Dynasore Blocks Ferroptosis through Combined Modulation of Iron Uptake and Inhibition of Mitochondrial Respiration



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines and Culture Conditions
2.3. Transfection with Small Interfering RNA (siRNA)
2.4. Cell Viability Assays
2.5. Fluorescence-Activated Cell Sorting (FACS) Assays
2.6. Western Blotting
2.7. Relative Intracelluar Iron Quantification
2.8. GSH Measurement
2.9. Lipid ROS Quantification
2.10. Mitochondrial ROS Quantification
2.11. General Cellular ROS Quantification
2.12. Seahorse Assay in Intact Cells
2.13. Seahorse Assay on Isolated Mitochondria
2.14. DPPH Assay
2.15. Time-Lapse Cell Death Assays
2.16. Quantification and Statistical Analysis
3. Results
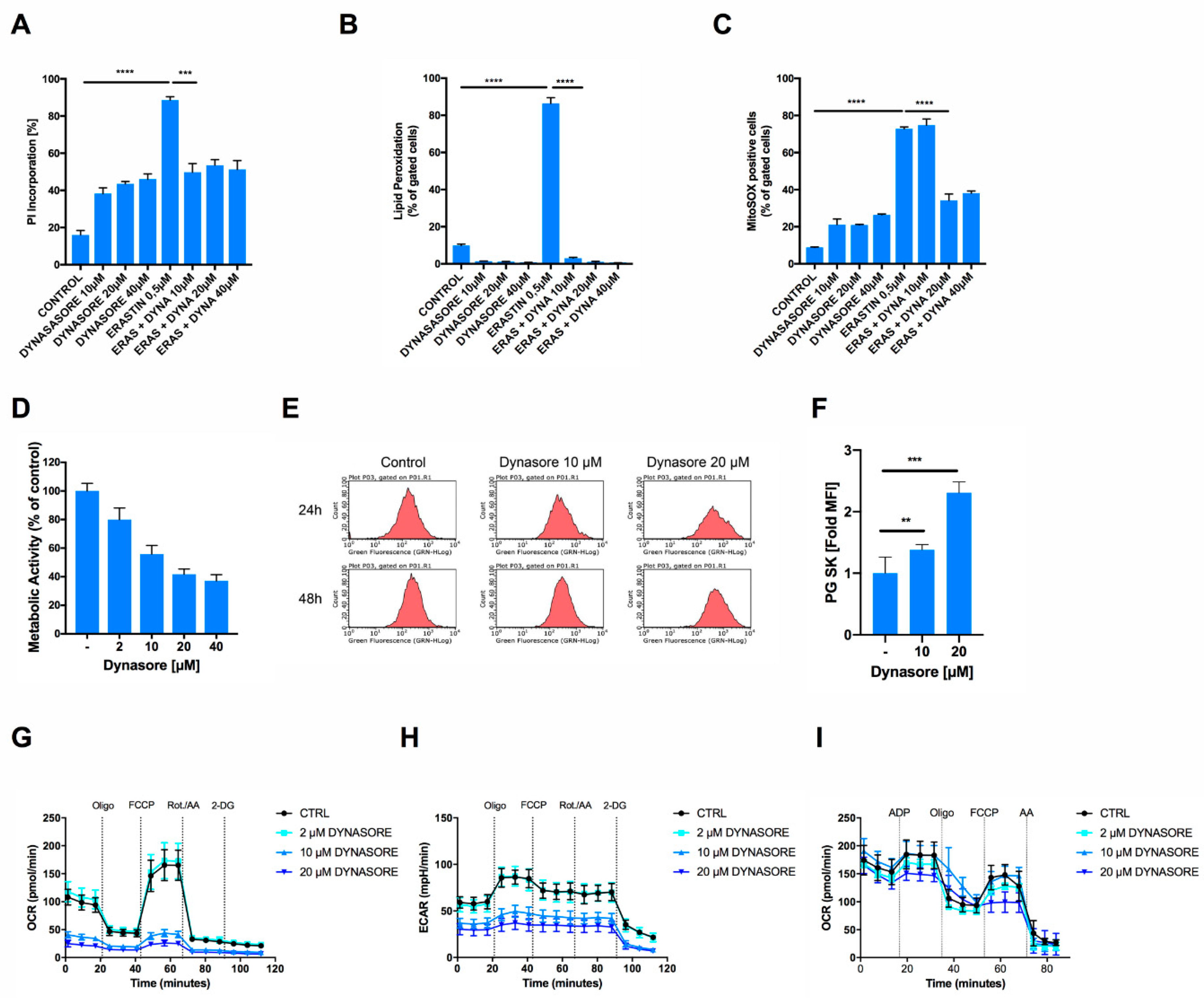
3.1. Dynasore Blocks Transferrin Receptor Uptake and Ferroptosis
3.2. Inhibition of Dynamin 1- and 2-Regulated Iron Uptake is Insufficient to Block Ferroptosis
3.3. Dynasore Protects Neuronal Cells from Ferroptosis and Blocks Mitochondrial ROS Formation and Respiration
3.4. Dynasore Functions as a Broadly Effective Radical-Trapping Agent
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Morrison, B., III; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agmon, E.; Solon, J.X.R.X.M.; Bassereau, P.; Stockwell, B.R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 2018, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Smith, R.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Basavarajappa, D.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; Panzilius, E.; Scheel, C.H.; Mourão, A.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Bassik, M.C.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [Green Version]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell. 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dam, E.M.; Stoorvogel, W. Dynamin-dependent transferrin receptor recycling by endosome-derived clathrin-coated vesicles. Mol. Biol. Cell. 2002, 13, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Shen, F.; Fan, Z.; Wang, Y.; Kong, X.; Yu, D.; Zhi, X.; Lv, G.; Cao, Y. Dynasore Improves Motor Function Recovery via Inhibition of Neuronal Apoptosis and Astrocytic Proliferation after Spinal Cord Injury in Rats. Mol. Neurobiol. 2017, 54, 7471–7482. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zhang, L.; Dhillon, R.; Hong, T.T.; Shaw, R.M.; Zhu, J. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS ONE 2013, 8, e60967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, S.; Tanaka, S.; Hsiao, H.M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Invest. 2019, 129, 2293–2304. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Weinlich, R.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [Green Version]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Michels, S.; Dolga, A.M.; Braun, M.D.; Kisko, T.M.; Sungur, A.Ö.; Witt, S.H.; Rietschel, M.; Dempfle, A.; Wöhr, M.; Schwarting, R.K.W.; et al. Interaction of the Psychiatric Risk Gene Cacna1c With Post-weaning Social Isolation or Environmental Enrichment Does Not Affect Brain Mitochondrial Bioenergetics in Rats. Front. Cell Neurosci. 2019, 13, 483. [Google Scholar] [CrossRef] [Green Version]
- Cocucci, E.; Gaudin, R.; Kirchhausen, T. Dynamin recruitment and membrane scission at the neck of a clathrin-coated pit. Mol. Biol. Cell. 2014, 25, 3595–3609. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Stockwell, B.R.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. ELife Sci. 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wagner, B.A.; Buettner, G.R.; Cullen, J.J. Role of labile iron in the toxicity of pharmacological ascorbate. Free Radic. Biol. Med. 2015, 84, 289–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossarizza, A.; Ferraresi, R.; Troiano, L.; Roat, E.; Gibellini, L.; Bertoncelli, L.; Nasi, M.; Pinti, M. Simultaneous analysis of reactive oxygen species and reduced glutathione content in living cells by polychromatic flow cytometry. Nat. Protoc. 2009, 4, 1790–1797. [Google Scholar] [CrossRef]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef]
- Yao, X.; Zhang, Y.; Hao, J.; Duan, H.Q.; Zhao, C.X.; Sun, C.; Li, B.; Fan, B.-Y.; Wang, X.; Fu, X.H.; et al. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen Res. 2019, 14, 532–541. [Google Scholar]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Francis, T.C.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Kareem, H.S.; Ariffin, A.; Nordin, N.; Heidelberg, T.; Abdul-Aziz, A.; Kong, K.W.; Yehye, W.A. Correlation of antioxidant activities with theoretical studies for new hydrazone compounds bearing a 3,4,5-trimethoxy benzyl moiety. Eur. J. Med. Chem. 2015, 20, 497–505. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Bebber, C.M.; Müller, F.; Prieto Clemente, L.; Weber, J.; von Karstedt, S. Ferroptosis in Cancer Cell Biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devos, D.; Moreau, C.; Kyheng, M.; Garçon, G.; Rolland, A.S.; Blasco, H.; Lenglet, T.T.; Duce, J.A.; Otto, M.; Dutheil, M.; et al. A ferroptosis-based panel of prognostic biomarkers for Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.-B.; Kalkhoran, S.B.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. Eur. J. Pharmacol. 2015, 763, 104–114. [Google Scholar]
- Stockwell, B.R.; Angeli JP, F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Hatzios, S.K.; Kagan, V.E.; Noel, K.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Morel, I.; Cillard, J.; Lescoat, G.; Sergent, O.; Pasdeloup, N.; Ocaktan, A.Z.; Abdallah, M.A.; Brissot, P.; Cillard, P. Antioxidant and free radical scavenging activities of the iron chelators pyoverdin and hydroxypyrid-4-ones in iron-loaded hepatocyte cultures: Comparison of their mechanism of protection with that of desferrioxamine. Free Radic. Biol. Med. 1992, 13, 499–508. [Google Scholar] [CrossRef]
- Yanatori, I.; Yasui, Y.; Noguchi, Y.; Kishi, F. Inhibition of iron uptake by ferristatin II is exerted through internalization of DMT1 at the plasma membrane. Cell Biol. Int. 2015, 39, 427–434. [Google Scholar] [CrossRef]
- Lane DJ, R.; Merlot, A.M.; Huang, M.H.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [Green Version]
- Preta, G.; Cronin, J.G.; Sheldon, I.M. Dynasore-not just a dynamin inhibitor. Cell Commun. Signal. 2015, 13, 24–27. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Latunde-Dada2. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H. Increased mitochondrial fission and neuronal dysfunction in Huntington’s disease: Implications for molecular inhibitors of excessive mitochondrial fission. Drug Discov. Today 2014, 19, 951–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, A.; Chintala, S.K.; Kim, J.; Ngan, M.; Itakura, T.; Panjwani, N.; Argüeso, P.; Barr, J.T.; Jeong, S.; Fini, M.E. Dynasore protects the ocular surface against damaging oxidative stress. PLoS ONE 2018, 13, e0204288. [Google Scholar] [CrossRef] [PubMed]
- Huo, H.; Zhou, Z.; Qin, J.; Liu, W.; Wang, B.; Gu, Y. Erastin Disrupts Mitochondrial Permeability Transition Pore (mPTP) and Induces Apoptotic Death of Colorectal Cancer Cells. PLoS ONE 2016, 11, e0154605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Yang, M.; Deng, J.; Li, P.; Su, W.; Jiang, R. Upregulation and activation of p53 by erastin-induced reactive oxygen species contribute to cytotoxic and cytostatic effects in A549 lung cancer cells. Oncol. Rep. 2018, 40, 2363–2370. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Stein, H.A.; Turschner, S.; Toegel, I.; Mora, R.; Jennewein, N.; Eils, R.; Brady, N.R. Artesunate activates mitochondrial apoptosis in breast cancer cells via iron-catalyzed lysosomal reactive oxygen species production. J. Biol. Chem. 2011, 286, 6587–6601. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clemente, L.P.; Rabenau, M.; Tang, S.; Stanka, J.; Cors, E.; Stroh, J.; Culmsee, C.; von Karstedt, S. Dynasore Blocks Ferroptosis through Combined Modulation of Iron Uptake and Inhibition of Mitochondrial Respiration. Cells 2020, 9, 2259. https://doi.org/10.3390/cells9102259
Clemente LP, Rabenau M, Tang S, Stanka J, Cors E, Stroh J, Culmsee C, von Karstedt S. Dynasore Blocks Ferroptosis through Combined Modulation of Iron Uptake and Inhibition of Mitochondrial Respiration. Cells. 2020; 9(10):2259. https://doi.org/10.3390/cells9102259
Chicago/Turabian StyleClemente, Laura Prieto, Malena Rabenau, Stephan Tang, Josefina Stanka, Eileen Cors, Jenny Stroh, Carsten Culmsee, and Silvia von Karstedt. 2020. "Dynasore Blocks Ferroptosis through Combined Modulation of Iron Uptake and Inhibition of Mitochondrial Respiration" Cells 9, no. 10: 2259. https://doi.org/10.3390/cells9102259
APA StyleClemente, L. P., Rabenau, M., Tang, S., Stanka, J., Cors, E., Stroh, J., Culmsee, C., & von Karstedt, S. (2020). Dynasore Blocks Ferroptosis through Combined Modulation of Iron Uptake and Inhibition of Mitochondrial Respiration. Cells, 9(10), 2259. https://doi.org/10.3390/cells9102259