miR-9-5p Exerts a Dual Role in Cervical Cancer and Targets Transcription Factor TWIST1

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Clinical Specimens
2.2. RNA/DNA Isolation and Bisulfite Treatment
2.3. Quantitative Methylation-Specific PCR (qMSP)
2.4. Quantitative Reverse Transcription-PCR (qRT-PCR)
2.4.1. miRNA qRT-PCR
2.4.2. mRNA qRT-PCR
2.5. External Data
2.6. Transfection with miRNA Inhibitors and Mimics
2.7. Cell Viability and Anchorage-Independent Growth
2.8. Construction of a pmirGLO Reporter Vector
2.9. Luciferase Dual-Reporter Assays for miR-9-5p Target Validation
2.10. Statistical Analysis
3. Results
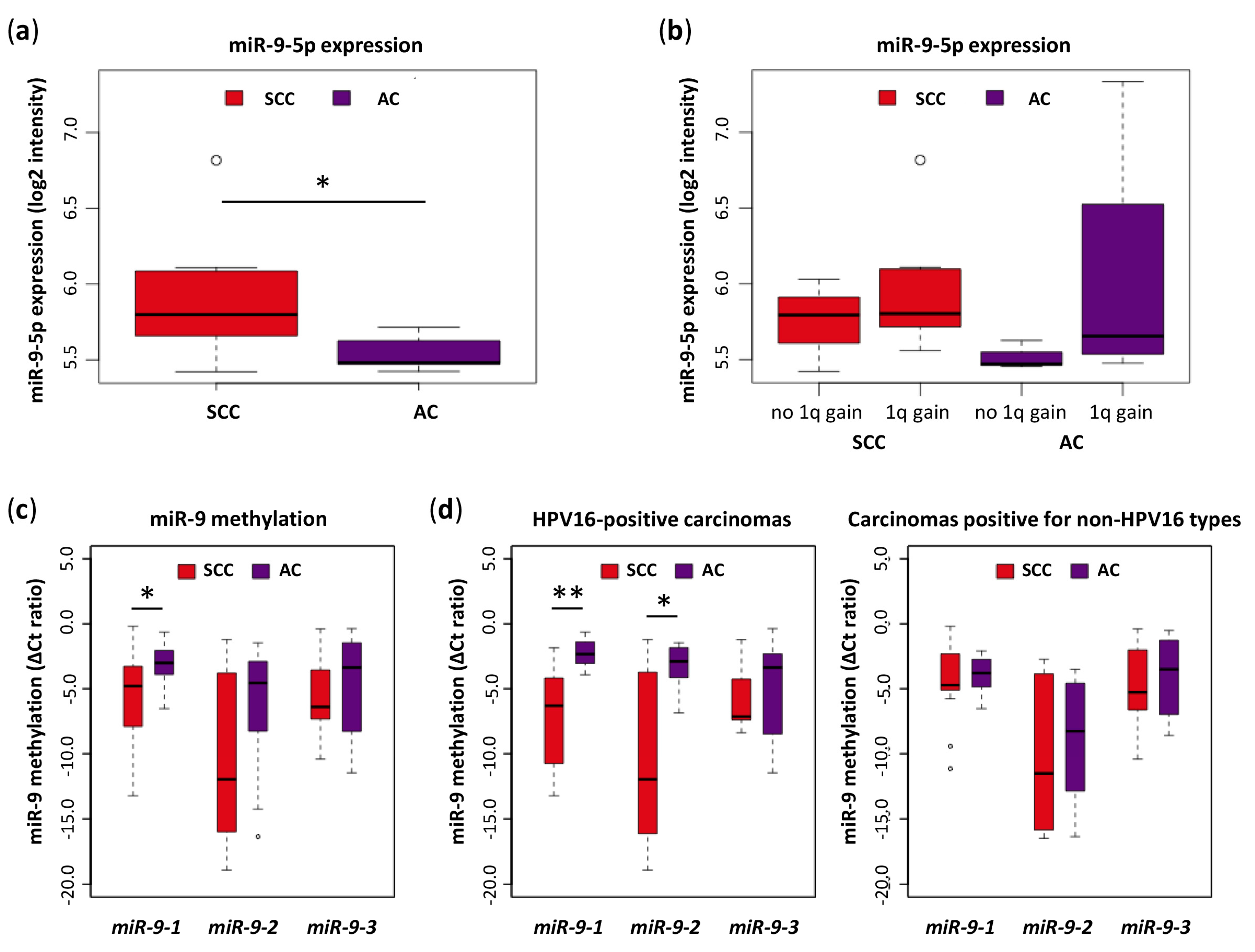
3.1. Low miR-9-5p Expression in AC Is Associated with DNA Hypermethylation
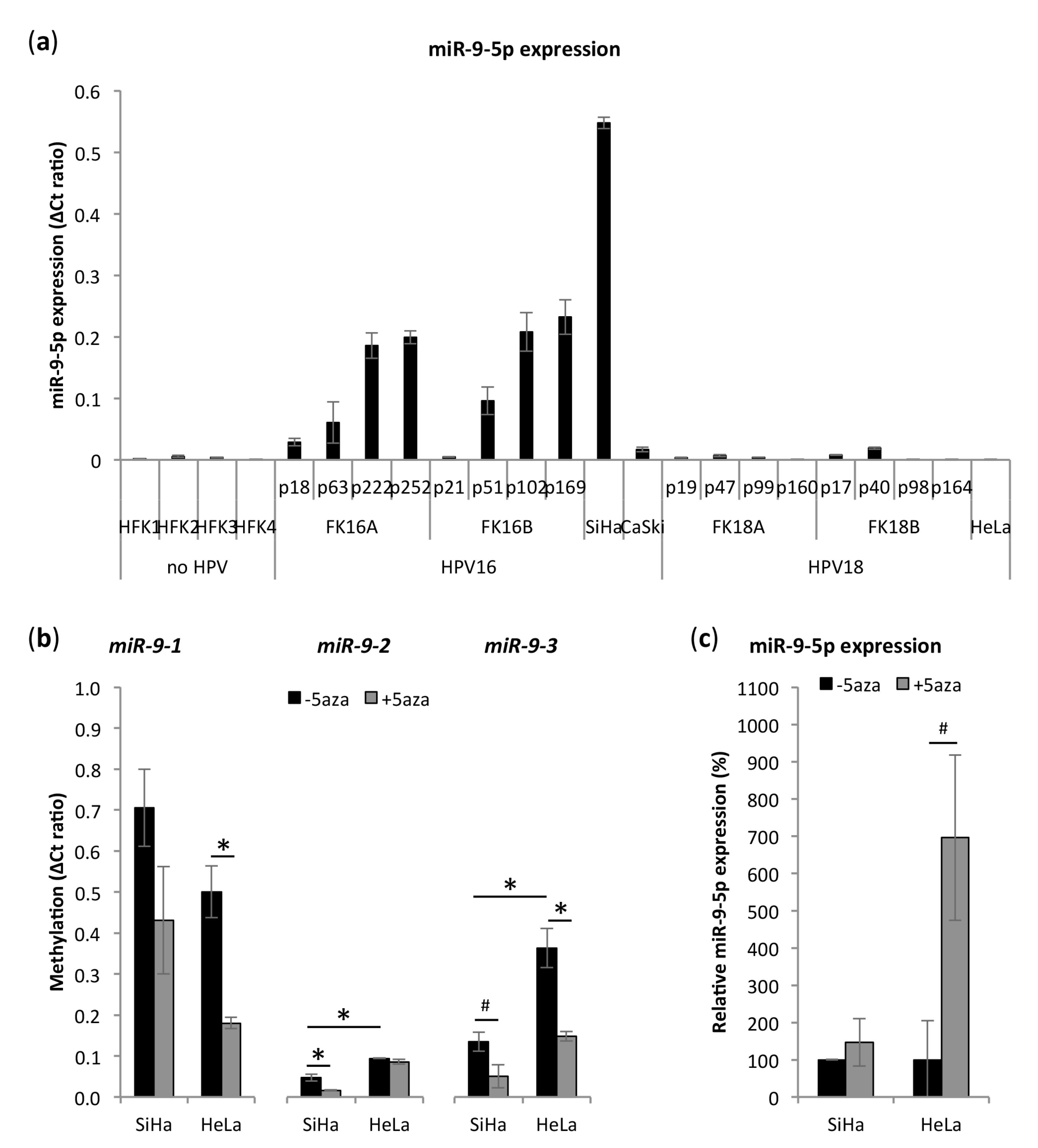
3.2. miR-9-5p Expression Is Increased in HPV16-Positive But Not in HPV18-Positive Cells
3.3. miR-9-5p Expression Is Regulated by DNA Methylation
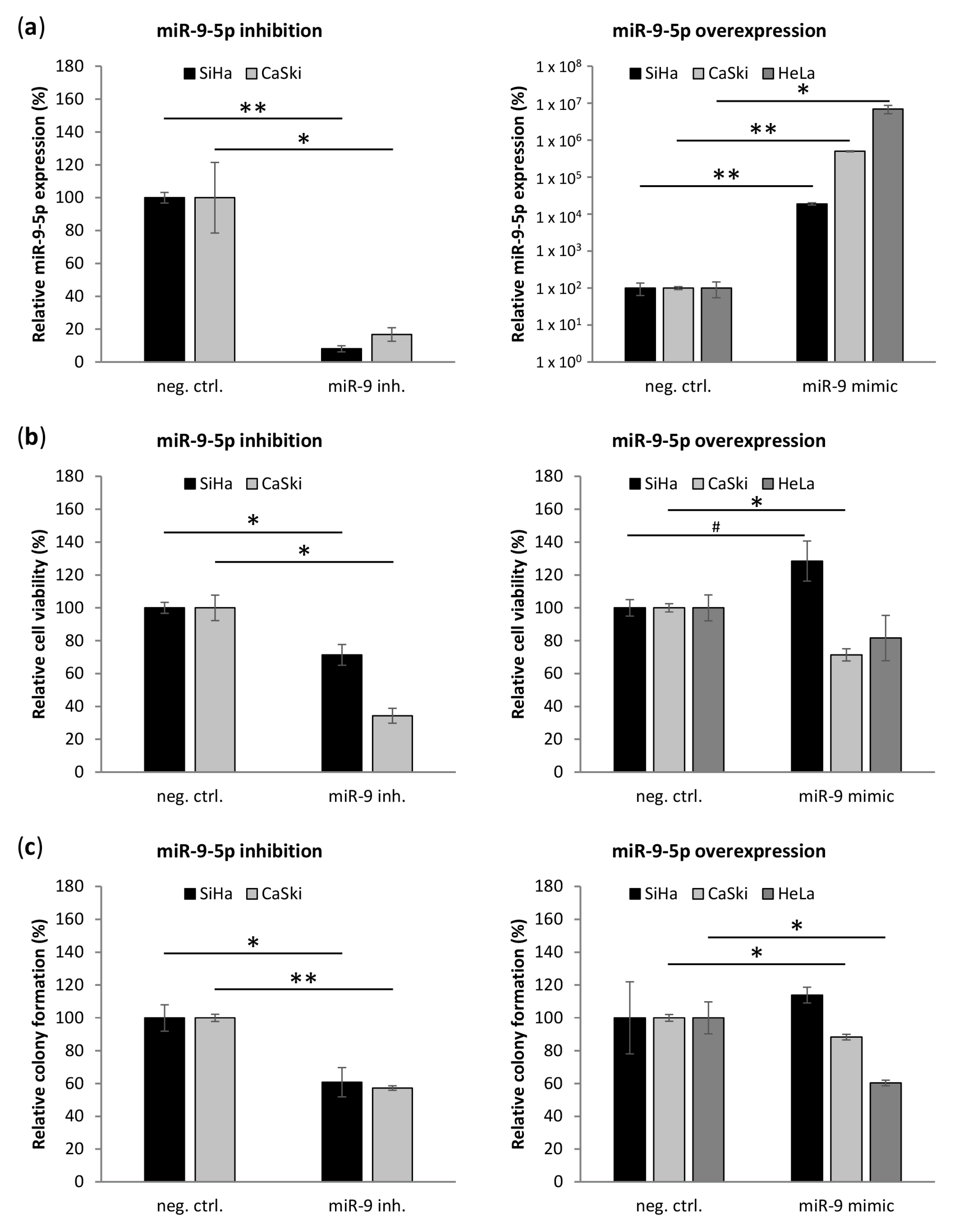
3.4. miR-9-5p Plays Opposing Functional Roles Cervical Cancer Cells
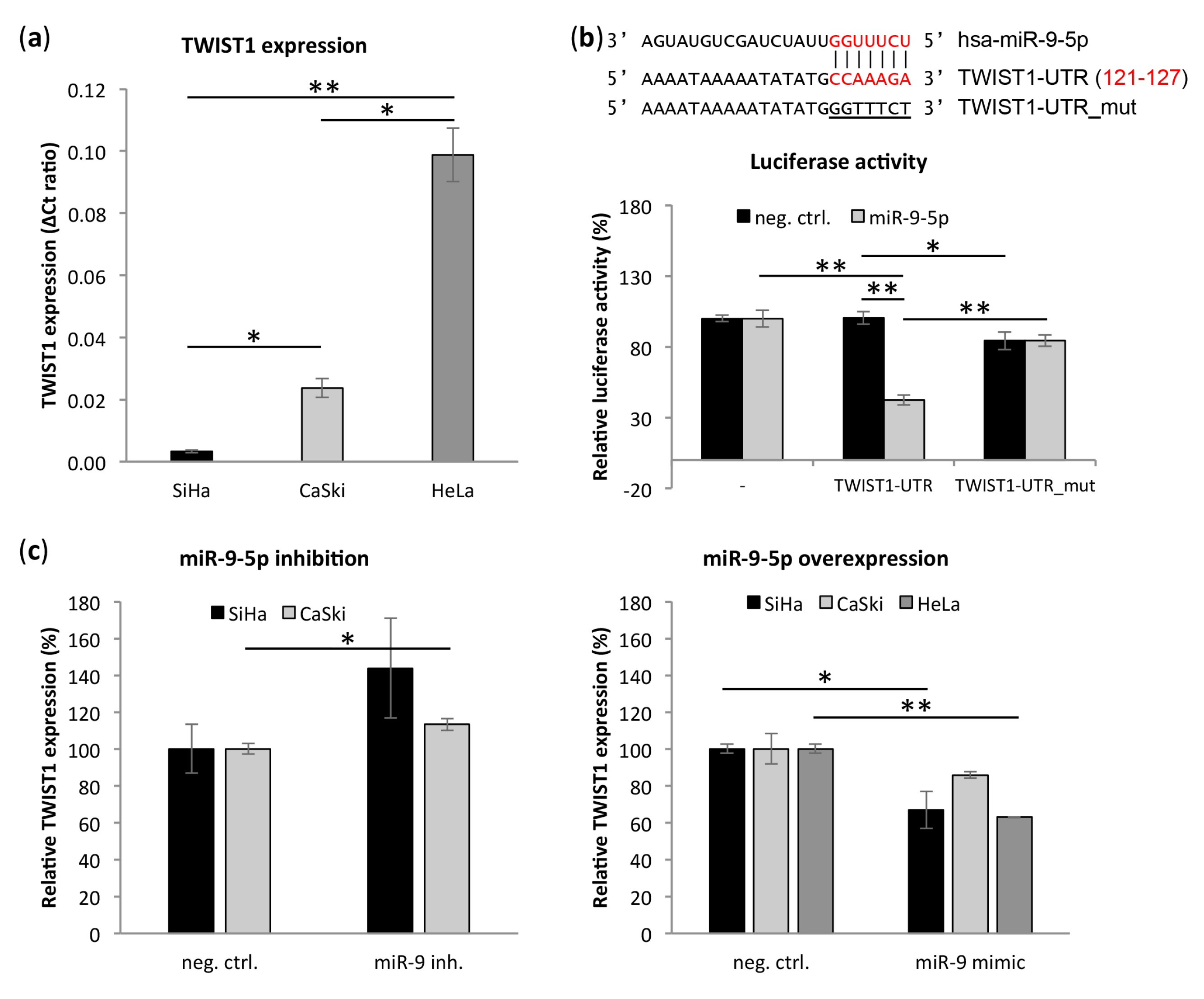
3.5. Transcription Factor TWIST1 Is a Direct Target of miR-9-5p
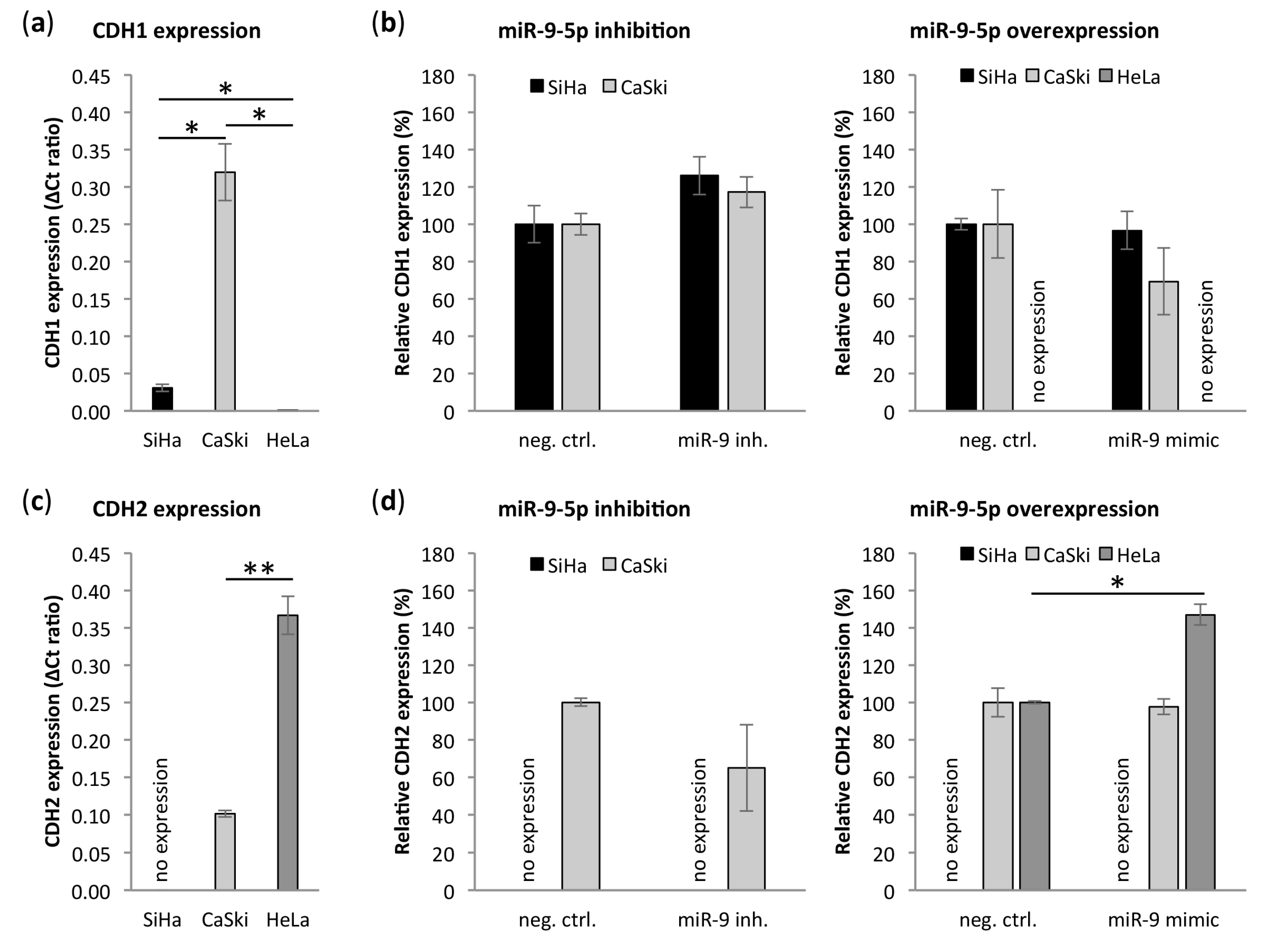
3.6. miR-9-5p Regulates Expression of CDH1 and CDH2
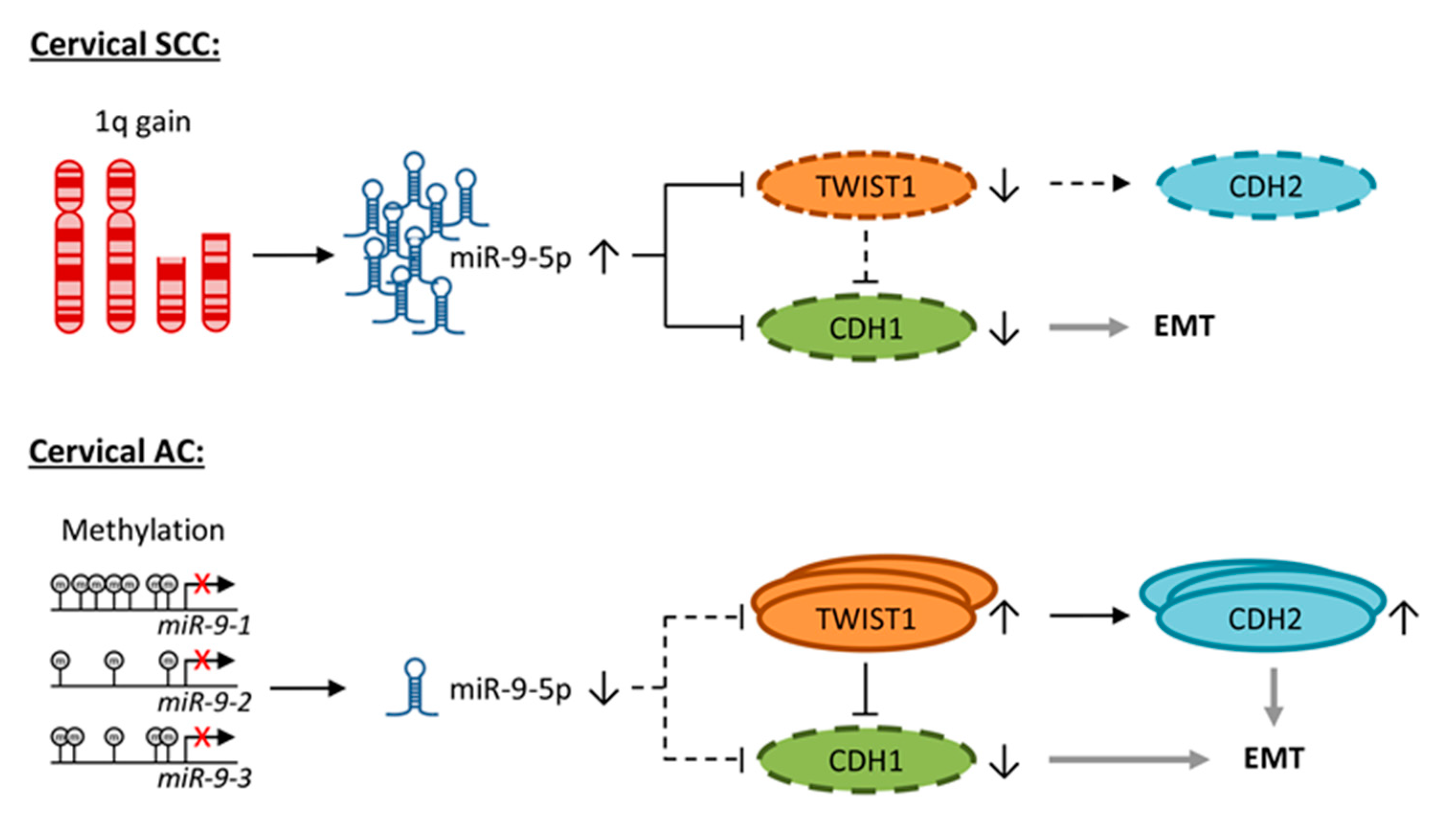
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Steenbergen, R.D.M.; Snijders, P.J.F.; Heideman, D.A.M.; Meijer, C.J.L.M. Clinical implications of (epi)genetic changes in HPV-induced cervical precancerous lesions. Nat. Rev. Cancer 2014, 14, 395–405. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Fu, Y.S.; Reagan, J.W. Pathology of the Uterine Cervix, Vagina, and Vulva; Saunders Philadelphia: Philadelphia, PA, USA, 1989; ISBN 0721674933. [Google Scholar]
- Pisani, P.; Bray, F.; Parkin, D.M. Estimates of the world-wide prevalence of cancer for 25 sites in the adult population. Int. J. cancer 2002, 97, 72–81. [Google Scholar] [CrossRef]
- Burk, R.D.; Terai, M.; Gravitt, P.E.; Brinton, L.A.; Kurman, R.J.; Barnes, W.A.; Greenberg, M.D.; Hadjimichael, O.C.; Fu, L.; McGowan, L.; et al. Distribution of human papillomavirus types 16 and 18 variants in squamous cell carcinomas and adenocarcinomas of the cervix. Cancer Res. 2003, 63, 7215–7220. [Google Scholar]
- Chen, A.A.; Gheit, T.; Franceschi, S.; Tommasino, M.; Clifford, G.M. IARC HPV Variant Study Group Human Papillomavirus 18 Genetic Variation and Cervical Cancer Risk Worldwide. J. Virol. 2015, 89, 10680–10687. [Google Scholar] [CrossRef] [Green Version]
- Wilting, S.M.; Snijders, P.J.F.; Verlaat, W.; Jaspers, A.; van de Wiel, M.A.; van Wieringen, W.N.; Meijer, G.A.; Kenter, G.G.; Yi, Y.; le Sage, C.; et al. Altered microRNA expression associated with chromosomal changes contributes to cervical carcinogenesis. Oncogene 2013, 32, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Dua, P.; Agarwal, S.M. A Comprehensive Review of Dysregulated miRNAs Involved in Cervical Cancer. Curr. Genom. 2014, 15, 310–323. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, F.; Xu, J.; Ye, F.; Shen, Y.; Zhou, J.; Lu, W.; Wan, X.; Ma, D.; Xie, X. Progressive miRNA expression profiles in cervical carcinogenesis and identification of HPV-related target genes for miR-29. J. Pathol. 2011, 224, 484–495. [Google Scholar] [CrossRef]
- Zeng, K.; Zheng, W.; Mo, X.; Liu, F.; Li, M.; Liu, Z.; Zhang, W.; Hu, X. Dysregulated microRNAs involved in the progression of cervical neoplasm. Arch. Gynecol. Obstet. 2015, 292, 905–9013. [Google Scholar] [CrossRef]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Zu, Y.; Zhu, Z.; Lin, M.; Xu, D.; Liang, Y.; Wang, Y.; Qiao, Z.; Cao, T.; Yang, D.; Gao, L.; et al. MiR-9 Promotes Apoptosis Suppressing SMC1A Expression in GBM Cell Lines. Curr. Chem. Genom. Trans. Med. 2017, 11, 31–40. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Zhang, L.; Wang, M.; Wang, W. miR-9 functions as a tumor inhibitor of cell proliferation in epithelial ovarian cancer through targeting the SDF-1/CXCR4 pathway. Exp. Ther. Med. 2017, 13, 1203–1208. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Liu, X.; Han, L.; Shen, H.; Liu, L.; Shu, Y. Up-regulation of miR-9 expression as a poor prognostic biomarker in patients with non-small cell lung cancer. Clin. Trans. Oncol. 2014, 16, 469–475. [Google Scholar] [CrossRef]
- Seashols-Williams, S.J.; Budd, W.; Clark, G.C.; Wu, Q.; Daniel, R.; Dragoescu, E.; Zehner, Z.E. miR-9 Acts as an OncomiR in Prostate Cancer through Multiple Pathways That Drive Tumour Progression and Metastasis. PLoS ONE 2016, 11, e0159601. [Google Scholar] [CrossRef] [Green Version]
- Nowek, K.; Wiemer, E.A.C.; Jongen-Lavrencic, M. The versatile nature of miR-9/9* in human cancer. Oncotarget 2018, 9, 20838–20854. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, Z.; Wang, S.; Zhang, S.; Bi, J. The mechanisms involved in miR-9 regulated apoptosis in cervical cancer by targeting FOXO3. Biomed. Pharmacother. 2018, 102, 626–632. [Google Scholar] [CrossRef]
- Babion, I.; Snoek, B.C.; Novianti, P.W.; Jaspers, A.; van Trommel, N.; Heideman, D.A.M.; Meijer, C.J.L.M.; Snijders, P.J.F.; Steenbergen, R.D.M.; Wilting, S.M. Triage of high-risk HPV-positive women in population-based screening by miRNA expression analysis in cervical scrapes; a feasibility study. Clin. Epigenet. 2018, 10, 76. [Google Scholar] [CrossRef]
- Wilting, S.; Snijders, P.; Meijer, G.; Ylstra, B.; van den IJssel, P.; Snijders, A.; Albertson, D.; Coffa, J.; Schouten, J.; van de Wiel, M.; et al. Increased gene copy numbers at chromosome 20q are frequent in both squamous cell carcinomas and adenocarcinomas of the cervix. J. Pathol. 2006, 209, 220–230. [Google Scholar] [CrossRef]
- Thomas, L.K.; Bermejo, J.L.; Vinokurova, S.; Jensen, K.; Bierkens, M.; Steenbergen, R.; Bergmann, M.; von Knebel Doeberitz, M.; Reuschenbach, M. Chromosomal gains and losses in human papillomavirus-associated neoplasia of the lower genital tract—A systematic review and meta-analysis. Eur. J. Cancer 2014, 50, 85–98. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, J.; Zhao, L.; Li, X.; Xie, Q.; Chen, X.; Wang, J.; Lu, F. Down-regulation of microRNA-9 leads to activation of IL-6/Jak/STAT3 pathway through directly targeting IL-6 in HeLa cell. Mol. Carcinog. 2016, 55, 732–742. [Google Scholar] [CrossRef]
- Snellenberg, S.; Cillessen, S.A.G.M.; van Criekinge, W.; Bosch, L.; Meijer, C.J.L.M.; Snijders, P.J.F.; Steenbergen, R.D.M. Methylation-mediated repression of PRDM14 contributes to apoptosis evasion in HPV-positive cancers. Carcinogenesis 2014, 35, 2611–2618. [Google Scholar] [CrossRef] [Green Version]
- Steenbergen, R.D.; Walboomers, J.M.; Meijer, C.J.; van der Raaij-Helmer, E.M.; Parker, J.N.; Chow, L.T.; Broker, T.R.; Snijders, P.J. Transition of human papillomavirus type 16 and 18 transfected human foreskin keratinocytes towards immortality: Activation of telomerase and allele losses at 3p, 10p, 11q and/or 18q. Oncogene 1996, 13, 1249–1257. [Google Scholar]
- Steenbergen, R.D.; Kramer, D.; Meijer, C.J.; Walboomers, J.M.; Trott, D.A.; Cuthbert, A.P.; Newbold, R.F.; Overkamp, W.J.; Zdzienicka, M.Z.; Snijders, P.J. Telomerase suppression by chromosome 6 in a human papillomavirus type 16-immortalized keratinocyte cell line and in a cervical cancer cell line. J. Natl. Cancer Inst. 2001, 93, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.V.; Snijders, P.J.; van den Brule, A.J.; Helmerhorst, T.J.; Meijer, C.J.; Walboomers, J.M. A general primer GP5+/GP6(+)-mediated PCR-enzyme immunoassay method for rapid detection of 14 high-risk and 6 low-risk human papillomavirus genotypes in cervical scrapings. J. Clin. Microbiol. 1997, 35, 791–795. [Google Scholar]
- Geraets, D.T.; Cuschieri, K.; de Koning, M.N.C.; van Doorn, L.J.; Snijders, P.J.F.; Meijer, C.J.L.M.; Quint, W.G.V.; Arbyn, M. Clinical evaluation of a GP5+/6+-based luminex assay having full high-risk human papillomavirus genotyping capability and an internal control. J. Clin. Microbiol. 2014, 52, 3996–4002. [Google Scholar] [CrossRef] [Green Version]
- Federa Federa. Human Tissue and Medical Research: Code of Conduct for Responsible Use. 2011. Available online: http://www.federa.org/sites/default/files/digital_version_first_part_code_of_conduct_in_uk_2011_12092012.pdf (accessed on 27 November 2019).
- van Zeeburg, H.J.T.; Snijders, P.J.F.; Pals, G.; Hermsen, M.A.J.A.; Rooimans, M.A.; Bagby, G.; Soulier, J.; Gluckman, E.; Wennerberg, J.; Leemans, C.R.; et al. Generation and Molecular Characterization of Head and Neck Squamous Cell Lines of Fanconi Anemia Patients. Cancer Res. 2005, 65, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Snellenberg, S.; De Strooper, L.M.A.; Hesselink, A.T.; Meijer, C.J.L.M.; Snijders, P.J.F.; Heideman, D.M.; Steenbergen, R.D.M. Development of a multiplex methylation-specific PCR as candidate triage test for women with an HPV-positive cervical scrape. BMC Cancer 2012, 12, 551. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Burk, R.D.; Chen, Z.Z.; Saller, C.; Tarvin, K.; Carvalho, A.L.; Scapulatempo-Neto, C.; Silveira, H.C.; Fregnani, J.H.; Creighton, C.J.; Anderson, M.L.; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar]
- Steenbergen, R.D.M.; Kramer, D.; Braakhuis, B.J.M.; Stern, P.L.; Verheijen, R.H.M.; Meijer, C.J.L.M.; Snijders, P.J.F. TSLC1 gene silencing in cervical cancer cell lines and cervical neoplasia. J. Natl. Cancer Inst. 2004, 96, 294–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henken, F.E.; Wilting, S.M.; Overmeer, R.M.; van Rietschoten, J.G.I.; Nygren, A.O.H.; Errami, A.; Schouten, J.P.; Meijer, C.J.L.M.; Snijders, P.J.F.; Steenbergen, R.D.M. Sequential gene promoter methylation during HPV-induced cervical carcinogenesis. Br. J. Cancer 2007, 97, 1457–1464. [Google Scholar] [CrossRef] [Green Version]
- Smeets, S.J.; van der Plas, M.; Schaaij-Visser, T.B.M.; van Veen, E.A.M.; van Meerloo, J.; Braakhuis, B.J.M.; Steenbergen, R.D.M.; Brakenhoff, R.H. Immortalization of oral keratinocytes by functional inactivation of the p53 and pRb pathways. Int. J. Cancer 2011, 128, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Guadamillas, M.C.; Cerezo, A.; del Pozo, M.A. Overcoming anoikis—Pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124, 3189–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinde, J.; Frankel, D.; Perrin, S.; Delecourt, V.; Lévy, N.; Barlesi, F.; Astoul, P.; Roll, P.; Kaspi, E. Lamins in Lung Cancer: Biomarkers and Key Factors for Disease Progression through miR-9 Regulation? Cells 2018, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.-H.; Huang, C.-C.; Pan, M.-R.; Chen, H.-H.; Hung, W.-C. Prospero Homeobox 1 Promotes Epithelial-Mesenchymal Transition in Colon Cancer Cells by Inhibiting E-cadherin via miR-9. Clin. Cancer Res. 2012, 18, 6416–6425. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Li, J.; Zhu, Y.; Dai, Y.; Zeng, T.; Liu, L.; Li, J.; Wang, H.; Qin, Y.; Zeng, M.; et al. MicroRNA-9 promotes tumor metastasis via repressing E-cadherin in esophageal squamous cell carcinoma. Oncotarget 2014, 5, 11669–11680. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wu, Q.; Zhang, Y.; Zhang, H.-N.; Wang, Y.-B.; Wang, W. TGF-β1-induced epithelial–mesenchymal transition in lung cancer cells involves upregulation of miR-9 and downregulation of its target, E-cadherin. Cell. Mol. Biol. Lett. 2017, 22, 22. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.-Z.; Li, X.-A.; Luo, Y.; Liu, J.-F.; Wu, H.-W.; Huang, G. MiR-9 promotes synovial sarcoma cell migration and invasion by directly targeting CDH1. Int. J. Biochem. Cell Biol. 2019, 112, 61–71. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.-S.; Tam, W.-L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A Pattern-Based Method for the Identification of MicroRNA Binding Sites and Their Corresponding Heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesuna, F.; van Diest, P.; Chen, J.H.; Raman, V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem. Biophys. Res. Commun. 2008, 367, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhou, B.P. Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer 2011, 11, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Sun, L.; Li, Q.; Han, X.; Lei, L.; Zhang, H.; Shang, Y. SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012, 31, 110–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.-Q.; Ma, C.; Wang, Q.; Song, Y.; Lv, T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumor Biol. 2016, 37, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.B.Y.; Lin, A.; Xu, W.; Waldron, L.; Perez-Ordonez, B.; Weinreb, I.; Shi, W.; Bruce, J.; Huang, S.H.; O’Sullivan, B.; et al. Potentially Prognostic miRNAs in HPV-Associated Oropharyngeal Carcinoma. Clin. Cancer Res. 2013, 19, 2154–2162. [Google Scholar] [CrossRef] [Green Version]
- Božinović, K.; Sabol, I.; Dediol, E.; Milutin Gašperov, N.; Manojlović, S.; Vojtechova, Z.; Tachezy, R.; Grce, M. Genome-wide miRNA profiling reinforces the importance of miR-9 in human papillomavirus associated oral and oropharyngeal head and neck cancer. Sci. Rep. 2019, 9, 2306. [Google Scholar]
- Harden, M.E.; Prasad, N.; Griffiths, A.; Munger, K. Modulation of microRNA-mRNA Target Pairs by Human Papillomavirus 16 Oncoproteins. MBio 2017, 8, e02170-16. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Gao, G.; Hu, X.; Wang, Y.; Schwarz, J.K.; Chen, J.J.; Grigsby, P.W.; Wang, X. Activation of miR-9 by human papillomavirus in cervical cancer. Oncotarget 2014, 5, 11620–11630. [Google Scholar] [CrossRef]
- Nilsen, A.; Jonsson, M.; Aarnes, E.-K.; Kristensen, G.B.; Lyng, H. Reference MicroRNAs for RT-qPCR Assays in Cervical Cancer Patients and Their Application to Studies of HPV16 and Hypoxia Biomarkers. Transl. Oncol. 2019, 12, 576–584. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial—Mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Arora, H.; Rizvi, M.A. EMT in cervical cancer: Its role in tumour progression and response to therapy. Cancer Lett. 2015, 356, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Chen, A.; Chao, X.; Li, X.; Cui, Y.; Xu, C.; Cao, J.; Ning, Y. Chrysin Inhibits Proinflammatory FactorInduced EMT Phenotype and Cancer Stem Cell-Like Features in HeLa Cells by Blocking the NF-κB/Twist Axis. Cell. Physiol. Biochem. 2019, 52, 1236–1250. [Google Scholar]
- Zhu, K.; Chen, L.; Han, X.; Wang, J.J.; Wang, J.J. Short hairpin RNA targeting Twist1 suppresses cell proliferation and improves chemosensitivity to cisplatin in HeLa human cervical cancer cells. Oncol. Rep. 2012, 27, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Qian, W.; Zhang, J.; Dong, Y.; Shi, C.; Liu, Z.; Wu, S. The indicative function of Twist2 and E-cadherin in HPV oncogene-induced epithelial-mesenchymal transition of cervical cancer cells. Oncol. Rep. 2015, 33, 639–650. [Google Scholar] [CrossRef]
- Liu, M.; Liu, J.; Yang, B.; Gao, X.; Gao, L.; Kong, Q.; Zhang, P.; Li, H. Inversed Expression Patterns of S100A4 and E-Cadherin in Cervical Cancers: Implication in Epithelial–Mesenchymal Transition. Anat. Rec. 2017, 300, 2184–2191. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Yue, Z.; Tian, Z.; Xie, Y.; Zhang, J.; She, Y.; Yang, B.; Ye, Y.; Yang, Y. Expression of Yin Yang 1 in cervical cancer and its correlation with E-cadherin expression and HPV16 E6. PLoS ONE 2018, 13, e0193340. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babion, I.; Jaspers, A.; van Splunter, A.P.; van der Hoorn, I.A.E.; Wilting, S.M.; Steenbergen, R.D.M. miR-9-5p Exerts a Dual Role in Cervical Cancer and Targets Transcription Factor TWIST1. Cells 2020, 9, 65. https://doi.org/10.3390/cells9010065
Babion I, Jaspers A, van Splunter AP, van der Hoorn IAE, Wilting SM, Steenbergen RDM. miR-9-5p Exerts a Dual Role in Cervical Cancer and Targets Transcription Factor TWIST1. Cells. 2020; 9(1):65. https://doi.org/10.3390/cells9010065
Chicago/Turabian StyleBabion, Iris, Annelieke Jaspers, Annina P. van Splunter, Iris A.E. van der Hoorn, Saskia M. Wilting, and Renske D.M. Steenbergen. 2020. "miR-9-5p Exerts a Dual Role in Cervical Cancer and Targets Transcription Factor TWIST1" Cells 9, no. 1: 65. https://doi.org/10.3390/cells9010065
APA StyleBabion, I., Jaspers, A., van Splunter, A. P., van der Hoorn, I. A. E., Wilting, S. M., & Steenbergen, R. D. M. (2020). miR-9-5p Exerts a Dual Role in Cervical Cancer and Targets Transcription Factor TWIST1. Cells, 9(1), 65. https://doi.org/10.3390/cells9010065