TRPC Channels in Proteinuric Kidney Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Burden of Chronic Kidney Disease (CKD)
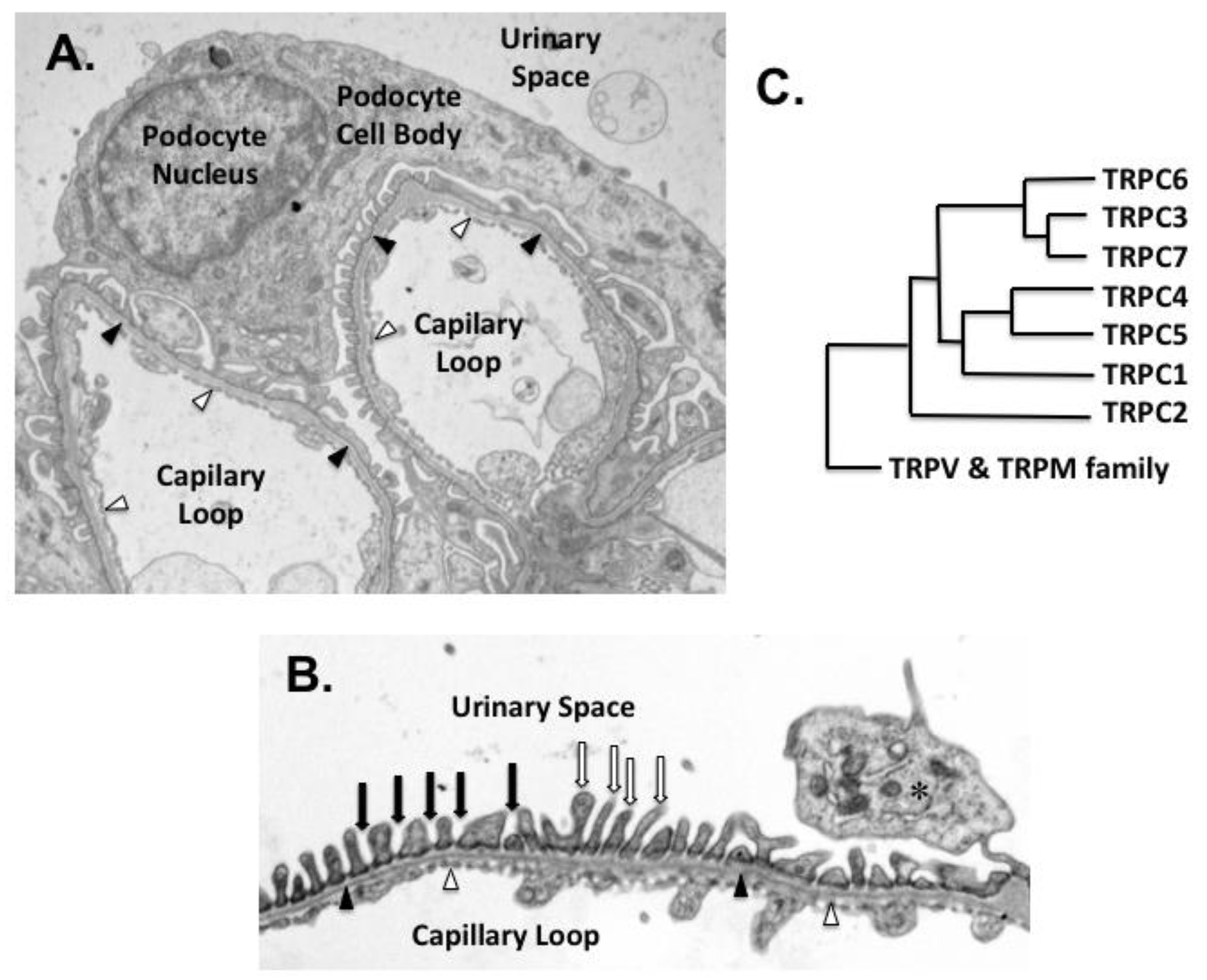
2. The Glomerular Podocyte Plays a Key Role in Proteinuric Kidney Diseases
3. TRPC Family Members
4. TRPC6 in Familial Forms of Nephrosis
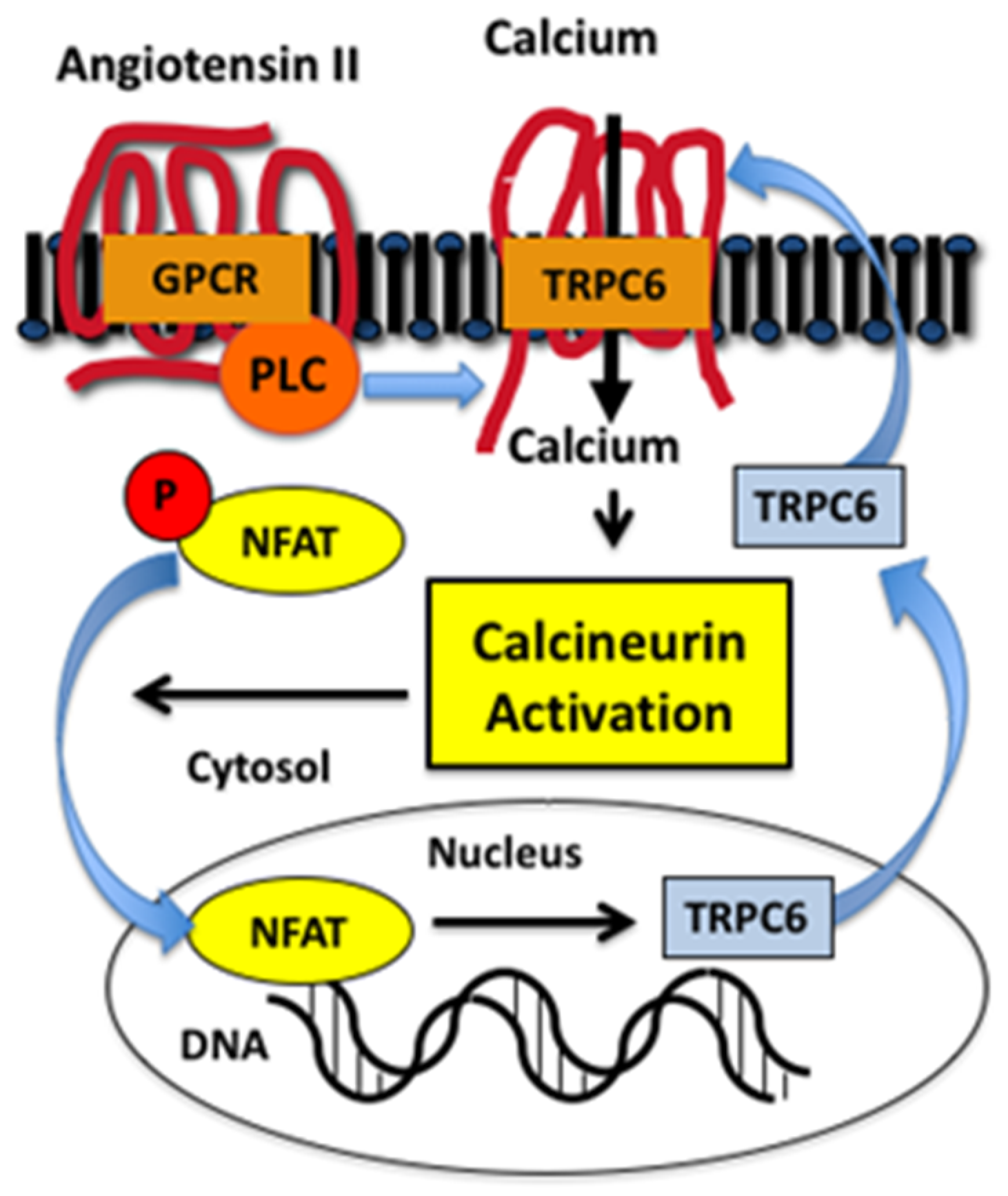
5. TRPC Family Members in Glomerular Diseases: Mechanisms of Renal Injury
6. Targeting TRPC6 to Treat FSGS
7. Targeting TRPC Family Members to Treat Diabetic Nephropathy
8. Targeting TRPC6 in Immune-Mediated Glomerular Diseases
9. Targeting TRPC5 in Proteinuric Kidney Diseases
10. Targeting TRPC Family Members in Other Acquired Kidney Diseases
11. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.; Cheung, A.K. Primer on Kidney Diseases, 4th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2005; p. 608. [Google Scholar]
- Foley, R.N.; Collins, A.J. The USRDS: What you need to know about what it can and can’t tell us about ESRD. Clin. J. Am. Soc. Nephrol. 2013, 8, 845–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Gretz, N.; Lemley, K.V. Progression of glomerular diseases: Is the podocyte the culprit? Kidney Int. 1998, 54, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Pavenstadt, H.; Kriz, W.; Kretzler, M. Cell biology of the glomerular podocyte. Physiol. Rev. 2003, 83, 253–307. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.E.; Jones, N. Nephrin Signaling in the Podocyte: An Updated View of Signal Regulation at the Slit Diaphragm and Beyond. Front. Endocrinol. (Lausanne) 2018, 9, 302. [Google Scholar] [CrossRef]
- Neal, C.R. Podocytes … What’s Under Yours? (Podocytes and Foot Processes and How They Change in Nephropathy). Front. Endocrinol. (Lausanne) 2015, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, K.; Simons, M.; Reiser, J.; Saleem, M.A.; Faul, C.; Kriz, W.; Shaw, A.S.; Holzman, L.B.; Mundel, P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J. Clin. Investig. 2001, 108, 1621–1629. [Google Scholar] [CrossRef]
- Harita, Y.; Kurihara, H.; Kosako, H.; Tezuka, T.; Sekine, T.; Igarashi, T.; Ohsawa, I.; Ohta, S.; Hattori, S. Phosphorylation of Nephrin Triggers Ca2+ Signaling by Recruitment and Activation of Phospholipase C-{gamma}1. J. Biol. Chem. 2009, 284, 8951–8962. [Google Scholar] [CrossRef] [Green Version]
- Jones, N.; Blasutig, I.M.; Eremina, V.; Ruston, J.M.; Bladt, F.; Li, H.; Huang, H.; Larose, L.; Li, S.S.; Takano, T.; et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 2006, 440, 818–823. [Google Scholar] [CrossRef]
- Li, H.; Lemay, S.; Aoudjit, L.; Kawachi, H.; Takano, T. SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J. Am. Soc. Nephrol. 2004, 15, 3006–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, R.; Wharram, B.; Kovari, I.; Kunkel, R.; Nihalani, D.; Wary, K.K.; Wiggins, R.C.; Killen, P.; Holzman, L.B. Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J. Biol. Chem. 2003, 278, 20716–20723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, T.B.; Kottgen, M.; Schilling, B.; Walz, G.; Benzing, T. Interaction with podocin facilitates nephrin signaling. J. Biol. Chem. 2001, 276, 41543–41546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, T.B.; Hartleben, B.; Kim, J.; Schmidts, M.; Schermer, B.; Keil, A.; Egger, L.; Lecha, R.L.; Borner, C.; Pavenstadt, H.; et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol. Cell. Biol. 2003, 23, 4917–4928. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M.R.; Quaggin, S.E.; Hoenig, M.P.; Dworkin, L.D. The glomerulus: The sphere of influence. Clin. J. Am. Soc. Nephrol. 2014, 9, 1461–1469. [Google Scholar] [CrossRef] [Green Version]
- Kriz, W.; Lemley, K.V. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J. Am. Soc. Nephrol. 2015, 26, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Kriz, W.; Lemley, K.V. Potential relevance of shear stress for slit diaphragm and podocyte function. Kidney Int. 2017, 91, 1283–1286. [Google Scholar] [CrossRef]
- Lin, J.S.; Jeon, J.S.; Fan, Q.; Wong, H.N.; Palmer, M.B.; Holzman, L.B. ARF6 mediates nephrin tyrosine phosphorylation-induced podocyte cellular dynamics. PLoS ONE 2017, 12, e0184575. [Google Scholar] [CrossRef] [Green Version]
- George, B.; Verma, R.; Soofi, A.A.; Garg, P.; Zhang, J.; Park, T.J.; Giardino, L.; Ryzhova, L.; Johnstone, D.B.; Wong, H.; et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. J. Clin. Investig. 2012, 122, 674–692. [Google Scholar] [CrossRef] [Green Version]
- Verma, R.; Venkatareddy, M.; Kalinowski, A.; Patel, S.R.; Garg, P. Integrin Ligation Results in Nephrin Tyrosine Phosphorylation In Vitro. PLoS ONE 2016, 11, e0148906. [Google Scholar] [CrossRef] [Green Version]
- Wiggins, R.C. The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 2007, 71, 1205–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, G.; Chen, S.; Ziyadeh, F.N. From the periphery of the glomerular capillary wall toward the center of disease: Podocyte injury comes of age in diabetic nephropathy. Diabetes 2005, 54, 1626–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Goyal, M.; Kurnit, D.; Wharram, B.; Wiggins, J.; Holzman, L.; Kershaw, D.; Wiggins, R. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int. 2001, 60, 957–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wharram, B.L.; Goyal, M.; Wiggins, J.E.; Sanden, S.K.; Hussain, S.; Filipiak, W.E.; Saunders, T.L.; Dysko, R.C.; Kohno, K.; Holzman, L.B.; et al. Podocyte depletion causes glomerulosclerosis: Diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J. Am. Soc. Nephrol. 2005, 16, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef] [Green Version]
- Nishizuka, Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 1992, 258, 607–614. [Google Scholar] [CrossRef]
- Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Martinac, B.; Hamill, O.P. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat. Cell Biol. 2005, 7, 179–185. [Google Scholar] [CrossRef]
- Spassova, M.A.; Hewavitharana, T.; Xu, W.; Soboloff, J.; Gill, D.L. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl. Acad. Sci. USA 2006, 103, 16586–16591. [Google Scholar] [CrossRef] [Green Version]
- Gomis, A.; Soriano, S.; Belmonte, C.; Viana, F. Hypoosmotic- and pressure-induced membrane stretch activate TRPC5 channels. J. Physiol. 2008, 586, 5633–5649. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Wong, C.O.; Lau, O.C.; Woo, T.; Bai, S.; Huang, Y.; Yao, X. Plasma membrane mechanical stress activates TRPC5 channels. PLoS ONE 2015, 10, e0122227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, Y.; Iribe, G.; Nishida, M.; Naruse, K. Role of TRPC3 and TRPC6 channels in the myocardial response to stretch: Linking physiology and pathophysiology. Prog. Biophys. Mol. Biol. 2017, 130, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Dryer, S.E. A mutation in TRPC6 channels abolishes their activation by hypoosmotic stretch but does not affect activation by diacylglycerol or G protein signaling cascades. Am. J. Physiol. Ren. Physiol. 2014, 306, F1018–F1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dryer, S.E.; Roshanravan, H.; Kim, E.Y. TRPC channels: Regulation, dysregulation and contributions to chronic kidney disease. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 1041–1066. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Kim, E.Y.; Hagmann, H.; Benzing, T.; Dryer, S.E. Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by membrane stretch or diacylglycerol. Am. J. Physiol. Cell Physiol. 2013, 305, C276–C289. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, Y.A.; Cox, C.D.; Ridone, P.; Rohde, P.R.; Cordero-Morales, J.F.; Vasquez, V.; Laver, D.R.; Martinac, B. Mammalian TRP ion channels are insensitive to membrane stretch. J. Cell. Sci. 2019. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Inoue, R.; Morii, T.; Takahashi, N.; Yamamoto, S.; Hara, Y.; Tominaga, M.; Shimizu, S.; Sato, Y.; Mori, Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2006, 2, 596–607. [Google Scholar] [CrossRef]
- Yamamoto, S.; Takahashi, N.; Mori, Y. Chemical physiology of oxidative stress-activated TRPM2 and TRPC5 channels. Prog. Biophys. Mol. Biol. 2010, 103, 18–27. [Google Scholar] [CrossRef]
- Ding, Y.; Winters, A.; Ding, M.; Graham, S.; Akopova, I.; Muallem, S.; Wang, Y.; Hong, J.H.; Gryczynski, Z.; Yang, S.H.; et al. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J. Biol. Chem. 2011, 286, 31799–31809. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.; Roshanravan, H.; Khine, J.; Dryer, S.E. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J. Cell. Physiol. 2014, 229, 434–442. [Google Scholar] [CrossRef]
- Kim, E.Y.; Anderson, M.; Wilson, C.; Hagmann, H.; Benzing, T.; Dryer, S.E. NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: Essential role of podocin in formation of this complex. Am. J. Physiol. Cell Physiol. 2013, 305, C960–C971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshanravan, H.; Dryer, S.E. ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: Role of podocin and reactive oxygen species. Am. J. Physiol. Ren. Physiol. 2014, 306, F1088–F1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiser, J.; Polu, K.R.; Moller, C.C.; Kenlan, P.; Altintas, M.M.; Wei, C.; Faul, C.; Herbert, S.; Villegas, I.; Avila-Casado, C.; et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 2005, 37, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Huber, T.B.; Schermer, B.; Muller, R.U.; Hohne, M.; Bartram, M.; Calixto, A.; Hagmann, H.; Reinhardt, C.; Koos, F.; Kunzelmann, K.; et al. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc. Natl. Acad. Sci. USA 2006, 103, 17079–17086. [Google Scholar] [CrossRef] [Green Version]
- Faul, C.; Asanuma, K.; Yanagida-Asanuma, E.; Kim, K.; Mundel, P. Actin up: Regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007, 17, 428–437. [Google Scholar] [CrossRef]
- Drenckhahn, D.; Franke, R.P. Ultrastructural organization of contractile and cytoskeletal proteins in glomerular podocytes of chicken, rat, and man. Lab. Investig. 1988, 59, 673–682. [Google Scholar]
- Riehle, M.; Buscher, A.K.; Gohlke, B.O.; Kassmann, M.; Kolatsi-Joannou, M.; Brasen, J.H.; Nagel, M.; Becker, J.U.; Winyard, P.; Hoyer, P.F.; et al. TRPC6 G757D Loss-of-Function Mutation Associates with FSGS. J. Am. Soc. Nephrol. 2016, 27, 2771–2783. [Google Scholar] [CrossRef] [Green Version]
- Santin, S.; Ars, E.; Rossetti, S.; Salido, E.; Silva, I.; Garcia-Maset, R.; Gimenez, I.; Ruiz, P.; Mendizabal, S.; Luciano Nieto, J.; et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2009, 24, 3089–3096. [Google Scholar] [CrossRef] [Green Version]
- Mir, S.; Yavascan, O.; Berdeli, A.; Sozeri, B. TRPC6 gene variants in Turkish children with steroid-resistant nephrotic syndrome. Nephrol. Dial. Transplant. 2012, 27, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Buscher, A.K.; Kranz, B.; Buscher, R.; Hildebrandt, F.; Dworniczak, B.; Pennekamp, P.; Kuwertz-Broking, E.; Wingen, A.M.; John, U.; Kemper, M.; et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2010, 5, 2075–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigante, M.; Caridi, G.; Montemurno, E.; Soccio, M.; d’Apolito, M.; Cerullo, G.; Aucella, F.; Schirinzi, A.; Emma, F.; Massella, L.; et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin. J. Am. Soc. Nephrol. 2011, 6, 1626–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeringa, S.F.; Moller, C.C.; Du, J.; Yue, L.; Hinkes, B.; Chernin, G.; Vlangos, C.N.; Hoyer, P.F.; Reiser, J.; Hildebrandt, F. A novel TRPC6 mutation that causes childhood FSGS. PLoS ONE 2009, 4, e7771. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Chen, N.; Wang, Z.H.; Pan, X.X.; Ren, H.; Zhang, W.; Wang, W.M. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat. Res. 2009, 664, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Hofstra, J.M.; Lainez, S.; van Kuijk, W.H.; Schoots, J.; Baltissen, M.P.; Hoefsloot, L.H.; Knoers, N.V.; Berden, J.H.; Bindels, R.J.; van der Vlag, J.; et al. New TRPC6 gain-of-function mutation in a non-consanguineous Dutch family with late-onset focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2013, 28, 1830–1838. [Google Scholar] [CrossRef] [Green Version]
- Buscher, A.K.; Konrad, M.; Nagel, M.; Witzke, O.; Kribben, A.; Hoyer, P.F.; Weber, S. Mutations in podocyte genes are a rare cause of primary FSGS associated with ESRD in adult patients. Clin. Nephrol. 2012, 78, 47–53. [Google Scholar] [CrossRef]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Howell, D.N.; Slotterbeck, B.D.; Smith, A.H.; Graham, F.L.; Bembe, M.; Quarles, L.D.; Pericak-Vance, M.A.; et al. Linkage of a gene causing familial focal segmental glomerulosclerosis to chromosome 11 and further evidence of genetic heterogeneity. Genomics 1999, 58, 113–120. [Google Scholar] [CrossRef]
- Moller, C.C.; Flesche, J.; Reiser, J. Sensitizing the Slit Diaphragm with TRPC6 ion channels. J. Am. Soc. Nephrol. 2009, 20, 950–953. [Google Scholar] [CrossRef] [Green Version]
- El Hindi, S.; Reiser, J. TRPC channel modulation in podocytes-inching toward novel treatments for glomerular disease. Pediatr. Nephrol. 2011, 26, 1057–1064. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Jirka, G.; Rosenberg, P.B.; Buckley, A.F.; Gomez, J.A.; Fields, T.A.; Winn, M.P.; Spurney, R.F. Gq signaling causes glomerular injury by activating TRPC6. J. Clin. Investig. 2015, 125, 1913–1926. [Google Scholar] [CrossRef] [Green Version]
- Moller, C.C.; Wei, C.; Altintas, M.M.; Li, J.; Greka, A.; Ohse, T.; Pippin, J.W.; Rastaldi, M.P.; Wawersik, S.; Schiavi, S.; et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J. Am. Soc. Nephrol. 2007, 18, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Krall, P.; Canales, C.P.; Kairath, P.; Carmona-Mora, P.; Molina, J.; Carpio, J.D.; Ruiz, P.; Mezzano, S.A.; Li, J.; Wei, C.; et al. Podocyte-specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLoS ONE 2010, 5, e12859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonneveld, R.; Hoenderop, J.G.; Isidori, A.M.; Henique, C.; Dijkman, H.B.; Berden, J.H.; Tharaux, P.L.; van der Vlag, J.; Nijenhuis, T. Sildenafil Prevents Podocyte Injury via PPAR-gamma-Mediated TRPC6 Inhibition. J. Am. Soc. Nephrol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Sonneveld, R.; van der Vlag, J.; Baltissen, M.P.; Verkaart, S.A.; Wetzels, J.F.; Berden, J.H.; Hoenderop, J.G.; Nijenhuis, T. Glucose specifically regulates TRPC6 expression in the podocyte in an AngII-dependent manner. Am. J. Pathol. 2014, 184, 1715–1726. [Google Scholar] [CrossRef]
- Ma, R.; Liu, L.; Jiang, W.; Yu, Y.; Song, H. FK506 ameliorates podocyte injury in type 2 diabetic nephropathy by down-regulating TRPC6 and NFAT expression. Int. J. Clin. Exp. Pathol. 2015, 8, 14063–14074. [Google Scholar]
- Ilatovskaya, D.V.; Levchenko, V.; Lowing, A.; Shuyskiy, L.S.; Palygin, O.; Staruschenko, A. Podocyte injury in diabetic nephropathy: Implications of angiotensin II-dependent activation of TRPC channels. Sci. Rep. 2015, 5, 17637. [Google Scholar] [CrossRef]
- Ma, R.; Xu, Y.; Zhou, H.; Zhang, D.; Yao, D.; Song, L.; Liu, Y. Participation of the AngII/TRPC6/NFAT axis in the pathogenesis of podocyte injury in rats with type 2 diabetes. Mol. Med. Rep. 2019, 19, 2421–2430. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.Y.; Liu, B.C.; Cao, Y.Z.; Song, C.; Su, H.; Chen, G.; Klein, J.D.; Zhang, H.X.; Wang, L.; Ma, H.P. High glucose reduces the expression of podocin in cultured human podocytes by stimulating TRPC6. Am. J. Physiol. Ren. Physiol. 2019. [Google Scholar] [CrossRef]
- Kistler, A.D.; Singh, G.; Altintas, M.M.; Yu, H.; Fernandez, I.C.; Gu, C.; Wilson, C.; Srivastava, S.K.; Dietrich, A.; Walz, K.; et al. Transient receptor potential channel 6 (TRPC6) protects podocytes during complement-mediated glomerular disease. J. Biol. Chem. 2013, 288, 36598–36609. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Shotorbani, P.Y.; Dryer, S.E. TRPC6 inactivation does not affect loss of renal function in nephrotoxic serum glomerulonephritis in rats, but reduces severity of glomerular lesions. Biochem. Biophys. Rep. 2019, 17, 139–150. [Google Scholar] [CrossRef]
- Sonneveld, R.; Ferre, S.; Hoenderop, J.G.; Dijkman, H.B.; Berden, J.H.; Bindels, R.J.; Wetzels, J.F.; van der Vlag, J.; Nijenhuis, T. Vitamin D down-regulates TRPC6 expression in podocyte injury and proteinuric glomerular disease. Am. J. Pathol. 2013, 182, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
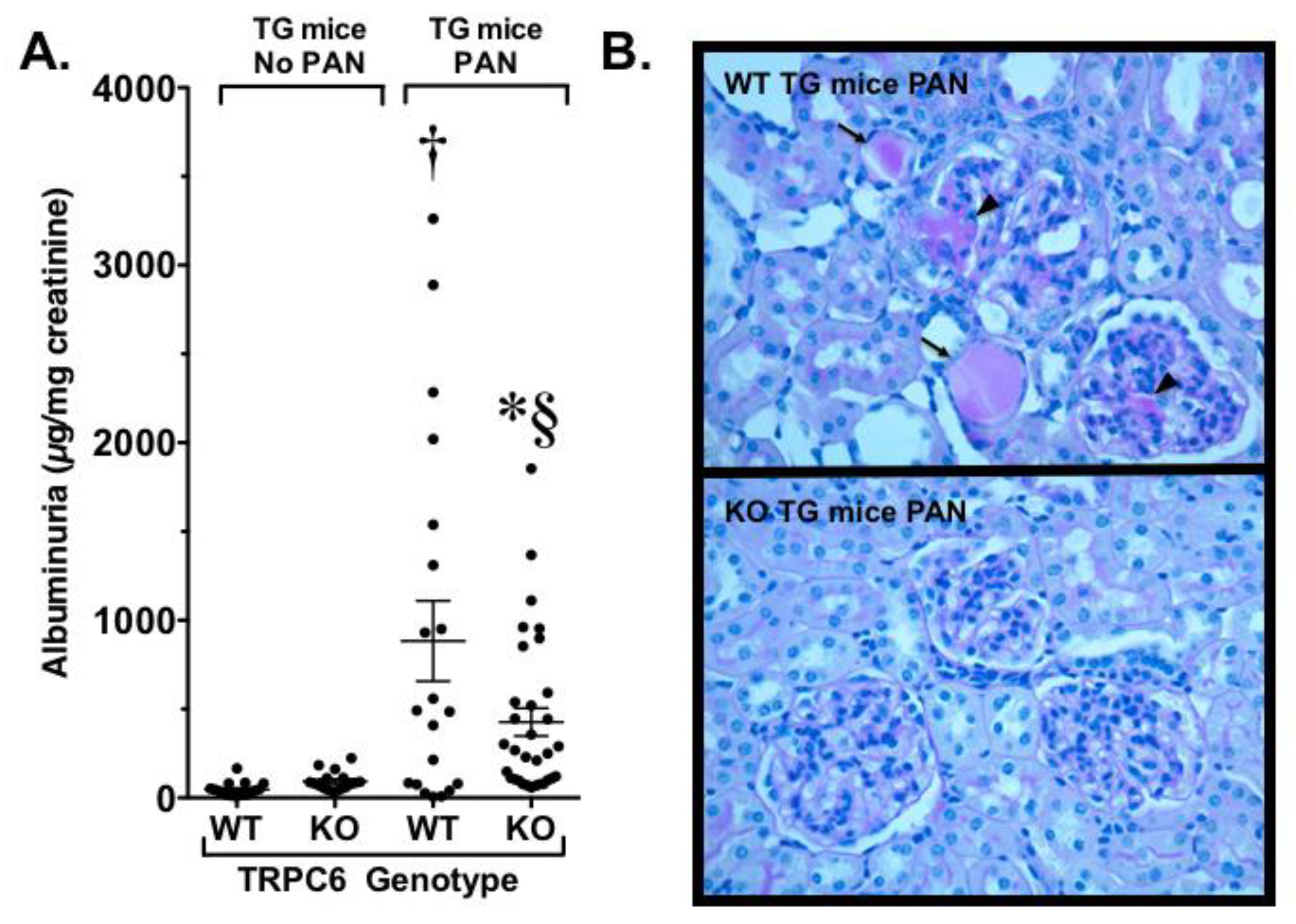
- Kim, E.Y.; Yazdizadeh Shotorbani, P.; Dryer, S.E. Trpc6 inactivation confers protection in a model of severe nephrosis in rats. J. Mol. Med. (Berl.) 2018, 96, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijenhuis, T.; Sloan, A.J.; Hoenderop, J.G.; Flesche, J.; van Goor, H.; Kistler, A.D.; Bakker, M.; Bindels, R.J.; de Boer, R.A.; Moller, C.C.; et al. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am. J. Pathol. 2011, 179, 1719–1732. [Google Scholar] [CrossRef] [PubMed]
- Saliba, Y.; Karam, R.; Smayra, V.; Aftimos, G.; Abramowitz, J.; Birnbaumer, L.; Fares, N. Evidence of a Role for Fibroblast Transient Receptor Potential Canonical 3 Ca2+ Channel in Renal Fibrosis. J. Am. Soc. Nephrol. 2015, 26, 1855–1876. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.L.; Xie, J.; An, S.W.; Oliver, N.; Barrezueta, N.X.; Lin, M.H.; Birnbaumer, L.; Huang, C.L. Inhibition of TRPC6 channels ameliorates renal fibrosis and contributes to renal protection by soluble klotho. Kidney Int. 2017, 91, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, K.; Wang, Y.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Hill, J.A.; Olson, E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Investig. 2006, 116, 3114–3126. [Google Scholar] [CrossRef]
- Bush, E.W.; Hood, D.B.; Papst, P.J.; Chapo, J.A.; Minobe, W.; Bristow, M.R.; Olson, E.N.; McKinsey, T.A. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J. Biol. Chem. 2006, 281, 33487–33496. [Google Scholar] [CrossRef] [Green Version]
- Onohara, N.; Nishida, M.; Inoue, R.; Kobayashi, H.; Sumimoto, H.; Sato, Y.; Mori, Y.; Nagao, T.; Kurose, H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006, 25, 5305–5316. [Google Scholar] [CrossRef]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes. Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.A.; Spurney, R.F. Twenty years after ACEIs and ARBs: Emerging treatment strategies for diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2015, 309, F807–F820. [Google Scholar] [CrossRef] [Green Version]
- Horsley, V.; Pavlath, G.K. NFAT: Ubiquitous regulator of cell differentiation and adaptation. J. Cell Biol. 2002, 156, 771–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, B.J.; Dai, Y.S.; Bueno, O.F.; Parsons, S.A.; Xu, J.; Plank, D.M.; Jones, F.; Kimball, T.R.; Molkentin, J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2004, 94, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jarad, G.; Tripathi, P.; Pan, M.; Cunningham, J.; Martin, D.R.; Liapis, H.; Miner, J.H.; Chen, F. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J. Am. Soc. Nephrol. 2010, 21, 1657–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlondorff, J.; Del Camino, D.; Carrasquillo, R.; Lacey, V.; Pollak, M.R. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am. J. Physiol. Cell Physiol. 2009, 296, C558–C569. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Rao, A.; Hogan, P.G. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 2011, 21, 91–103. [Google Scholar] [CrossRef] [Green Version]
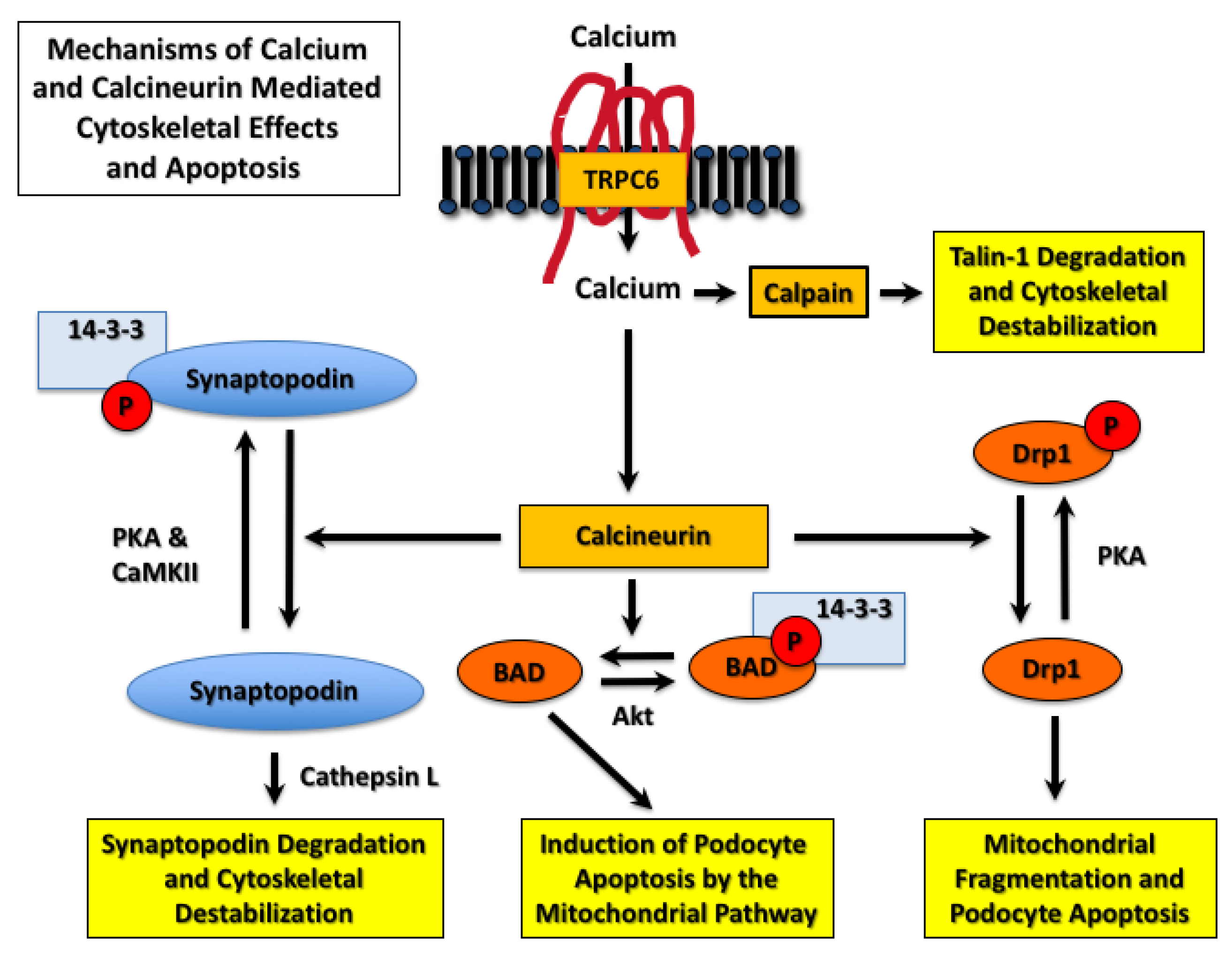
- Yu, H.; Kistler, A.; Faridi, M.H.; Meyer, J.O.; Tryniszewska, B.; Mehta, D.; Yue, L.; Dryer, S.; Reiser, J. Synaptopodin Limits TRPC6 Podocyte Surface Expression and Attenuates Proteinuria. J. Am. Soc. Nephrol. 2016, 27, 3308–3319. [Google Scholar] [CrossRef]
- Faul, C.; Donnelly, M.; Merscher-Gomez, S.; Chang, Y.H.; Franz, S.; Delfgaauw, J.; Chang, J.M.; Choi, H.Y.; Campbell, K.N.; Kim, K.; et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 2008, 14, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chang, J.H.; Paik, S.Y.; Tang, Y.; Eisner, W.; Spurney, R.F. Calcineurin (CN) activation promotes apoptosis of glomerular podocytes both in vitro and in vivo. Mol. Endocrinol. 2011, 25, 1376–1386. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.C.; Song, X.; Lu, X.Y.; Li, D.T.; Eaton, D.C.; Shen, B.Z.; Li, X.Q.; Ma, H.P. High glucose induces podocyte apoptosis by stimulating TRPC6 via elevation of reactive oxygen species. Biochim. Biophys. Acta 2013, 1833, 1434–1442. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Shibasaki, F.; Hallin, U.; Uchino, H. Calcineurin as a multifunctional regulator. J. Biochem. 2002, 131, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.Y.; Qi, R. Role of bad in podocyte apoptosis induced by puromycin aminonucleoside. Transplant. Proc. 2013, 45, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.B.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Mechanisms of renal apoptosis in health and disease. J. Am. Soc. Nephrol. 2008, 19, 1634–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheijden, K.A.T.; Sonneveld, R.; Bakker-van Bebber, M.; Wetzels, J.F.M.; van der Vlag, J.; Nijenhuis, T. The Calcium-Dependent Protease Calpain-1 Links TRPC6 Activity to Podocyte Injury. J. Am. Soc. Nephrol. 2018, 29, 2099–2109. [Google Scholar] [CrossRef] [Green Version]
- Farmer, L.K.; Rollason, R.; Whitcomb, D.J.; Ni, L.; Goodliff, A.; Lay, A.C.; Birnbaumer, L.; Heesom, K.J.; Xu, S.Z.; Saleem, M.A.; et al. TRPC6 Binds to and Activates Calpain, Independent of Its Channel Activity, and Regulates Podocyte Cytoskeleton, Cell Adhesion, and Motility. J. Am. Soc. Nephrol. 2019, 30, 1910–1924. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.L.; Mattson, M.P. Caspase and calpain substrates: Roles in synaptic plasticity and cell death. J. Neurosci. Res. 1999, 58, 167–190. [Google Scholar] [CrossRef]
- Tian, X.; Kim, J.J.; Monkley, S.M.; Gotoh, N.; Nandez, R.; Soda, K.; Inoue, K.; Balkin, D.M.; Hassan, H.; Son, S.H.; et al. Podocyte-associated talin1 is critical for glomerular filtration barrier maintenance. J. Clin. Investig. 2014, 124, 1098–1113. [Google Scholar] [CrossRef] [Green Version]
- Tian, D.; Jacobo, S.M.; Billing, D.; Rozkalne, A.; Gage, S.D.; Anagnostou, T.; Pavenstadt, H.; Hsu, H.H.; Schlondorff, J.; Ramos, A.; et al. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci. Signal. 2010, 3, ra77. [Google Scholar] [CrossRef] [Green Version]
- Greka, A.; Mundel, P. Balancing calcium signals through TRPC5 and TRPC6 in podocytes. J. Am. Soc. Nephrol. 2011, 22, 1969–1980. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Ding, J.; Tsai, H.; Li, L.; Feng, Q.; Miao, J.; Fan, Q. Over-expressing transient receptor potential cation channel 6 in podocytes induces cytoskeleton rearrangement through increases of intracellular Ca2+ and RhoA activation. Exp. Biol. Med. (Maywood) 2011, 236, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.; Spurney, R.F. Losing their footing: Rac1 signaling causes podocyte detachment and FSGS. Kidney Int. 2017, 92, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Gupta, I.R.; Baldwin, C.; Auguste, D.; Ha, K.C.; El Andalousi, J.; Fahiminiya, S.; Bitzan, M.; Bernard, C.; Akbari, M.R.; Narod, S.A.; et al. ARHGDIA: A novel gene implicated in nephrotic syndrome. J. Med. Genet. 2013, 50, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robins, R.; Baldwin, C.; Aoudjit, L.; Cote, J.F.; Gupta, I.R.; Takano, T. Rac1 activation in podocytes induces the spectrum of nephrotic syndrome. Kidney Int. 2017, 92, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Akilesh, S.; Suleiman, H.; Yu, H.; Stander, M.C.; Lavin, P.; Gbadegesin, R.; Antignac, C.; Pollak, M.; Kopp, J.B.; Winn, M.P.; et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J. Clin. Investig. 2011, 121, 4127–4137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouawad, F.; Tsui, H.; Takano, T. Role of Rho-GTPases and their regulatory proteins in glomerular podocyte function. Can. J. Physiol. Pharmacol. 2013, 91, 773–782. [Google Scholar] [CrossRef]
- Schaldecker, T.; Kim, S.; Tarabanis, C.; Tian, D.; Hakroush, S.; Castonguay, P.; Ahn, W.; Wallentin, H.; Heid, H.; Hopkins, C.R.; et al. Inhibition of the TRPC5 ion channel protects the kidney filter. J. Clin. Investig. 2013, 123, 5298–5309. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Castonguay, P.; Sidhom, E.H.; Clark, A.R.; Dvela-Levitt, M.; Kim, S.; Sieber, J.; Wieder, N.; Jung, J.Y.; Andreeva, S.; et al. A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science 2017, 358, 1332–1336. [Google Scholar] [CrossRef] [Green Version]
- Ilatovskaya, D.V.; Staruschenko, A. TRPC6 channel as an emerging determinant of the podocyte injury susceptibility in kidney diseases. Am. J. Physiol. Ren. Physiol. 2015, 309, F393–F397. [Google Scholar] [CrossRef] [Green Version]
- Eckel, J.; Lavin, P.J.; Finch, E.A.; Mukerji, N.; Burch, J.; Gbadegesin, R.; Wu, G.; Bowling, B.; Byrd, A.; Hall, G.; et al. TRPC6 enhances angiotensin II-induced albuminuria. J. Am. Soc. Nephrol. 2011, 22, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Spires, D.; Ilatovskaya, D.V.; Levchenko, V.; North, P.E.; Geurts, A.M.; Palygin, O.; Staruschenko, A. Protective role of Trpc6 knockout in the progression of diabetic kidney disease. Am. J. Physiol. Ren. Physiol. 2018, 315, F1091–F1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, A.; Mederos, Y.S.M.; Gollasch, M.; Gross, V.; Storch, U.; Dubrovska, G.; Obst, M.; Yildirim, E.; Salanova, B.; Kalwa, H.; et al. Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol. Cell. Biol. 2005, 25, 6980–6989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullich, G.; Domingo-Gallego, A.; Vargas, I.; Ruiz, P.; Lorente-Grandoso, L.; Furlano, M.; Fraga, G.; Madrid, A.; Ariceta, G.; Borregan, M.; et al. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 2018, 94, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Kestila, M.; Lenkkeri, U.; Mannikko, M.; Lamerdin, J.; McCready, P.; Putaala, H.; Ruotsalainen, V.; Morita, T.; Nissinen, M.; Herva, R.; et al. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol. Cell 1998, 1, 575–582. [Google Scholar] [CrossRef]
- Boute, N.; Gribouval, O.; Roselli, S.; Benessy, F.; Lee, H.; Fuchshuber, A.; Dahan, K.; Gubler, M.C.; Niaudet, P.; Antignac, C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet. 2000, 24, 349–354. [Google Scholar] [CrossRef]
- Shih, N.Y.; Li, J.; Karpitskii, V.; Nguyen, A.; Dustin, M.L.; Kanagawa, O.; Miner, J.H.; Shaw, A.S. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 1999, 286, 312–315. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Wu, H.; Green, G.; Winkler, C.A.; Kopp, J.B.; Miner, J.H.; Unanue, E.R.; Shaw, A.S. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science 2003, 300, 1298–1300. [Google Scholar] [CrossRef]
- Kaplan, J.M.; Kim, S.H.; North, K.N.; Rennke, H.; Correia, L.A.; Tong, H.Q.; Mathis, B.J.; Rodriguez-Perez, J.C.; Allen, P.G.; Beggs, A.H.; et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000, 24, 251–256. [Google Scholar] [CrossRef]
- Staffel, J.; Valletta, D.; Federlein, A.; Ehm, K.; Volkmann, R.; Fuchsl, A.M.; Witzgall, R.; Kuhn, M.; Schweda, F. Natriuretic Peptide Receptor Guanylyl Cyclase-A in Podocytes is Renoprotective but Dispensable for Physiologic Renal Function. J. Am. Soc. Nephrol. 2017, 28, 260–277. [Google Scholar] [CrossRef] [Green Version]
- Korbet, S.M. Treatment of primary FSGS in adults. J. Am. Soc. Nephrol. 2012, 23, 1769–1776. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Lin, H.; Geshi, N.; Mori, Y.; Kawarabayashi, Y.; Takami, N.; Mori, M.X.; Honda, A.; Inoue, R. Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J. Physiol. 2008, 586, 4209–4223. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Kuwahara, K.; Nishida, M.; Jian, Z.; Rong, X.; Kiyonaka, S.; Kuwabara, Y.; Kurose, H.; Inoue, R.; Mori, Y.; et al. Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart. Circ. Res. 2010, 106, 1849–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, M.; Watanabe, K.; Sato, Y.; Nakaya, M.; Kitajima, N.; Ide, T.; Inoue, R.; Kurose, H. Phosphorylation of TRPC6 channels at Thr69 is required for anti-hypertrophic effects of phosphodiesterase 5 inhibition. J. Biol. Chem. 2010, 285, 13244–13253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, G.; Rowell, J.; Farinelli, F.; Gbadegesin, R.A.; Lavin, P.; Wu, G.; Homstad, A.; Malone, A.; Lindsey, T.; Jiang, R.; et al. Phosphodiesterase 5 inhibition ameliorates angiontensin II-induced podocyte dysmotility via the protein kinase G-mediated downregulation of TRPC6 activity. Am. J. Physiol. Ren. Physiol. 2014, 306, F1442–F1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Champion, H.C.; Li, M.; Belardi, D.; Ren, S.; Rodriguez, E.R.; Bedja, D.; Gabrielson, K.L.; Wang, Y.; Kass, D.A. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 2005, 11, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef]
- Scheele, W.; Diamond, S.; Gale, J.; Clerin, V.; Tamimi, N.; Le, V.; Walley, R.; Grover-Paez, F.; Perros-Huguet, C.; Rolph, T.; et al. Phosphodiesterase Type 5 Inhibition Reduces Albuminuria in Subjects with Overt Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Molitch, M.E.; DeFronzo, R.A.; Franz, M.J.; Keane, W.F.; Mogensen, C.E.; Parving, H.H.; Steffes, M.W. Nephropathy in diabetes. Diabetes Care 2004, 27, S79–S83. [Google Scholar]
- Yang, H.; Zhao, B.; Liao, C.; Zhang, R.; Meng, K.; Xu, J.; Jiao, J. High glucose-induced apoptosis in cultured podocytes involves TRPC6-dependent calcium entry via the RhoA/ROCK pathway. Biochem. Biophys. Res. Commun. 2013, 434, 394–400. [Google Scholar] [CrossRef]
- Zhang, X.; Song, Z.; Guo, Y.; Zhou, M. The novel role of TRPC6 in vitamin D ameliorating podocyte injury in STZ-induced diabetic rats. Mol. Cell. Biochem. 2015, 399, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Ilatovskaya, D.V.; Blass, G.; Palygin, O.; Levchenko, V.; Pavlov, T.S.; Grzybowski, M.N.; Winsor, K.; Shuyskiy, L.S.; Geurts, A.M.; Cowley, A.W., Jr.; et al. A NOX4/TRPC6 Pathway in Podocyte Calcium Regulation and Renal Damage in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 1917–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
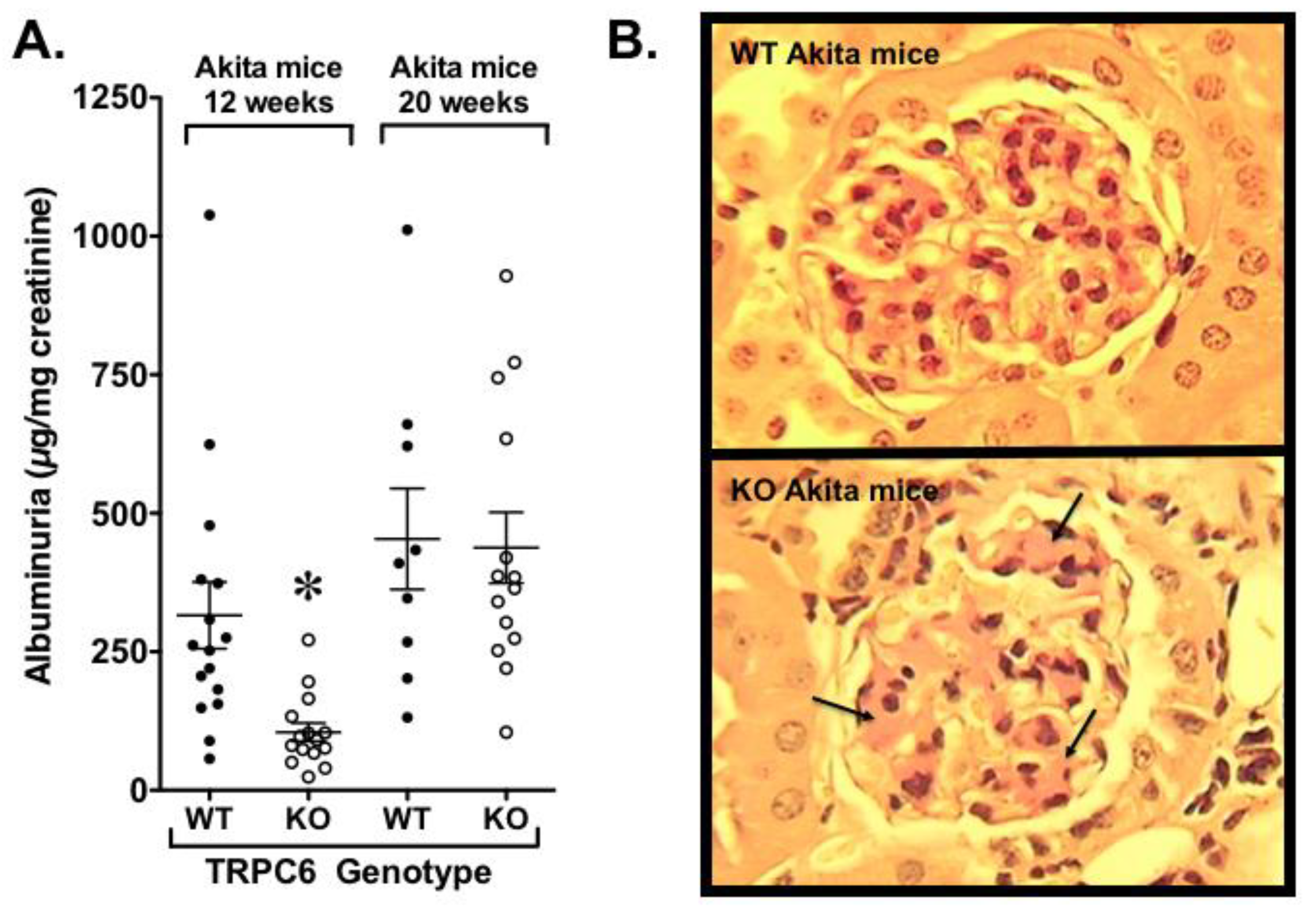
- Wang, L.; Chang, J.H.; Buckley, A.F.; Spurney, R.F. Knockout of TRPC6 promotes insulin resistance and exacerbates glomerular injury in Akita mice. Kidney Int. 2019, 95, 321–332. [Google Scholar] [CrossRef]
- Welsh, G.I.; Hale, L.J.; Eremina, V.; Jeansson, M.; Maezawa, Y.; Lennon, R.; Pons, D.A.; Owen, R.J.; Satchell, S.C.; Miles, M.J.; et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010, 12, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Coward, R.J.; Welsh, G.I.; Yang, J.; Tasman, C.; Lennon, R.; Koziell, A.; Satchell, S.; Holman, G.D.; Kerjaschki, D.; Tavare, J.M.; et al. The human glomerular podocyte is a novel target for insulin action. Diabetes 2005, 54, 3095–3102. [Google Scholar] [CrossRef] [Green Version]
- Isshiki, K.; Haneda, M.; Koya, D.; Maeda, S.; Sugimoto, T.; Kikkawa, R. Thiazolidinedione compounds ameliorate glomerular dysfunction independent of their insulin-sensitizing action in diabetic rats. Diabetes 2000, 49, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, S.B.; Kawano, Y.; Wakino, S.; Collins, A.R.; Hsueh, W.A. Expression and function of peroxisome proliferator-activated receptor-gamma in mesangial cells. Hypertension 2001, 37, 722–727. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Saha, J.; Byun, J.; Schin, M.; Lorenz, M.; Kennedy, R.T.; Kretzler, M.; Feldman, E.L.; Pennathur, S.; Brosius, F.C., 3rd. Rosiglitazone reduces renal and plasma markers of oxidative injury and reverses urinary metabolite abnormalities in the amelioration of diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2008, 295, F1071–F1081. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; He, X.; Li, S.; Xu, B.; Birnbaumer, L.; Liao, Y. Deletion of diacylglycerol-responsive TRPC genes attenuates diabetic nephropathy by inhibiting activation of the TGFbeta1 signaling pathway. Am. J. Transl. Res. 2017, 9, 5619–5630. [Google Scholar]
- Chen, N.Y.; Chen, W.Y.; Bellush, L.; Yang, C.W.; Striker, L.J.; Striker, G.E.; Kopchick, J.J. Effects of streptozotocin treatment in growth hormone (GH) and GH antagonist transgenic mice. Endocrinology 1995, 136, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, G.A.; Coletto, L.A.; Sciorati, C.; Bozzolo, E.P.; Manunta, P.; Rovere-Querini, P.; Manfredi, A.A. Ion Channels and Transporters in Inflammation: Special Focus on TRP Channels and TRPC6. Cells 2018, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, E.W.; Han, F.; Tauseef, M.; Birnbaumer, L.; Mehta, D.; Muller, W.A. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J. Exp. Med. 2015, 212, 1883–1899. [Google Scholar] [CrossRef] [PubMed]
- Riazanski, V.; Gabdoulkhakova, A.G.; Boynton, L.S.; Eguchi, R.R.; Deriy, L.V.; Hogarth, D.K.; Loaec, N.; Oumata, N.; Galons, H.; Brown, M.E.; et al. TRPC6 channel translocation into phagosomal membrane augments phagosomal function. Proc. Natl. Acad. Sci. USA 2015, 112, E6486–E6495. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, O.; Umlauf, D.; Frank, S.; Schimmelpfennig, S.; Bertrand, J.; Pap, T.; Hanley, P.J.; Fabian, A.; Dietrich, A.; Schwab, A. TRPC6 regulates CXCR2-mediated chemotaxis of murine neutrophils. J. Immunol. 2013, 190, 5496–5505. [Google Scholar] [CrossRef] [Green Version]
- Cybulsky, A.V.; Bonventre, J.V.; Quigg, R.J.; Lieberthal, W.; Salant, D.J. Cytosolic calcium and protein kinase C reduce complement-mediated glomerular epithelial injury. Kidney Int. 1990, 38, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Jiang, R.; Aoudjit, L.; Jones, N.; Takano, T. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 2011, 22, 1621–1630. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.P.; Hawley, S.P.; Ruston, J.; Du, J.; Brakebusch, C.; Jones, N.; Pawson, T. Podocyte-specific loss of Cdc42 leads to congenital nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Blattner, S.M.; Hodgin, J.B.; Nishio, M.; Wylie, S.A.; Saha, J.; Soofi, A.A.; Vining, C.; Randolph, A.; Herbach, N.; Wanke, R.; et al. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int. 2013, 84, 920–930. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Suleiman, H.; Kim, A.H.; Miner, J.H.; Dani, A.; Shaw, A.S.; Akilesh, S. Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol. Cell. Biol. 2013, 33, 4755–4764. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ellis, M.J.; Gomez, J.A.; Eisner, W.; Fennell, W.; Howell, D.N.; Ruiz, P.; Fields, T.A.; Spurney, R.F. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int. 2012, 81, 1075–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezzerides, V.J.; Ramsey, I.S.; Kotecha, S.; Greka, A.; Clapham, D.E. Rapid vesicular translocation and insertion of TRP channels. Nat. Cell Biol. 2004, 6, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Shi, J.; Zhu, Y.; Kustov, M.; Tian, J.B.; Stevens, A.; Wu, M.; Xu, J.; Long, S.; Yang, P.; et al. Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J. Biol. Chem. 2011, 286, 33436–33446. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.; Podlich, D.; Hahnel, B.; Kriz, W.; Gretz, N. Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J. Am. Soc. Nephrol. 2004, 15, 1475–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, J.M.; Schaefer, M.; Hill, K. Clemizole hydrochloride is a novel and potent inhibitor of transient receptor potential channel TRPC5. Mol. Pharmacol. 2014, 86, 514–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Dande, R.R.; Yu, H.; Samelko, B.; Miller, R.E.; Altintas, M.M.; Reiser, J. TRPC5 Does Not Cause or Aggravate Glomerular Disease. J. Am. Soc. Nephrol. 2018, 29, 409–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, H.; Yamamura, K.; Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199. [Google Scholar]
- Cheung, S.Y.; Henrot, M.; Al-Saad, M.; Baumann, M.; Muller, H.; Unger, A.; Rubaiy, H.N.; Mathar, I.; Dinkel, K.; Nussbaumer, P.; et al. TRPC4/TRPC5 channels mediate adverse reaction to the cancer cell cytotoxic agent (-)-Englerin A. Oncotarget 2018, 9, 29634–29643. [Google Scholar] [CrossRef] [Green Version]
- Sourbier, C.; Scroggins, B.T.; Ratnayake, R.; Prince, T.L.; Lee, S.; Lee, M.J.; Nagy, P.L.; Lee, Y.H.; Trepel, J.B.; Beutler, J.A.; et al. Englerin A stimulates PKCtheta to inhibit insulin signaling and to simultaneously activate HSF1: Pharmacologically induced synthetic lethality. Cancer Cell 2013, 23, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Carson, C.; Raman, P.; Tullai, J.; Xu, L.; Henault, M.; Thomas, E.; Yeola, S.; Lao, J.; McPate, M.; Verkuyl, J.M.; et al. Englerin A Agonizes the TRPC4/C5 Cation Channels to Inhibit Tumor Cell Line Proliferation. PLoS ONE 2015, 10, e0127498. [Google Scholar] [CrossRef]
- Ilatovskaya, D.V.; Palygin, O.; Chubinskiy-Nadezhdin, V.; Negulyaev, Y.A.; Ma, R.; Birnbaumer, L.; Staruschenko, A. Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glomeruli. Kidney Int. 2014, 86, 506–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanyam, P.; Colecraft, H.M. Ion channel engineering: Perspectives and strategies. J. Mol. Biol. 2015, 427, 190–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyonaka, S.; Kato, K.; Nishida, M.; Mio, K.; Numaga, T.; Sawaguchi, Y.; Yoshida, T.; Wakamori, M.; Mori, E.; Numata, T.; et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Natl. Acad. Sci. USA 2009, 106, 5400–5405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; Del Camino, D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Cha, S.K.; An, S.W.; Kuro, O.M.; Birnbaumer, L.; Huang, C.L. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat. Commun. 2012, 3, 1238. [Google Scholar] [CrossRef] [Green Version]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef] [Green Version]
- Eder, P.; Molkentin, J.D. TRPC channels as effectors of cardiac hypertrophy. Circ. Res. 2011, 108, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Musah, S.; Mammoto, A.; Ferrante, T.C.; Jeanty, S.S.F.; Hirano-Kobayashi, M.; Mammoto, T.; Roberts, K.; Chung, S.; Novak, R.; Ingram, M.; et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip. Nat. Biomed. Eng. 2017, 1, 69. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hall, G.; Wang, L.; Spurney, R.F. TRPC Channels in Proteinuric Kidney Diseases. Cells 2020, 9, 44. https://doi.org/10.3390/cells9010044
Hall G, Wang L, Spurney RF. TRPC Channels in Proteinuric Kidney Diseases. Cells. 2020; 9(1):44. https://doi.org/10.3390/cells9010044
Chicago/Turabian StyleHall, Gentzon, Liming Wang, and Robert F. Spurney. 2020. "TRPC Channels in Proteinuric Kidney Diseases" Cells 9, no. 1: 44. https://doi.org/10.3390/cells9010044
APA StyleHall, G., Wang, L., & Spurney, R. F. (2020). TRPC Channels in Proteinuric Kidney Diseases. Cells, 9(1), 44. https://doi.org/10.3390/cells9010044