On ATG4B as Drug Target for Treatment of Solid Tumours—The Knowns and the Unknowns



Abstract
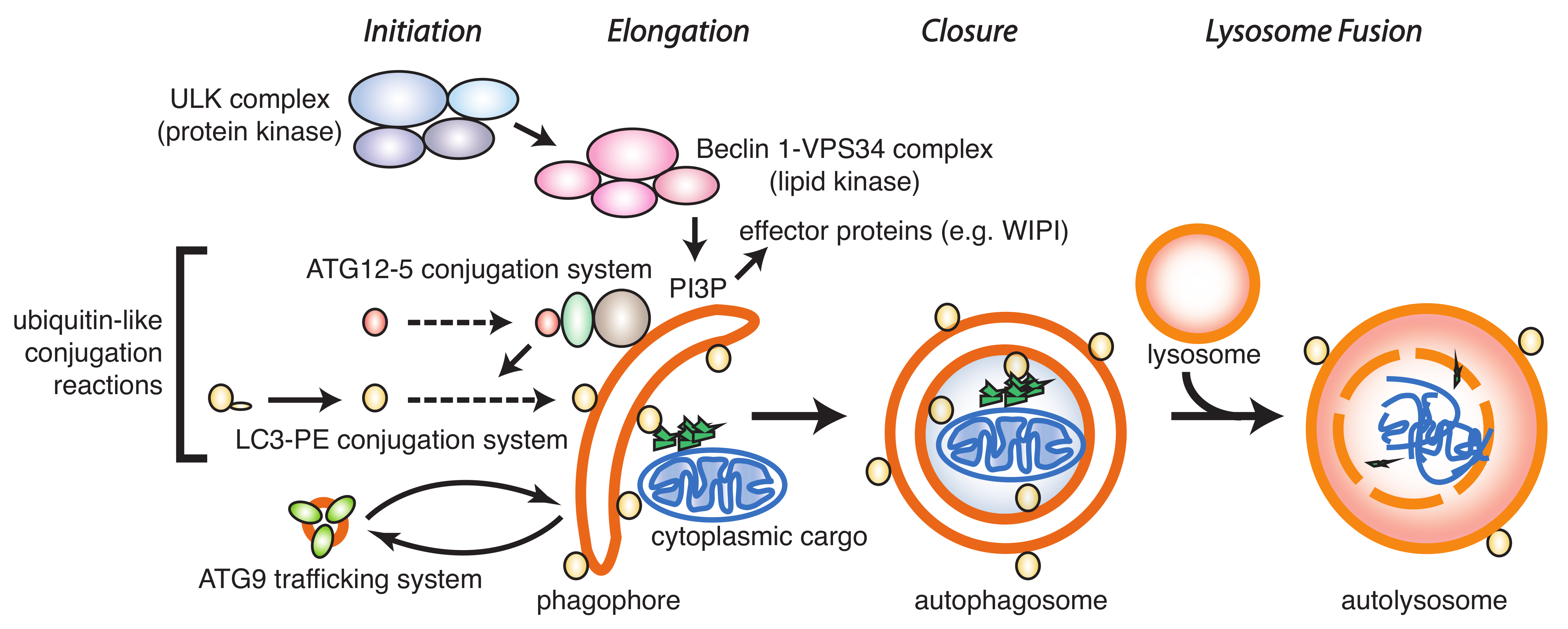
:1. Introduction
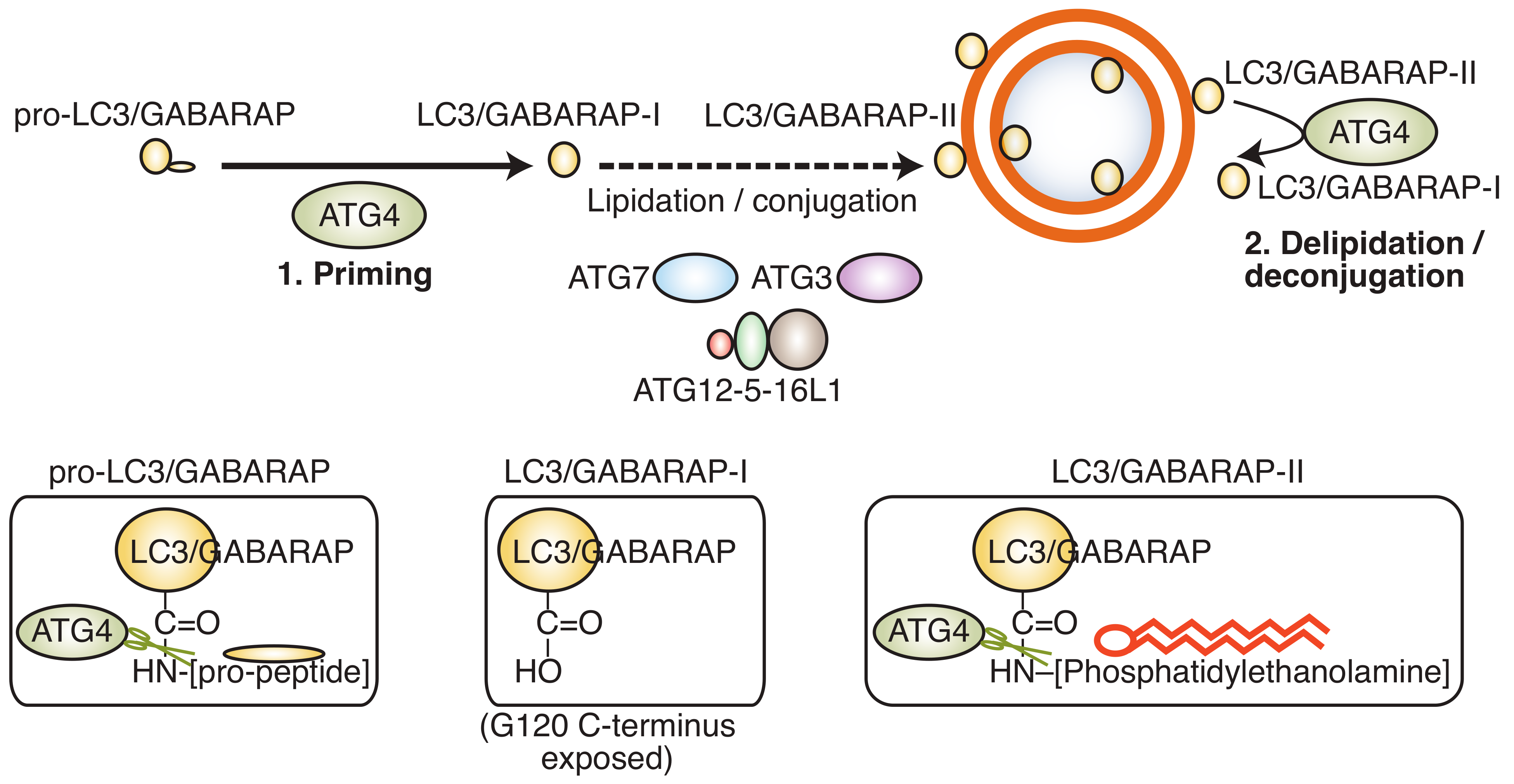
2. ATG4B as Drug Target
3. ATG4B in Cancer
3.1. Breast Cancer
3.2. Pancreatic Cancer
3.3. Lung Cancer
3.4. Colorectal Cancer
3.5. Prostate Cancer
3.6. Other Cancers
4. Screening Assays and Tool Compounds
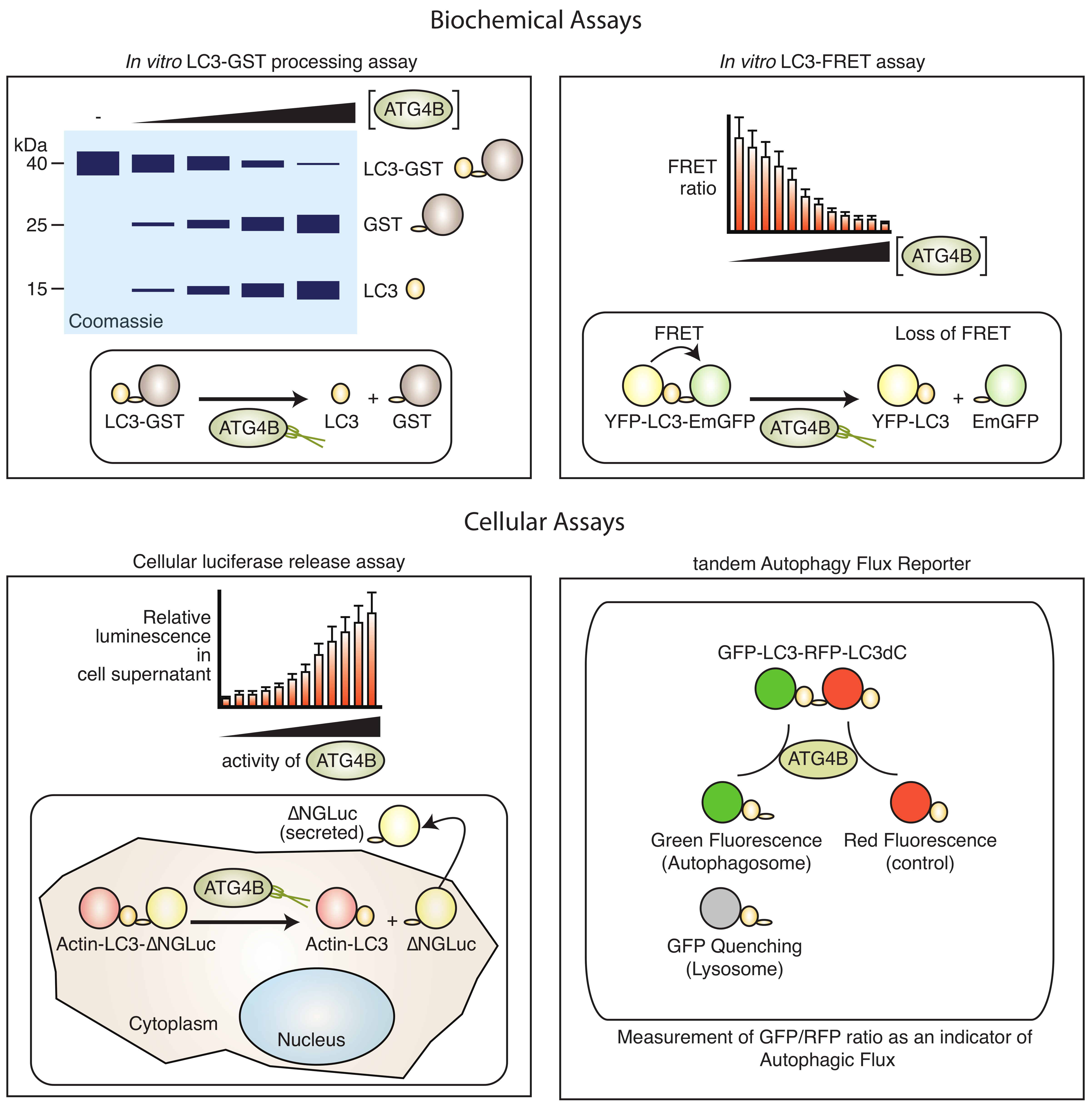
4.1. Screening Assays
4.1.1. Biochemical Assays
4.1.2. Computational Assays
4.1.3. Cell-Based Assays
4.2. Tool Compounds
4.2.1. ATG4B Inhibitors
4.2.2. ATG4B Activators
4.2.3. Conclusion
5. Biomarkers, Assays for In Vivo Activity, and Target Engagement
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Debnath, J. The Evolving, Multifaceted Roles of Autophagy in Cancer. Adv. Cancer Res. 2016, 130, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [Green Version]
- Costa, J.R.; Prak, K.; Aldous, S.; Gewinner, C.A.; Ketteler, R. Autophagy gene expression profiling identifies a defective microtubule-associated protein light chain 3A mutant in cancer. Oncotarget 2016, 7, 41203–41216. [Google Scholar] [CrossRef] [Green Version]
- Lebovitz, C.B.; Robertson, A.G.; Goya, R.; Jones, S.J.; Morin, R.D.; Marra, M.A.; Gorski, S.M. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 2015, 11, 1668–1687. [Google Scholar] [CrossRef] [Green Version]
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019, 33, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pengo, N.; Agrotis, A.; Prak, K.; Jones, J.; Ketteler, R. A reversible phospho-switch mediated by ULK1 regulates the activity of autophagy protease ATG4B. Nat. Commun. 2017, 8, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Wandelmer, J.; Kriegenburg, F.; Rohringer, S.; Schuschnig, M.; Gomez-Sanchez, R.; Zens, B.; Abreu, S.; Hardenberg, R.; Hollenstein, D.; Gao, J.; et al. Atg4 proteolytic activity can be inhibited by Atg1 phosphorylation. Nat. Commun. 2017, 8, 295. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; He, J.; Wu, Y.; Hu, C.; Dai, X.; Yan, X.; Li, B.; Li, X.; Xiong, H.; Li, Y.; et al. AKT-mediated phosphorylation of ATG4B impairs mitochondrial activity and enhances the Warburg effect in hepatocellular carcinoma cells. Autophagy 2018, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017, 32, 840–855. [Google Scholar] [CrossRef] [Green Version]
- Herhaus, L.; Bhaskara, R.M.; Lystad, A.H.; Gestal-Mato, U.; Covarrubias-Pinto, A.; Bonn, F.; Simonsen, A.; Hummer, G.; Dikic, I. TBK1-mediated phosphorylation of LC3C and GABARAP-L2 controls autophagosome shedding by ATG4 protease. EMBO Rep. 2019, e48317. [Google Scholar] [CrossRef]
- Jacquin, E.; Fletcher, K.; Florey, O. Imaging Noncanonical Autophagy and LC3-Associated Phagocytosis in Cultured Cells. Methods Mol. Biol. 2019, 1880, 295–303. [Google Scholar] [CrossRef]
- Jacquin, E.; Leclerc-Mercier, S.; Judon, C.; Blanchard, E.; Fraitag, S.; Florey, O. Pharmacological modulators of autophagy activate a parallel noncanonical pathway driving unconventional LC3 lipidation. Autophagy 2017, 13, 854–867. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Teubner, B.J.W.; Tummers, B.; Boada-Romero, E.; Harris, L.; Yang, M.; Guy, C.S.; Zakharenko, S.S.; Green, D.R. LC3-Associated Endocytosis Facilitates beta-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 2019, 178, 536–551. [Google Scholar] [CrossRef]
- Agrotis, A.; von Chamier, L.; Oliver, H.; Kiso, K.; Singh, T.; Ketteler, R. Human ATG4 autophagy proteases counteract attachment of ubiquitin-like LC3/GABARAP proteins to other cellular proteins. J. Biol. Chem. 2019, 294, 12610–12621. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zheng, Q.; Wang, Y.; Pang, Z.; Liu, J.; Yin, Z.; Lou, Z. Activity-Based Protein Profiling Identifies ATG4B as a Key Host Factor for EV71 Proliferation. J. Virol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Perlstein, E.O.; Imarisio, S.; Pineau, S.; Cordenier, A.; Maglathlin, R.L.; Webster, J.A.; Lewis, T.A.; O’Kane, C.J.; Schreiber, S.L.; et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 2007, 3, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yu, J.; Pan, H.; Hu, P.; Hao, Y.; Cai, W.; Zhu, H.; Yu, A.D.; Xie, X.; Ma, D.; et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc. Natl. Acad. Sci. USA 2007, 104, 19023–19028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [Green Version]
- Rebecca, V.W.; Nicastri, M.C.; Fennelly, C.; Chude, C.I.; Barber-Rotenberg, J.S.; Ronghe, A.; McAfee, Q.; McLaughlin, N.P.; Zhang, G.; Goldman, A.R.; et al. PPT1 Promotes Tumor Growth and Is the Molecular Target of Chloroquine Derivatives in Cancer. Cancer Discov. 2019, 9, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, P.; Strambi, A.; Zipoli, C.; Hagg-Olofsson, M.; Buoncervello, M.; Linder, S.; De Milito, A. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: Implications for cancer therapies. Autophagy 2014, 10, 562–571. [Google Scholar] [CrossRef] [Green Version]
- Rebecca, V.W.; Amaravadi, R.K. Emerging strategies to effectively target autophagy in cancer. Oncogene 2016, 35, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Florey, O.; Gammoh, N.; Kim, S.E.; Jiang, X.; Overholtzer, M. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 2015, 11, 88–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuma, A.; Komatsu, M.; Mizushima, N. Autophagy-monitoring and autophagy-deficient mice. Autophagy 2017, 13, 1619–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshii, S.R.; Kuma, A.; Mizushima, N. Transgenic rescue of Atg5-null mice from neonatal lethality with neuron-specific expression of ATG5: Systemic analysis of adult Atg5-deficient mice. Autophagy 2017, 13, 763–764. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, L.D.; Young, A.R.; Perez-Mancera, P.A.; Nimmervoll, B.; Jaulim, A.; Chen, H.C.; McIntyre, D.J.O.; Brais, R.; Ricketts, T.; Pacey, S.; et al. A novel Atg5-shRNA mouse model enables temporal control of Autophagy in vivo. Autophagy 2018, 14, 1256–1266. [Google Scholar] [CrossRef]
- Yang, A.; Herter-Sprie, G.; Zhang, H.; Lin, E.Y.; Biancur, D.; Wang, X.; Deng, J.; Hai, J.; Yang, S.; Wong, K.K.; et al. Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discov. 2018, 8, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Agrotis, A.; Pengo, N.; Burden, J.J.; Ketteler, R. Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 2019, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, K.J.; Yu, S.; Jin, J.; Mugo, B.; Nguyen, N.; O’Brien, A.; Nag, S.; Lystad, A.H.; Melia, T.J. Delipidation of mammalian Atg8-family proteins by each of the four ATG4 proteases. Autophagy 2018, 14, 992–1010. [Google Scholar] [CrossRef]
- Tanida, I.; Sou, Y.-s.S.; Ezaki, J.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J. Biol. Chem. 2004, 279, 36268–36276. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Hou, Y.; Wang, J.; Chen, X.; Shao, Z.M.; Yin, X.M. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J. Biol. Chem. 2011, 286, 7327–7338. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Sou, Y.S.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Atg8L/Apg8L is the fourth mammalian modifier of mammalian Atg8 conjugation mediated by human Atg4B, Atg7 and Atg3. FEBS J. 2006, 273, 2553–2562. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.; Lane, J.D. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 2009, 122, 2554–2566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 2008, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef]
- Vaites, L.P.; Paulo, J.A.; Huttlin, E.L.; Harper, J.W. Systematic Analysis of Human Cells Lacking ATG8 Proteins Uncovers Roles for GABARAPs and the CCZ1/MON1 Regulator C18orf8/RMC1 in Macroautophagic and Selective Autophagic Flux. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, K.; Shibata, M.; Koike, M.; Gotoh, K.; Fukaya, M.; Watanabe, M.; Uchiyama, Y. Effects of RNA interference of Atg4B on the limited proteolysis of LC3 in PC12 cells and expression of Atg4B in various rat tissues. Autophagy 2006, 2, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Yang, H.; Sutton, M.N.; Yang, M.; Clarke, C.H.; Liao, W.S.; Bast, R.C., Jr. ARHI (DIRAS3) induces autophagy in ovarian cancer cells by downregulating the epidermal growth factor receptor, inhibiting PI3K and Ras/MAP signaling and activating the FOXo3a-mediated induction of Rab7. Cell Death Differ. 2014, 21, 1275–1289. [Google Scholar] [CrossRef] [Green Version]
- Mammucari, C.; Schiaffino, S.; Sandri, M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy 2008, 4, 524–526. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.H.; Kim, H.P.; Sciurba, F.C.; Lee, S.J.; Feghali-Bostwick, C.; Stolz, D.B.; Dhir, R.; Landreneau, R.J.; Schuchert, M.J.; Yousem, S.A.; et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE 2008, 3, e3316. [Google Scholar] [CrossRef] [Green Version]
- Kenzelmann Broz, D.; Spano Mello, S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Zhou, L.; Gao, Q.; Zhang, A.; Wei, J.; Hong, D.; Chu, Y.; Duan, X.; Zhang, Y.; Xu, G. MicroRNA-144-3p inhibits autophagy activation and enhances Bacillus Calmette-Guerin infection by targeting ATG4a in RAW264.7 macrophage cells. PLoS ONE 2017, 12, e0179772. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Chen, Y.; Song, H.; Xu, Y.; Wang, R.; Chen, L. Mir-24-3p downregulation contributes to VP16-DDP resistance in small-cell lung cancer by targeting ATG4A. Oncotarget 2015, 6, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothe, K.; Lin, H.; Lin, K.B.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, D.L.; Gorski, S.M.; Jiang, X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Huang, X.; Xie, J.; Chen, Y.; Fu, J.; Wang, L. Let-7i-Induced Atg4B Suppression Is Essential for Autophagy of Placental Trophoblast in Preeclampsia. J. Cell. Physiol. 2017, 232, 2581–2589. [Google Scholar] [CrossRef]
- Li, Z.; Wang, G.; Feng, D.; Zu, G.; Li, Y.; Shi, X.; Zhao, Y.; Jing, H.; Ning, S.; Le, W.; et al. Targeting the miR-665-3p-ATG4B-autophagy axis relieves inflammation and apoptosis in intestinal ischemia/reperfusion. Cell Death Dis. 2018, 9, 483. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, G.; le Sage, C.; Tekirdag, K.A.; Agami, R.; Gozuacik, D. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy 2012, 8, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Frankel, L.B.; Wen, J.; Lees, M.; Hoyer-Hansen, M.; Farkas, T.; Krogh, A.; Jaattela, M.; Lund, A.H. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011, 30, 4628–4641. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; An, Y.; Wang, Y.; Zhang, C.; Zhang, H.; Huang, C.; Jiang, H.; Wang, X.; Li, X. miR-101 inhibits autophagy and enhances cisplatin-induced apoptosis in hepatocellular carcinoma cells. Oncol. Rep. 2013, 29, 2019–2024. [Google Scholar] [CrossRef] [Green Version]
- Kuang, E.; Okumura, C.Y.; Sheffy-Levin, S.; Varsano, T.; Shu, V.C.; Qi, J.; Niesman, I.R.; Yang, H.-J.J.; López-Otín, C.; Yang, W.Y.; et al. Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Huang, Z.; Hong, L.; Lu, J.H.; Feng, D.; Yin, X.M.; Li, M. Targeting ATG4 in Cancer Therapy. Cancers 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.K.; Park, N.Y.; Park, S.J.; Kim, B.G.; Shin, J.H.; Jo, D.S.; Bae, D.J.; Suh, Y.A.; Chang, J.H.; Lee, E.K.; et al. O-GlcNAcylation of ATG4B positively regulates autophagy by increasing its hydroxylase activity. Oncotarget 2016, 7, 57186–57196. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Wang, L.; Wang, P.; Xue, Y.; Li, X.; Qiao, X.; Zhang, X.; Xu, T.; Liu, G.; et al. Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy 2017, 13, 1145–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betin, V.M.; MacVicar, T.D.; Parsons, S.F.; Anstee, D.J.; Lane, J.D. A cryptic mitochondrial targeting motif in Atg4D links caspase cleavage with mitochondrial import and oxidative stress. Autophagy 2012, 8, 664–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Heintze, J.; Costa, J.R.; Weber, M.; Ketteler, R. Ribose 5-phosphate isomerase inhibits LC3 processing and basal autophagy. Cell. Signal. 2016, 28, 1380–1388. [Google Scholar] [CrossRef] [Green Version]
- Qiao, S.; Dennis, M.; Song, X.; Vadysirisack, D.D.; Salunke, D.; Nash, Z.; Yang, Z.; Liesa, M.; Yoshioka, J.; Matsuzawa, S.; et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat. Commun. 2015, 6, 7014. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wilkie-Grantham, R.P.; Yanagi, T.; Shu, C.W.; Matsuzawa, S.; Reed, J.C. ATG4B (Autophagin-1) phosphorylation modulates autophagy. J. Biol. Chem. 2015, 290, 26549–26561. [Google Scholar] [CrossRef] [Green Version]
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef]
- Kumanomidou, T.; Mizushima, T.; Komatsu, M.; Suzuki, A.; Tanida, I.; Sou, Y.-S.S.; Ueno, T.; Kominami, E.; Tanaka, K.; Yamane, T. The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J. Mol. Biol. 2006, 355, 612–618. [Google Scholar] [CrossRef]
- Satoo, K.; Noda, N.N.; Kumeta, H.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009, 28, 1341–1350. [Google Scholar] [CrossRef]
- Sugawara, K.; Suzuki, N.N.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. J. Biol. Chem. 2005, 280, 40058–40065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pengo, N.; Prak, K.; Costa, J.R.; Luft, C.; Agrotis, A.; Freeman, J.; Gewinner, C.A.; Chan, A.W.E.; Selwood, D.L.; Kriston-Vizi, J.; et al. Identification of Kinases and Phosphatases That Regulate ATG4B Activity by siRNA and Small Molecule Screening in Cells. Front. Cell Dev. Biol. 2018, 6, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skytte Rasmussen, M.; Mouilleron, S.; Kumar Shrestha, B.; Wirth, M.; Lee, R.; Bowitz Larsen, K.; Abudu Princely, Y.; O’Reilly, N.; Sjottem, E.; Tooze, S.A.; et al. ATG4B contains a C-terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8. Autophagy 2017, 13, 834–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abreu, S.; Kriegenburg, F.; Gomez-Sanchez, R.; Mari, M.; Sanchez-Wandelmer, J.; Skytte Rasmussen, M.; Soares Guimaraes, R.; Zens, B.; Schuschnig, M.; Hardenberg, R.; et al. Conserved Atg8 recognition sites mediate Atg4 association with autophagosomal membranes and Atg8 deconjugation. EMBO Rep. 2017, 18, 765–780. [Google Scholar] [CrossRef] [Green Version]
- Shu, C.W.; Drag, M.; Bekes, M.; Zhai, D.; Salvesen, G.S.; Reed, J.C. Synthetic substrates for measuring activity of autophagy proteases: Autophagins (Atg4). Autophagy 2010, 6, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, S.; Maciel, M.; Herrera, I.; Nava, T.; Vergara, F.; Gaxiola, M.; Lopez-Otin, C.; Selman, M.; Pardo, A. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy 2015, 11, 670–684. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, A.; Lopez-Alonso, I.; Gonzalez-Lopez, A.; Amado-Rodriguez, L.; Batalla-Solis, E.; Astudillo, A.; Blazquez-Prieto, J.; Fernandez, A.F.; Galvan, J.A.; dos Santos, C.C.; et al. Defective autophagy impairs ATF3 activity and worsens lung injury during endotoxemia. J. Mol. Med. 2014, 92, 665–676. [Google Scholar] [CrossRef]
- Cabrera, S.; Fernandez, A.F.; Marino, G.; Aguirre, A.; Suarez, M.F.; Espanol, Y.; Vega, J.A.; Laura, R.; Fueyo, A.; Fernandez-Garcia, M.S.; et al. ATG4B/autophagin-1 regulates intestinal homeostasis and protects mice from experimental colitis. Autophagy 2013, 9, 1188–1200. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.H.; Behrns, K.E.; Leeuwenburgh, C.; Kim, J.S. Critical role of autophage in ischemia/reperfusion injury to aged livers. Autophagy 2012, 8, 140–141. [Google Scholar] [CrossRef] [Green Version]
- Proenca, C.C.; Stoehr, N.; Bernhard, M.; Seger, S.; Genoud, C.; Roscic, A.; Paganetti, P.; Liu, S.; Murphy, L.O.; Kuhn, R.; et al. Atg4b-dependent autophagic flux alleviates Huntington’s disease progression. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Bortnik, S.; Choutka, C.; Horlings, H.M.; Leung, S.; Baker, J.H.; Lebovitz, C.; Dragowska, W.H.; Go, N.E.; Bally, M.B.; Minchinton, A.I.; et al. Identification of breast cancer cell subtypes sensitive to ATG4B inhibition. Oncotarget 2016, 7, 66970–66988. [Google Scholar] [CrossRef] [PubMed]
- Dower, C.M.; Bhat, N.; Wang, E.W.; Wang, H.G. Selective Reversible Inhibition of Autophagy in Hypoxic Breast Cancer Cells Promotes Pulmonary Metastasis. Cancer Res. 2017, 77, 646–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Guo, M.; Li, J.; Zheng, Y.; Zhang, S.; Xie, T.; Liu, B. Systems biology-based discovery of a potential Atg4B agonist (Flubendazole) that induces autophagy in breast cancer. Mol. Biosyst. 2015, 11, 2860–2866. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.W.; Choi, J.; Lee, S.Y.; Sung, S.; Yoo, H.J.; Kang, M.J.; Cheong, H.; Son, J. Autophagy is required for PDAC glutamine metabolism. Sci. Rep. 2016, 6, 37594. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 4945. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Kimmelman, A.C. Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status. Autophagy 2014, 10, 1683–1684. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.F.; Leung, C.M.; Chang, Y.H.; Cheng, J.S.; Chen, J.J.; Weng, C.J.; Tsai, K.W.; Hsu, C.J.; Liu, Y.C.; Hsu, P.C.; et al. ATG4B promotes colorectal cancer growth independent of autophagic flux. Autophagy 2014, 10, 1454–1465. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.F.; Tsai, K.L.; Hsu, C.J.; Tsai, W.L.; Cheng, J.S.; Chang, H.W.; Shiau, C.W.; Goan, Y.G.; Tseng, H.H.; Wu, C.H.; et al. Drug Repurposing Screening Identifies Tioconazole as an ATG4 Inhibitor that Suppresses Autophagy and Sensitizes Cancer Cells to Chemotherapy. Theranostics 2018, 8, 830–845. [Google Scholar] [CrossRef] [PubMed]
- Blessing, A.M.; Rajapakshe, K.; Reddy Bollu, L.; Shi, Y.; White, M.A.; Pham, A.H.; Lin, C.; Jonsson, P.; Cortes, C.J.; Cheung, E.; et al. Transcriptional regulation of core autophagy and lysosomal genes by the androgen receptor promotes prostate cancer progression. Autophagy 2017, 13, 506–521. [Google Scholar] [CrossRef]
- Mouratidis, P.X.; Colston, K.W.; Charolidi, N.; Pirianov, G. Differential role of apoptosis and autophagy associated with anticancer effect of lupulone (hop beta-acid) derivatives on prostate cancer cells. Anti-Cancer Agents Med. Chem. 2014, 14, 1169–1178. [Google Scholar] [CrossRef]
- Liao, H.; Xiao, Y.; Hu, Y.; Xiao, Y.; Yin, Z.; Liu, L.; Kang, X.; Chen, Y. Methylation-induced silencing of miR-34a enhances chemoresistance by directly upregulating ATG4B-induced autophagy through AMPK/mTOR pathway in prostate cancer. Oncol. Rep. 2016, 35, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Chow, A.; Goda, T.; Wong, A.; Blakely, K.; Rocha, M.; Taeb, S.; Hoang, V.C.; Liu, S.K.; Emmenegger, U. Context-dependent role of ATG4B as target for autophagy inhibition in prostate cancer therapy. Biochem. Biophys. Res. Commun. 2013, 441, 726–731. [Google Scholar] [CrossRef]
- Akin, D.; Wang, S.K.; Habibzadegah-Tari, P.; Law, B.; Ostrov, D.; Li, M.; Yin, X.M.; Kim, J.S.; Horenstein, N.; Dunn, W.A., Jr. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014, 10, 2021–2035. [Google Scholar] [CrossRef]
- Vezenkov, L.; Honson, N.S.; Kumar, N.S.; Bosc, D.; Kovacic, S.; Nguyen, T.G.; Pfeifer, T.A.; Young, R.N. Development of fluorescent peptide substrates and assays for the key autophagy-initiating cysteine protease enzyme, ATG4B. Bioorganic Med. Chem. 2015, 23, 3237–3247. [Google Scholar] [CrossRef]
- Nguyen, T.G.; Honson, N.S.; Arns, S.; Davis, T.L.; Dhe-Paganon, S.; Kovacic, S.; Kumar, N.S.; Pfeifer, T.A.; Young, R.N. Development of fluorescent substrates and assays for the key autophagy-related cysteine protease enzyme, ATG4B. Assay Drug Dev. Technol. 2014, 12, 176–189. [Google Scholar] [CrossRef]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2018, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleenewerck, M.; Grootaert, M.O.J.; Gladysz, R.; Adriaenssens, Y.; Roelandt, R.; Joossens, J.; Lambeir, A.M.; De Meyer, G.R.Y.; Declercq, W.; Augustyns, K.; et al. Inhibitor screening and enzymatic activity determination for autophagy target Atg4B using a gel electrophoresis-based assay. Eur. J. Med. Chem. 2016, 123, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.W.; Madiraju, C.; Zhai, D.; Welsh, K.; Diaz, P.; Sergienko, E.; Sano, R.; Reed, J.C. High-throughput fluorescence assay for small-molecule inhibitors of autophagins/Atg4. J. Biomol. Screen. 2011, 16, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Chen, X.; Ye, Q.-Z.Z.; Vogt, A.; Yin, X.-M.M. A high-throughput FRET-based assay for determination of Atg4 activity. Autophagy 2012, 8, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Xu, Z.; Han, L.; Liu, C.; Zhou, Z.; Qiu, Z.; Lin, X.; Tang, G.; Shen, H.; Aebi, J.; et al. Identification of New ATG4B Inhibitors Based on a Novel High-Throughput Screening Platform. Slas Discov. Adv. Life Sci. 2017, 22, 338–347. [Google Scholar] [CrossRef] [Green Version]
- Bosc, D.; Vezenkov, L.; Bortnik, S.; An, J.; Xu, J.; Choutka, C.; Hannigan, A.M.; Kovacic, S.; Loo, S.; Clark, P.G.K.; et al. A new quinoline-based chemical probe inhibits the autophagy-related cysteine protease ATG4B. Sci. Rep. 2018, 8, 11653. [Google Scholar] [CrossRef] [Green Version]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019. [Google Scholar] [CrossRef]
- Kraft, L.J.; Kenworthy, A.K. Imaging protein complex formation in the autophagy pathway: Analysis of the interaction of LC3 and Atg4B(C74A) in live cells using Förster resonance energy transfer and fluorescence recovery after photobleaching. J. Biomed. Opt. 2012, 17, 11008. [Google Scholar] [CrossRef]
- Ketteler, R.; Seed, B. Quantitation of autophagy by luciferase release assay. Autophagy 2008, 4, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Ketteler, R.; Sun, Z.; Kovacs, K.F.; He, W.W.; Seed, B. A pathway sensor for genome-wide screens of intracellular proteolytic cleavage. Genome Biol. 2008, 9, R64. [Google Scholar] [CrossRef] [Green Version]
- Luft, C.; Freeman, J.; Elliott, D.; Al-Tamimi, N.; Kriston-Vizi, J.; Heintze, J.; Lindenschmidt, I.; Seed, B.; Ketteler, R. Application of Gaussia luciferase in bicistronic and non-conventional secretion reporter constructs. BMC Biochem. 2014, 15, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurdinger, T.; Badr, C.; Pike, L.; de Kleine, R.; Weissleder, R.; Breakefield, X.O.; Tannous, B.A. A secreted luciferase for ex vivo monitoring of in vivo processes. Nat. Methods 2008, 5, 171–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, A.F.; Lopez-Otin, C. The functional and pathologic relevance of autophagy proteases. J. Clin. Investig. 2015, 125, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, J.; Ouyang, L.; Liu, B.; Cheng, Y. Unraveling the roles of Atg4 proteases from autophagy modulation to targeted cancer therapy. Cancer Lett. 2016, 373, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, A.; Cleenewerck, M.; Vangestel, C.; Lyssens, S.; Declercq, W.; Timmermans, J.P.; Stroobants, S.; Augustyns, K.; De Meyer, G.R.Y.; Van Der Veken, P.; et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem. Pharmacol. 2017, 138, 150–162. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhu, H.; Zhang, W.; Okon, I.; Wang, Q.; Li, H.; Le, Y.Z.; Xie, Z. 7-Ketocholesterol induces autophagy in vascular smooth muscle cells through Nox4 and Atg4B. Am. J. Pathol. 2013, 183, 626–637. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Kuhn, B.; Aebi, J.; Lin, X.; Ding, H.; Zhou, Z.; Xu, Z.; Xu, D.; Han, L.; Liu, C.; et al. Discovery of Fluoromethylketone-Based Peptidomimetics as Covalent ATG4B (Autophagin-1) Inhibitors. ACS Med. Chem. Lett. 2016, 7, 802–806. [Google Scholar] [CrossRef] [Green Version]
- Quintana, M.; Bilbao, A.; Comas-Barcelo, J.; Bujons, J.; Triola, G. Identification of benzo[cd]indol-2(1H)-ones as novel Atg4B inhibitors via a structure-based virtual screening and a novel AlphaScreen assay. Eur. J. Med. Chem. 2019, 178, 648–666. [Google Scholar] [CrossRef]
- Tanc, M.; Cleenewerck, M.; Kurdi, A.; Roelandt, R.; Declercq, W.; De Meyer, G.; Augustyns, K.; Martinet, W.; Van der Veken, P. Synthesis and evaluation of novel benzotropolones as Atg4B inhibiting autophagy blockers. Bioorganic Chem. 2019, 87, 163–168. [Google Scholar] [CrossRef]
- Guan, Y.; Fu, Y.; Liu, Y.; Wang, S.; Zhao, M.; Jin, Z.; Jiang, F.; Hong, L.; Huang, M.; Li, M.; et al. Quantitative bioanalytical LC-MS/MS assay for S130 in rat plasma-application to a pharmacokinetic study. Bioanalysis 2019, 11, 1469–1481. [Google Scholar] [CrossRef]
- Chu, J.; Fu, Y.; Xu, J.; Zheng, X.; Gu, Q.; Luo, X.; Dai, Q.; Zhang, S.; Liu, P.; Hong, L.; et al. ATG4B inhibitor FMK-9a induces autophagy independent on its enzyme inhibition. Arch. Biochem. Biophys. 2018, 644, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.W.; Liu, P.F.; Tsai, W.L.; Hu, W.H.; Hu, Y.C.; Yang, H.C.; Lin, W.Y.; Weng, J.R.; Shu, C.W. Xanthium strumarium Fruit Extract Inhibits ATG4B and Diminishes the Proliferation and Metastatic Characteristics of Colorectal Cancer Cells. Toxins 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Luo, Q.; Yuan, L.; Miao, C.; Mu, X.; Xiao, W.; Li, J.; Sun, T.; Ma, E. JNK-dependent Atg4 upregulation mediates asperphenamate derivative BBP-induced autophagy in MCF-7 cells. Toxicol. Appl. Pharmacol. 2012, 263, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Moulis, M.; Vindis, C. Methods for Measuring Autophagy in Mice. Cells 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef]
- Li, L.; Wang, Z.V.; Hill, J.A.; Lin, F. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J. Am. Soc. Nephrol. 2014, 25, 305–315. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Villarejo-Zori, B.; Ball, G.; Boya, P.; Ganley, I.G. A comparative map of macroautophagy and mitophagy in the vertebrate eye. Autophagy 2019, 15, 1296–1308. [Google Scholar] [CrossRef]
- Yazawa, R.; Nishida, Y.; Aoyama, S.; Tanida, I.; Miyatsuka, T.; Suzuki, L.; Himuro, M.; Haruna, H.; Takubo, N.; Shimizu, T.; et al. Establishment of a system for screening autophagic flux regulators using a modified fluorescent reporter and CRISPR/Cas9. Biochem. Biophys. Res. Commun. 2019, 516, 686–692. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef]
- Kaizuka, T.; Morishita, H.; Hama, Y.; Tsukamoto, S.; Matsui, T.; Toyota, Y.; Kodama, A.; Ishihara, T.; Mizushima, T.; Mizushima, N. An Autophagic Flux Probe that Releases an Internal Control. Mol. Cell 2016, 64, 835–849. [Google Scholar] [CrossRef] [Green Version]
- Bartok, E.; Bauernfeind, F.; Khaminets, M.G.; Jakobs, C.; Monks, B.; Fitzgerald, K.A.; Latz, E.; Hornung, V. iGLuc: A luciferase-based inflammasome and protease activity reporter. Nat. Methods 2013, 10, 147–154. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Therapeutic Modality | In Vivo Model | Biomarker | Reference |
|---|---|---|---|---|
| Breast cancer | siRNA ATG4B/Trastuzumab | MCF7 xenograft | HER2, ATG4B | [81] |
| Colorectal cancer | Tioconazole | HCT-116 Xenograft | none | [93] |
| Colorectal cancer | S130/ Caloric restriction | HCT-116 Xenograft | none | [101] |
| Colorectal cancer | UAMC2526/ oxaliplatin | HT-29 Xenograft | LC3 conversion | [115] |
| Glioblastoma | NSC185058/ Chloroquine | M83 glioma xenograft | none | [15] |
| Lung adenocarcinoma | Doxicylcin-inducible ATG4B C74A | GEMM | K-Ras mutation | [36] |
| Osteosarcoma | NSC185058/ starvation | SAOS Xenograft | none | [98] |
| Pancreatic ductal adenocarcinoma | Doxicyclin-inducible ATG4B C74A | GEMM | K-Ras mutation | [36] |
| Prostate cancer | ATG4B C74A/ doxorubicin | PC-3 Xenograft | none | [97] |
| Compound | Chemical Scaffold | Screening Assay | Cell Type | Cancer Type | Reference |
|---|---|---|---|---|---|
| 7-keto-cholesterol | Keto-cholesterol | LC3–PLA2 | HASMC | N.D. | [116] |
| Aurin-tricarboxylic acid | Polyphenole | FRET (YFP–LC3–EmGFP) | N.D. | N.D. | [100] |
| FMK9A | Methyl-ketone | TR–FRET | N.D. | N.D. | [117] |
| Hypericin | Anthra-quinone | FRET (YFP–LC3–EmGFP) | N.D. | N.D. | [100] |
| LV-320 | Styryl-quinoline | In silico | SKBR3, MCF7, JIMT1, MDA-MB-231 | Breast Cancer | [106] |
| NSC185058 | Pyridine-carbothioamide | In silico | SAOS M83 | Osteosarcoma Glioblastoma | [98] [15] |
| NSC611216 | Benzo-indolone | Alphascreen | HT-29 | Colorectal cancer | [118] |
| S130 | Dibenzo-quinoline | In silico | HCT-116 | Colorectal cancer | [101] |
| Tioconazole | Dichlorphenylethyl-imidazole | In silico, GABARPL2–PLA | HCT-116 | Colorectal cancer | [93] |
| UAMC2526 | Benzo-tropolone | LC3–GST | HT-29 | Colorectal cancer | [115] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agrotis, A.; Ketteler, R. On ATG4B as Drug Target for Treatment of Solid Tumours—The Knowns and the Unknowns. Cells 2020, 9, 53. https://doi.org/10.3390/cells9010053
Agrotis A, Ketteler R. On ATG4B as Drug Target for Treatment of Solid Tumours—The Knowns and the Unknowns. Cells. 2020; 9(1):53. https://doi.org/10.3390/cells9010053
Chicago/Turabian StyleAgrotis, Alexander, and Robin Ketteler. 2020. "On ATG4B as Drug Target for Treatment of Solid Tumours—The Knowns and the Unknowns" Cells 9, no. 1: 53. https://doi.org/10.3390/cells9010053
APA StyleAgrotis, A., & Ketteler, R. (2020). On ATG4B as Drug Target for Treatment of Solid Tumours—The Knowns and the Unknowns. Cells, 9(1), 53. https://doi.org/10.3390/cells9010053