang, S. Polyploid giant cancer cells with budding and the expression of cyclin E, S-phase kinase-associated protein 2, stathmin associated with the grading and metastasis in serous ovarian tumor. BMC Cancer 2014, 14, 576. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Mercado-Uribe, I.; Hanash, S.; Liu, J. iTRAQ-based proteomic analysis of polyploid giant cancer cells and budding progeny cells reveals several distinct pathways for ovarian cancer development. PLoS ONE 2013, 8, e80120. [Google Scholar] [CrossRef]
- Niu, N.; Zhang, J.; Zhang, N.; Mercado-Uribe, I.; Tao, F.; Han, Z.; Pathak, S.; Multani, A.S.; Kuang, J.; Yao, J.; et al. Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis 2016, 5, e281. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Carballo, D.; Saka, S.; Klein, J.; Rennkamp, T.; Acikelli, A.H.; Malak, S.; Jastrow, H.; Wennemuth, G.; Tempfer, C.; Schmitz, I.; et al. A Distinct Oncogenerative Multinucleated Cancer Cell Serves as a Source of Stemness and Tumor Heterogeneity. Cancer Res. 2018, 78, 2318–2331. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenzon, I.; Pellarin, I.; Pellizzari, I.; D’Andrea, S.; Belletti, B.; Sonego, M.; Baldassarre, G.; Schiappacassi, M. Identification and Characterization of a New Platinum-Induced TP53 Mutation in MDAH Ovarian Cancer Cells. Cells 2020, 9, 36. https://doi.org/10.3390/cells9010036
Lorenzon I, Pellarin I, Pellizzari I, D’Andrea S, Belletti B, Sonego M, Baldassarre G, Schiappacassi M. Identification and Characterization of a New Platinum-Induced TP53 Mutation in MDAH Ovarian Cancer Cells. Cells. 2020; 9(1):36. https://doi.org/10.3390/cells9010036
Chicago/Turabian StyleLorenzon, Ilaria, Ilenia Pellarin, Ilenia Pellizzari, Sara D’Andrea, Barbara Belletti, Maura Sonego, Gustavo Baldassarre, and Monica Schiappacassi. 2020. "Identification and Characterization of a New Platinum-Induced TP53 Mutation in MDAH Ovarian Cancer Cells" Cells 9, no. 1: 36. https://doi.org/10.3390/cells9010036
APA StyleLorenzon, I., Pellarin, I., Pellizzari, I., D’Andrea, S., Belletti, B., Sonego, M., Baldassarre, G., & Schiappacassi, M. (2020). Identification and Characterization of a New Platinum-Induced TP53 Mutation in MDAH Ovarian Cancer Cells. Cells, 9(1), 36. https://doi.org/10.3390/cells9010036