The Multi-Omics Architecture of Juvenile Idiopathic Arthritis


 , ,
, ,
Abstract
:1. Introduction
2. JIA as a Heterogeneous Group of Diseases
2.1. Systemic JIA (sJIA)
2.2. Oligoarthritis and Polyarthritis
2.3. Psoriatic Arthritis
2.4. Enthesitis-Related JIA
3. Genetic Studies of Monogenic Forms of JIA
3.1. LACC1
3.2. LRBA
3.3. NFIL3
3.4. UNC13D
4. Genetic Studies of Polygenic Forms of JIA
4.1. The Association with HLA
4.1.1. Oligoarthritis and Polyarthritis RF-Negative
4.1.2. Polyarthritis RF-Positive
4.1.3. sJIA
4.1.4. Psoriatic and Enthesitis-Related JIA
4.2. Association with Non-HLA Loci
4.2.1. Oligoarthritis and Polyarthritis RF Negative
4.2.2. Polyarthritis RF-Positive
4.2.3. sJIA
5. Transcriptome Study of JIA
5.1. Transcriptome Profiling of Neutrophils in JIA
5.2. Transcriptome Profiling of Macrophages in JIA
5.3. Transcriptome Profiling of Monocytes in JIA
5.4. Transcriptome Profiling of T Cells in JIA
6. Epigenomics Study of JIA
7. Conclusions
Funding
Conflicts of Interest
References
- Thierry, S.; Fautrel, B.; Lemelle, I.; Guillemin, F. Prevalence and incidence of juvenile idiopathic arthritis: A systematic review. Jt. Bone Spine 2014, 81, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Fink, C.W. Proposal for the development of classification criteria for idiopathic arthritides of childhood. J. Rheumatol. 1995, 22, 1566–1569. [Google Scholar] [PubMed]
- Thatayatikom, A.; De Leucio, A. Juvenile Idiopathic Arthritis (JIA); StatPearls, National Library of Medicine (NLM): Bethesda, MD, USA, 2020. [Google Scholar]
- Martini, A. It is time to rethink juvenile idiopathic arthritis classification and nomenclature. Ann. Rheum. Dis. 2012, 71, 1437–1439. [Google Scholar] [CrossRef] [PubMed]
- Martini, A.; Ravelli, A.; Avcin, T.; Beresford, M.W.; Burgos-Vargas, R.; Cuttica, R.; Ilowite, N.T.; Khubchandani, R.; Laxer, R.M.; Lovell, D.J. Toward New Classification Criteria for Juvenile Idiopathic Arthritis: First Steps, Pediatric Rheumatology International Trials Organization International Consensus. J. Rheumatol. 2018, 46, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.-M.; et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar]
- Guo, R.; Cao, L.; Kong, X.; Liu, X.; Xue, H.; Shen, L.; Li, X. Fever as an Initial Manifestation of Enthesitis-Related Arthritis Subtype of Juvenile Idiopathic Arthritis: Retrospective Study. PLoS ONE 2015, 10, e0128979. [Google Scholar] [CrossRef]
- Masters, S.L.; Simon, A.; Aksentijevich, I.; Kastner, D.L. Horror Autoinflammaticus: The Molecular Pathophysiology of Autoinflammatory Disease. Annu. Rev. Immunol. 2009, 27, 621–668. [Google Scholar] [CrossRef] [Green Version]
- De Jesus, A.A.; Goldbach-Mansky, R. Monogenic autoinflammatory diseases: Concept and clinical manifestations. Clin. Immunol. 2013, 147, 155–174. [Google Scholar] [CrossRef] [Green Version]
- Hersh, A.O.; Prahalad, S. Immunogenetics of juvenile idiopathic arthritis: A comprehensive review. J. Autoimmun. 2015, 64, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Cader, M.Z.; Boroviak, K.; Zhang, Q.; Assadi, G.; Kempster, S.L.; Sewell, G.W.; Saveljeva, S.; Ashcroft, J.W.; Clare, S.; Mukhopadhyay, S.; et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat. Immunol. 2016, 17, 1046–1056. [Google Scholar] [CrossRef] [Green Version]
- Kallinich, T.; Thorwarth, A.; Von Stuckrad, S.-L.; Rösen-Wolff, A.; Luksch, H.; Hundsdoerfer, P.; Minden, K.; Krawitz, P.M. Juvenile arthritis caused by a novel FAMIN (LACC1) mutation in two children with systemic and extended oligoarticular course. Pediatr. Rheumatol. Online J. 2016, 14, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabionet, R.; Remesal, A.; Mensa-Vilaró, A.; Murías, S.; Alcobendas, R.; González-Roca, E.; Ruiz-Ortiz, E.; Antón, J.; Iglesias, E.; Modesto, C. Biallelic loss-of-function LACC1/FAMIN mutations presenting as rheumatoid factor-negative polyarticular juvenile idiopathic arthritis. Sci. Rep. 2019, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Karacan, I.; Uğurlu, S.; Şahin, S.; Everest, E.; Kasapçopur, Ö.; Tolun, A.; Özdoğan, H.; Turanlı, E.T.; Tolun, A. LACC1 Gene Defects in Familial Form of Juvenile Arthritis. J. Rheumatol. 2018, 45, 726–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakil, S.M.; Monies, D.M.; Abouelhoda, M.; Altassan, N.A.; Al-Dusery, H.; Naim, E.A.; Al-Younes, B.; Shinwari, J.; Al-Mohanna, F.A.; Meyer, B.F.; et al. Association of a Mutation inLACC1With a Monogenic Form of Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2015, 67, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, A.; Hedl, M.; Yan, J.; Abraham, C. Human LACC1 increases innate receptor-induced responses and a LACC1 disease-risk variant modulates these outcomes. Nat. Commun. 2017, 8, 15614. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, A.M.; Ombrello, M. Using genes to triangulate the pathophysiology of granulomatous autoinflammatory disease: NOD2, PLCG2 and LACC1. Int. Immunol. 2018, 30, 205–213. [Google Scholar] [CrossRef]
- Skon-Hegg, C.; Zhang, J.; Wu, X.; Sagolla, M.; Ota, N.; Wuster, A.; Tom, J.; Doran, E.; Ramamoorthi, N.; Caplazi, P.; et al. LACC1 Regulates TNF and IL-17 in Mouse Models of Arthritis and Inflammation. J. Immunol. 2019, 202, 183–193. [Google Scholar] [CrossRef]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Irwanto, A.; Fu, X.; Yu, G.; Yu, Y.; Sun, Y.; Wang, C.; Wang, Z.; Okada, Y.; Low, H.; et al. Discovery of six new susceptibility loci and analysis of pleiotropic effects in leprosy. Nat. Genet. 2015, 47, 267. [Google Scholar] [CrossRef]
- Takeuchi, M.; Mizuki, N.; Meguro, A.; Ombrello, M.J.; Kirino, Y.; Satorius, C.; Le, J.; Blake, M.; Erer, B.; Kawagoe, T. Dense genotyping of immune-related loci implicates host responses to microbial exposure in Behçet’s disease susceptibility. Nature Genet. 2017, 49, 438. [Google Scholar] [CrossRef]
- Wang, J.-W.; Howson, J.; Haller, E.; Kerr, W.G. Identification of a Novel Lipopolysaccharide-Inducible Gene with Key Features of Both a Kinase Anchor Proteins and chs1/beige Proteins. J. Immunol. 2001, 166, 4586–4595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, B.; Fritz, J.M.; Su, H.C.; Uzel, G.; Jordan, M.B.; Lenardo, M.J. CHAI and LATAIE: New genetic diseases of CTLA-4 checkpoint insufficiency. Blood J. Am. Soc. Hematol. 2016, 128, 1037–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alegre, M.-L.; Frauwirth, K.A.; Thompson, C.B. T-cell regulation by CD28 and CTLA-4. Nat. Rev. Immunol. 2001, 1, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Van Der Merwe, P.A.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. CD80 (B7-1) Binds Both CD28 and CTLA-4 with a Low Affinity and Very Fast Kinetics. J. Exp. Med. 1997, 185, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Ostrov, D.A.; Shi, W.; Schwartz, J.C.; Almo, S.C.; Nathenson, S.G. Structure of Murine CTLA-4 and Its Role in Modulating T Cell Responsiveness. Science 2000, 290, 816–819. [Google Scholar] [CrossRef]
- Cribbs, A.P.; Kennedy, A.; Penn, H.; Read, J.E.; Amjadi, P.; Green, P.; Syed, K.; Manka, S.W.; Brennan, F.M.; Gregory, B.; et al. Treg Cell Function in Rheumatoid Arthritis Is Compromised by CTLA-4 Promoter Methylation Resulting in a Failure to Activate the Indoleamine 2,3-Dioxygenase Pathway. Arthritis Rheumatol. 2014, 66, 2344–2354. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, H.; Guan, H.; Liu, H. Study of the association between CD28/CTLA-4 expression and disease activity in juvenile idiopathic arthritis. Exp. Ther. Med. 2015, 9, 1733–1738. [Google Scholar] [CrossRef] [Green Version]
- Azizi, G.; Kiaee, F.; Hedayat, E.; Yazdani, R.; Dolatshahi, E.; Alinia, T.; Sharifi, L.; Mohammadi, H.; Kavosi, H.; Jadidi-Niaragh, F.; et al. Rheumatologic complications in a cohort of 227 patients with common variable immunodeficiency. Scand. J. Immunol. 2018, 87, e12663. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Herrera, G.; Tampella, G.; Pan-Hammarström, Q.; Herholz, P.; Trujillo-Vargas, C.M.; Phadwal, K.; Simon, A.K.; Moutschen, M.; Etzioni, A.; Mory, A.; et al. Deleterious Mutations in LRBA Are Associated with a Syndrome of Immune Deficiency and Autoimmunity. Am. J. Hum. Genet. 2012, 90, 986–1001. [Google Scholar] [CrossRef] [Green Version]
- Oz, R.S.; Tesher, M.S. Arthritis in children with LRBA deficiency—Case report and literature review. Pediatr. Rheumatol. Online J. 2019, 17, 1–6. [Google Scholar] [CrossRef]
- Gámez-Díaz, L.; Neumann, J.; Jäger, F.; Proietti, M.; Felber, F.; Soulas-Sprauel, P.; Perruzza, L.; Grassi, F.; Kögl, T.; Aichele, P.; et al. Immunological phenotype of the murine Lrba knockout. Immunol. Cell Biol. 2017, 95, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Kashiwada, M.; Cassel, S.L.; Colgan, J.D.; Rothman, P.B. NFIL3/E4BP4 controls type 2 T helper cell cytokine expression. EMBO J. 2011, 30, 2071–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Matsuoka, K.; Sheikh, S.Z.; Elloumi, H.Z.; Kamada, N.; Hisamatsu, T.; Hansen, J.J.; Doty, K.R.; Pope, S.D.; Smale, S.T.; et al. NFIL3 Is a Regulator of IL-12 p40 in Macrophages and Mucosal Immunity. J. Immunol. 2011, 186, 4649–4655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlenner, S.; Pasciuto, E.; Lagou, V.; Burton, O.; Prezzemolo, T.; Junius, S.; Roca, C.P.; Seillet, C.; Louis, C.; Dooley, J.; et al. NFIL3 mutations alter immune homeostasis and sensitise for arthritis pathology. Ann. Rheum. Dis. 2018, 78, 342–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmann, J.; Callebaut, I.; Raposo, G.; Certain, S.; Bacq, D.; Dumont, C.; Lambert, N.; Ouachée-Chardin, M.; Chedeville, G.; Tamary, H.; et al. Munc13-4 Is Essential for Cytolytic Granules Fusion and Is Mutated in a Form of Familial Hemophagocytic Lymphohistiocytosis (FHL3). Cell 2003, 115, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Schulert, G.S.; Zhang, M.; Husami, A.; Fall, N.; Brunner, H.; Zhang, K.; Cron, R.Q.; Grom, A.A. Brief Report: Novel UNC13D Intronic Variant Disrupting an NF-κB Enhancer in a Patient With Recurrent Macrophage Activation Syndrome and Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2018, 70, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Biroschak, J.; Glass, D.N.; Thompson, S.D.; Finkel, T.; Passo, M.H.; Binstadt, B.A.; Filipovich, A.; Grom, A.A. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis is associated with MUNC13-4 polymorphisms. Arthritis Rheum. 2008, 58, 2892–2896. [Google Scholar] [CrossRef] [Green Version]
- Hazen, M.M.; Woodward, A.L.; Hofmann, I.; Degar, B.A.; Grom, A.; Filipovich, A.H.; Binstadt, B.A. Mutations of the hemophagocytic lymphohistiocytosis–associated geneUNC13D in a patient with systemic juvenile idiopathic arthritis. Arthritis Rheum. 2008, 58, 567–570. [Google Scholar] [CrossRef]
- Moncrieffe, H.; Prahalad, S.; Thompson, S.D. Genetics of juvenile idiopathic arthritis: New tools bring new approaches. Curr. Opin. Rheumatol. 2014, 26, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Savolainen, A.; Saila, H.; Kotaniemi, K.; Kaipiainen-Seppan, O.; Leirisalo-Repo, M.; Aho, K. Magnitude of the genetic component in juvenile idiopathic arthritis. Ann. Rheum. Dis. 2000, 59, 1001. [Google Scholar] [CrossRef] [Green Version]
- Hinks, A.; Registry, B.C.J.; Cobb, J.; Marion, M.C.; Prahalad, S.; Sudman, M.; Bowes, J.; Martin, P.; Comeau, M.E.; Sajuthi, S.; et al. Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat. Genet. 2013, 45, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Førre, Ä.; Smerdel, A. Genetic epidemiology of juvenile idiopathic arthritis. Scand. J. Rheumatol. 2002, 31, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Solberg, O.D.; Mack, S.J.; Lancaster, A.K.; Single, R.M.; Tsai, Y.; Sanchez-Mazas, A.; Thomson, G. Balancing selection and heterogeneity across the classical human leukocyte antigen loci: A meta-analytic review of 497 population studies. Hum. Immunol. 2008, 69, 443–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaiz-Villena, A.; Gomez-Reino, J.J.; Gamir, M.L.; Regueiro, J.R.; Vicario, J.L.; Góamez-Reino, F.J.; Alonso, A.; Fernandez-Dapica, M.P.; Irigoyen, M.V.; Mateo, I.; et al. Dr, C4, and BF allotypes in juvenile rheumatoid arthritis. Arthritis Rheum. 1984, 27, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.J.; Burman, S.J.; Laurent, M.R.; Briggs, D.; Venning, H.E.; Leak, A.M.; Bedford, P.A.; Ansell, B.M. Genetic susceptibility to early onset pauciarticular juvenile chronic arthritis: A study of HLA and complement markers in 158 British patients. Ann. Rheum. Dis. 1986, 45, 464–474. [Google Scholar] [CrossRef] [Green Version]
- Vicario, J.L.; Martinez-Laso, J.; Gomez-Reino, J.J.; Gomez-Reino, F.J.; Regueiro, J.R.; Corell, A.; Segurado, O.G.; Arnaiz-Villena, A. Both HLA class II and class III DNA polymorphisms are linked to juvenile rheumatoid arthritis susceptibility. Clin. Immunol. Immunopathol. 1990, 56, 22–28. [Google Scholar] [CrossRef]
- De Silvestri, A.; Capittini, C.; Poddighe, D.; Marseglia, G.L.; Mascaretti, L.; Bevilacqua, E.; Scotti, V.; Rebuffi, C.; Pasi, A.; Martinetti, M.; et al. HLA-DRB1 alleles and juvenile idiopathic arthritis: Diagnostic clues emerging from a meta-analysis. Autoimmun. Rev. 2017, 16, 1230–1236. [Google Scholar] [CrossRef]
- Hollenbach, J.A.; Thompson, S.D.; Bugawan, T.L.; Ryan, M.; Sudman, M.; Marion, M.; Langefeld, C.D.; Thomson, G.; Erlich, H.A.; Glass, D.N. Juvenile idiopathic arthritis and HLA Class I and Class II interactions and age-at-onset effects. Arthritis Rheum. 2010, 62, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Paul, C.; Schoenwald, U.; Truckenbrodt, H.; Bettinotti, M.; Brünnler, G.; Keller, E.; Nevinny-Stickel, C.; Yao, Z.; Albert, E.D. HLA-DP/DR interaction in early onset pauciarticular juvenile chronic arthritis. Immunogenetics 1993, 37, 442–448. [Google Scholar] [CrossRef]
- Hinks, A.; Bowes, J.; Cobb, J.; Ainsworth, H.C.; Marion, M.C.; Comeau, M.E.; Sudman, M.; Han, B.; Becker, M.L.; Bohnsack, J.F.; et al. Fine-mapping the MHC locus in juvenile idiopathic arthritis (JIA) reveals genetic heterogeneity corresponding to distinct adult inflammatory arthritic diseases. Ann. Rheum. Dis. 2016, 76, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Vehe, R.K.; Begovich, A.B.; Nepom, B.S. HLA susceptibility genes in rheumatoid factor positive juvenile rheumatoid arthritis. J. Rheumatol. Suppl. 1990, 26, 11–15. [Google Scholar] [PubMed]
- Barron, K.S.; Silverman, E.D.; Gonzales, J.C.; Owerbach, D.; Reveille, J.D. DNA analysis of HLA-DR, DQ, and DP alleles in children with polyarticular juvenile rheumatoid arthritis. J. Rheumatol. 1992, 1, 1611–1616. [Google Scholar]
- Thomson, W.; Barrett, J.H.; Donn, R.; Pepper, L.; Kennedy, L.J.; Ollier, W.E.R.; Silman, A.J.S.; Woo, P.; Southwood, T. Juvenile idiopathic arthritis classified by the ILAR criteria: HLA associations in UK patients. Rheumatology 2002, 41, 1183–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prahalad, S.; Thompson, S.D.; Conneely, K.N.; Jiang, Y.; Leong, T.; Prozonic, J.; Brown, M.R.; Ponder, L.A.; Angeles-Han, S.T.; Vogler, L.B.; et al. Hierarchy of risk of childhood-onset rheumatoid arthritis conferred by HLA-DRB1 alleles encoding the shared epitope. Arthritis Rheum. 2012, 64, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Hisa, K.; Yanagimachi, M.; Naruto, T.; Miyamae, T.; Kikuchi, M.; Hara, R.; Imagawa, T.; Yokota, S.; Mori, M. PADI4 and the HLA-DRB1 shared epitope in juvenile idiopathic arthritis. PLoS ONE 2017, 12, e0171961. [Google Scholar] [CrossRef] [PubMed]
- Hinks, A.; Marion, M.C.; Cobb, J.; Comeau, M.E.; Sudman, M.; Ainsworth, H.C.; Bowes, J.; Becker, M.L.; Bohnsack, J.F.; Haas, J.-P.; et al. Brief Report: The Genetic Profile of Rheumatoid Factor–Positive Polyarticular Juvenile Idiopathic Arthritis Resembles That of Adult Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 957–962. [Google Scholar] [CrossRef] [PubMed]
- De Bakker, P.I.; McVean, G.; Sabeti, P.C.; Miretti, M.M.; Green, T.; Marchini, J.; Ke, X.; Monsuur, A.J.; Whittaker, P.; Delgado, M.; et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 2006, 38, 1166–1172. [Google Scholar] [CrossRef]
- Mahmud, S.A.; Binstadt, B.A. Autoantibodies in the Pathogenesis, Diagnosis, and Prognosis of Juvenile Idiopathic Arthritis. Front. Immunol. 2019, 9, 3168. [Google Scholar] [CrossRef] [Green Version]
- Ombrello, M.; Remmers, E.F.; Tachmazidou, I.; Grom, A.; Foell, D.; Haas, J.-P.; Martini, A.; Gattorno, M.; Özen, S.; Prahalad, S.; et al. HLA-DRB1*11 and variants of the MHC class II locus are strong risk factors for systemic juvenile idiopathic arthritis. Proc. Natl. Acad. Sci. USA 2015, 112, 15970–15975. [Google Scholar] [CrossRef] [Green Version]
- Stanevicha, V.; Eglite, J.; Zavadska, D.; Sochnevs, A.; Lazareva, A.; Guseinova, D.; Šantere, R.; Gardovska, D. HLA B27 allele types in homogeneous groups of juvenile idiopathic arthritis patients in Latvia. Pediatr. Rheumatol. Online J. 2010, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.; Phatak, S.; Yadav, A.; Bajpai, P.; Aggarwal, A. HLA B27 typing in 511 children with juvenile idiopathic arthritis from India. Rheumatol. Int. 2016, 36, 1407–1411. [Google Scholar] [CrossRef] [PubMed]
- Kavadichanda, C.; Seth, G.; Kumar, G.; Gulati, R.; Negi, V.S. Clinical correlates of HLA-B*27 and its subtypes in enthesitis-related arthritis variant of juvenile idiopathic arthritis in south Indian Tamil patients. Int. J. Rheum. Dis. 2019, 22, 1289–1296. [Google Scholar] [CrossRef]
- Shih, Y.-J.; Yang, Y.-H.; Lin, C.-Y.; Chang, C.-L.; Chiang, B.-L. Enthesitis-related arthritis is the most common category of juvenile idiopathic arthritis in Taiwan and presents persistent active disease. Pediatr. Rheumatol. Online J. 2019, 17, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, R.; Agnihotry, S.; Aggarwal, R.; Bajpai, P.; Aggarwal, A. HLA-B27 subtypes in enthesitis-related arthritis category of juvenile idiopathic arthritis and ankylosing spondylitis in northern India. Clin. Exp. Rheumatol. 2015, 33, 931–935. [Google Scholar] [PubMed]
- Eyre, S.; Bowes, J.; Diogo, D.; Lee, A.; Barton, A.; Martin, P.; Zhernakova, A.; Stahl, E.; Viatte, S.; McAllister, K.; et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 2012, 44, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Hinks, A.; Barton, A.; Shephard, N.; Eyre, S.; Bowes, J.; Cargill, M.; Wang, E.; Ke, X.; Kennedy, G.C.; John, S.; et al. Identification of a novel susceptibility locus for juvenile idiopathic arthritis by genome-wide association analysis. Arthritis Rheum. 2009, 60, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T.H.; Li, J.; Wei, Z.; Wang, W.; Zhang, H.; Behrens, E.M.; Reuschel, E.L.; Limou, S.; Wise, C.; Punaro, M.; et al. Variants in CXCR4 associate with juvenile idiopathic arthritis susceptibility. BMC Med. Genet. 2016, 17, 24. [Google Scholar] [CrossRef] [Green Version]
- Hinks, A.; Barton, A.; John, S.; Bruce, I.; Hawkins, C.; Griffiths, C.E.M.; Donn, R.; Thomson, W.; Silman, A.; Worthington, J. Association between thePTPN22 gene and rheumatoid arthritis and juvenile idiopathic arthritis in a UK population: Further support thatPTPN22 is an autoimmunity gene. Arthritis Rheum. 2005, 52, 1694–1699. [Google Scholar] [CrossRef]
- Wiede, F.; Shields, B.J.; Chew, S.H.; Kyparissoudis, K.; Van Vliet, C.; Galic, S.; Tremblay, M.L.; Russell, S.M.; Godfrey, D.I.; Tiganis, T. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J. Clin. Investig. 2011, 121, 4758–4774. [Google Scholar] [CrossRef] [Green Version]
- Chiang, G.G.; Sefton, B.M. Specific Dephosphorylation of the Lck Tyrosine Protein Kinase at Tyr-394 by the SHP-1 Protein-tyrosine Phosphatase. J. Biol. Chem. 2001, 276, 23173–23178. [Google Scholar] [CrossRef] [Green Version]
- Brownlie, R.J.; A Miosge, L.; Vassilakos, D.; Svensson, L.M.; Cope, A.P.; Zamoyska, R. Lack of the Phosphatase PTPN22 Increases Adhesion of Murine Regulatory T Cells to Improve Their Immunosuppressive Function. Sci. Signal. 2012, 5, ra87. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Martin, F.; Huang, G.; Tumas, D.; Diehl, L.; Chan, A.C. PEST Domain-Enriched Tyrosine Phosphatase (PEP) Regulation of Effector/Memory T Cells. Science 2004, 303, 685–689. [Google Scholar] [CrossRef]
- Salmond, R.J.; Brownlie, R.J.; Zamoyska, R. Multifunctional roles of the autoimmune disease-associated tyrosine phosphatase PTPN22 in regulating T cell homeostasis. Cell Cycle 2015, 14, 705–711. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.-H.; Kim, H.-O.; Ju, Y.-J.; Kye, Y.-C.; Lee, G.-W.; Lee, S.-W.; Yun, C.-H.; Bottini, N.; Webster, K.; Goodnow, C.C.; et al. CD45-mediated control of TCR tuning in naïve and memory CD8+ T cells. Nat. Commun. 2016, 7, 13373. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.R.; Li, J.; Zhao, S.D.; Bradfield, J.P.; Mentch, F.D.; Maggadottir, S.M.; Hou, C.; Abrams, D.J.; Chang, D.; Gao, F.; et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med. 2015, 21, 1018–1027. [Google Scholar] [CrossRef]
- Thompson, S.D.; Sudman, M.; Ramos, P.S.; Marion, M.C.; Ryan, M.; Tsoras, M.; Weiler, T.; Wagner, M.; Keddache, M.; Haas, J.-P.; et al. The susceptibility loci juvenile idiopathic arthritis shares with other autoimmune diseases extend to PTPN2, COG6, and ANGPT1. Arthritis Rheum. 2010, 62, 3265–3276. [Google Scholar] [CrossRef]
- Thompson, S.D.; Marion, M.C.; Sudman, M.; Ryan, M.; Tsoras, M.; Howard, T.D.; Barnes, M.G.; Ramos, P.S.; Thomson, W.; Hinks, A.; et al. Genome-wide association analysis of juvenile idiopathic arthritis identifies a new susceptibility locus at chromosomal region 3q13. Arthritis Rheum. 2012, 64, 2781–2791. [Google Scholar] [CrossRef] [Green Version]
- Cortes, A.; A Brown, M. Promise and pitfalls of the Immunochip. Arthritis Res. Ther. 2011, 13, 101. [Google Scholar] [CrossRef] [Green Version]
- Ba, L.A.M.; Ma, M.C.M.; Sudman, M.; Ma, M.E.C.; Becker, M.L.; Bohnsack, J.F.; Fingerlin, T.E.; Griffin, T.A.; Haas, J.P.; Lovell, D.J.; et al. Genome-Wide Association Meta-Analysis Reveals Novel Juvenile Idiopathic Arthritis Susceptibility Loci. Arthritis Rheumatol. 2017, 69, 2222–2232. [Google Scholar] [CrossRef] [Green Version]
- Redmond, W.L.; Ruby, C.E.; Weinberg, A.D. The Role of OX40-Mediated Co-stimulation in T-Cell Activation and Survival. Crit. Rev. Immunol. 2009, 29, 187–201. [Google Scholar] [CrossRef]
- Zervou, M.I.; Dimopoulou, D.G.; Eliopoulos, E.; Trachana, M.; Pratsidou-Gkertsi, P.; Andreou, A.; Sidiropoulos, P.; Spandidos, D.A.; Garyfallos, A.; Goulielmos, G.N. Τhe genetics of juvenile idiopathic arthritis: Searching for new susceptibility loci. Mol. Med. Rep. 2017, 16, 8793–8798. [Google Scholar] [CrossRef] [Green Version]
- Couturier, N.; Bucciarelli, F.; Nurtdinov, R.N.; Debouverie, M.; Lebrun, C.; Defer, G.; Moreau, T.; Confavreux, C.; Vukusic, S.; Cournu-Rebeix, I.; et al. Tyrosine kinase 2 variant influences T lymphocyte polarization and multiple sclerosis susceptibility. Brain 2011, 134, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Omar, A.; Abo-Elyoun, I.; Hussein, H.; Nabih, M.; Atwa, H.; Gad, S.; Emad, Y. Anti-cyclic citrullinated peptide (anti-CCP) antibody in juvenile idiopathic arthritis (JIA): Correlations with disease activity and severity of joint damage (a multicenter trial). Jt. Bone Spine 2013, 80, 38–43. [Google Scholar] [CrossRef]
- Prahalad, S.; Conneely, K.N.; Jiang, Y.; Sudman, M.; Wallace, C.A.; Brown, M.R.; Ponder, L.A.; Rohani-Pichavant, M.; Zwick, M.E.; Cutler, D.J.; et al. Susceptibility to childhood-onset rheumatoid arthritis: Investigation of a weighted genetic risk score that integrates cumulative effects of variants at five genetic loci. Arthritis Rheum. 2013, 65, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Fishman, D.; Faulds, G.; Jeffery, R.; Mohamed-Ali, V.; Yudkin, J.S.; Humphries, S.; Woo, P. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J. Clin. Investig. 1998, 102, 1369–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogilvie, E.M.; Fife, M.S.; Thompson, S.D.; Twine, N.; Tsoras, M.; Moroldo, M.; Fisher, S.A.; Lewis, C.M.; Prieur, A.-M.; Glass, D.N.; et al. The −174G allele of the interleukin-6 gene confers susceptibility to systemic arthritis in children: A multicenter study using simplex and multiplex juvenile idiopathic arthritis families. Arthritis Rheum. 2003, 48, 3202–3206. [Google Scholar] [CrossRef] [PubMed]
- Donn, R.P.; Shelley, E.; Ollier, W.E.R.; Thomson, W.; British Paediatric Rheumatology Study Group. A novel 5′-flanking region polymorphism of macrophage migration inhibitory factor is associated with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2001, 44, 1782–1785. [Google Scholar] [CrossRef]
- De Benedetti, F.; Meazza, C.; Vivarelli, M.; Rossi, F.; Pistorio, A.; Lamb, R.; Lunt, M.; Thomson, W.; Ravelli, A.; Donn, R.; et al. Functional and prognostic relevance of the −173 polymorphism of the macrophage migration inhibitory factor gene in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2003, 48, 1398–1407. [Google Scholar] [CrossRef]
- Fife, M.; Gutierrez, A.; Ogilvie, E.M.; Stock, C.J.; Samuel, J.M.; Thomson, W.; Mack, L.F.; Lewis, C.M.; Woo, P. Novel IL10 gene family associations with systemic juvenile idiopathic arthritis. Arthritis Res. Ther. 2006, 8, R148. [Google Scholar] [CrossRef] [Green Version]
- Omoyinmi, E.; Forabosco, P.; Hamaoui, R.; Bryant, A.; Hinks, A.; Ursu, S.; Wedderburn, L.R.; Thomson, W.; Lewis, C.M.; Woo, P. Association of the IL-10 Gene Family Locus on Chromosome 1 with Juvenile Idiopathic Arthritis (JIA). PLoS ONE 2012, 7, e47673. [Google Scholar] [CrossRef]
- Stock, C.J.; Ogilvie, E.M.; Samuel, J.M.; Fife, M.; Lewis, C.M.; Woo, P. Comprehensive association study of genetic variants in the IL-1 gene family in systemic juvenile idiopathic arthritis. Genes Immun. 2008, 9, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinks, A.; Martin, P.; Thompson, S.D.; Sudman, M.; Stock, C.J.; Thomson, W.; Day, T.G.; Packham, J.; Ramanan, A.V.; Donn, R. Autoinflammatory gene polymorphisms and susceptibility to UK juvenile idiopathic arthritis. Pediatr. Rheumatol. Online J. 2013, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheibel, I.; Veit, T.; Neves, A.G.; Souza, L.; Prezzi, S.; Machado, S.; Kohem, C.; Icarelli, M.; Xavier, R.M.; Brenol, J.C.; et al. Differential CCR5Δ32 allelic frequencies in juvenile idiopathic arthritis subtypes: Evidence for different regulatory roles of CCR5 in rheumatological diseases. Scand. J. Rheumatol. 2008, 37, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Thomson, W.; British Society of Paediatric and Adolescent Rheumatology; Ogilvie, E.M.; Donn, R. Positive association ofSLC26A2 gene polymorphisms with susceptibility to systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2007, 56, 1286–1291. [Google Scholar] [CrossRef] [PubMed]
- Bukulmez, H.; Fife, M.; Tsoras, M.; Thompson, S.D.; Twine, N.A.; Woo, P.; Olson, J.M.; Elston, R.C.; Glass, D.N.; Colbert, R.A. Tapasin gene polymorphism in systemic onset juvenile rheumatoid arthritis: A family-based case–control study. Arthritis Res. Ther. 2005, 7, R285–R290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ombrello, M.; Arthur, V.L.; Remmers, E.F.; Hinks, A.; Tachmazidou, I.; A Grom, A.; Foell, D.; Martini, A.; Gattorno, M.; Özen, S.; et al. Genetic architecture distinguishes systemic juvenile idiopathic arthritis from other forms of juvenile idiopathic arthritis: Clinical and therapeutic implications. Ann. Rheum. Dis. 2016, 76, 906–913. [Google Scholar] [CrossRef]
- Bharti, S.; Handrow-Metzmacher, H.; Zickenheiner, S.; Zeitvogel, A.; Baumann, R.; Starzinski-Powitz, A. Novel Membrane Protein shrew-1 Targets to Cadherin-Mediated Junctions in Polarized Epithelial Cells. Mol. Biol. Cell 2004, 15, 397–406. [Google Scholar] [CrossRef]
- Ruperto, N.; Lovell, D.J.; Quartier, P.; Paz, E.; Rubio-Pérez, N.; Silva, C.A.; Abud-Mendoza, C.; Burgos-Vargas, R.; Gerloni, V.; Melo-Gomes, J.A.; et al. Abatacept in children with juvenile idiopathic arthritis: A randomised, double-blind, placebo-controlled withdrawal trial. Lancet 2008, 372, 383–391. [Google Scholar] [CrossRef]
- Record, J.L.; Beukelman, T.; Cron, R.Q.; Hasegawa, M.; Segawa, T.; Maeda, M.; Yoshida, T.; Sudo, A. Combination Therapy of Abatacept and Anakinra in Children with Refractory Systemic Juvenile Idiopathic Arthritis: A Retrospective Case Series. J. Rheumatol. 2011, 38, 180–181. [Google Scholar] [CrossRef]
- Arthur, V.L.; Shuldiner, E.; Remmers, E.F.; Hinks, A.; Grom, A.A.; Foell, D.; Martini, A.; Gattorno, M.; Ozen, S.; Prahalad, S.; et al. IL1RN Variation Influences Both Disease Susceptibility and Response to Recombinant Human Interleukin-1 Receptor Antagonist Therapy in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2018, 70, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Mo, A.; Marigorta, U.M.; Gulick, D.; Chan, L.H.K.; Ponder, L.; Jang, S.R.; Prince, J.; Kugathasan, S.; Prahalad, S.; Gibson, G. Disease-specific regulation of gene expression in a comparative analysis of juvenile idiopathic arthritis and inflammatory bowel disease. Genome Med. 2018, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogilvie, E.M.; Khan, A.; Hubank, M.; Kellam, P.; Woo, P. Specific gene expression profiles in systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007, 56, 1954–1965. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.A.; Barnes, M.G.; Ilowite, N.T.; Olson, J.C.; Sherry, D.D.; Gottlieb, B.S.; Aronow, B.J.; Pavlidis, P.; Hinze, C.H.; Thornton, S.; et al. Gene expression signatures in polyarticular juvenile idiopathic arthritis demonstrate disease heterogeneity and offer a molecular classification of disease subsets. Arthritis Rheum. 2009, 60, 2113–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ter Haar, N.M.; Tak, T.; Mokry, M.; Scholman, R.C.; Meerding, J.M.; De Jager, W.; Verwoerd, A.; Foell, D.; Vogl, T.; Roth, J.; et al. Reversal of Sepsis-Like Features of Neutrophils by Interleukin-1 Blockade in Patients With Systemic-Onset Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2018, 70, 943–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.; Henderlight, M.; Do, T.; Yasin, S.; Grom, A.A.; DeLay, M.; Thornton, S.; Schulert, G.S. Neutrophils From Children With Systemic Juvenile Idiopathic Arthritis Exhibit Persistent Proinflammatory Activation Despite Long-Standing Clinically Inactive Disease. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Bleesing, J.; Prada, A.; Siegel, D.M.; Villanueva, J.; Olson, J.; Ilowite, N.T.; Brunner, H.I.; Griffin, T.; Graham, T.B.; Sherry, D.D.; et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor α-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007, 56, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Schulert, G.S.; Fall, N.; Harley, J.B.; Shen, N.; Lovell, D.J.; Thornton, S.; Grom, A.A. Monocyte MicroRNA Expression in Active Systemic Juvenile Idiopathic Arthritis Implicates MicroRNA-125a-5p in Polarized Monocyte Phenotypes. Arthritis Rheumatol. 2016, 68, 2300–2313. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, Y.; Kawada, J.-I.; Kawano, Y.; Torii, Y.; Kawabe, S.; Iwata, N.; Ito, Y. Serum microRNAs as Potential Biomarkers of Juvenile Idiopathic Arthritis. Clin. Rheumatol. 2015, 34, 1705–1712. [Google Scholar] [CrossRef]
- Do, T.; Tan, R.; Bennett, M.F.; Medvedovic, M.; Grom, A.A.; Shen, N.; Thornton, S.; Schulert, G.S. MicroRNA networks associated with active systemic juvenile idiopathic arthritis regulate CD163 expression and anti-inflammatory functions in macrophages through two distinct mechanisms. J. Leukoc. Biol. 2017, 103, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Moncrieffe, H.; Bennett, M.F.; Tsoras, M.; Luyrink, L.K.; Johnson, A.L.; Xu, H.; Dare, J.; Becker, M.L.; Prahalad, S.; Rosenkranz, M.; et al. Transcriptional profiles of JIA patient blood with subsequent poor response to methotrexate. Rheumatology 2017, 56, 1542–1551. [Google Scholar] [CrossRef] [Green Version]
- Schulert, G.S.; Yasin, S.; Carey, B.; Chalk, C.; Do, T.; Schapiro, A.H.; Husami, A.; Watts, A.; Brunner, H.I.; Huggins, J.; et al. Systemic Juvenile Idiopathic Arthritis–Associated Lung Disease: Characterization and Risk Factors. Arthritis Rheumatol. 2019, 71, 1943–1954. [Google Scholar] [CrossRef] [Green Version]
- Leong, J.Y.; Chen, P.; Yeo, J.G.; Ally, F.; Chua, C.; Hazirah, S.N.; Poh, S.L.; Pan, L.; Lai, L.; Lee, E.S.C.; et al. Immunome perturbation is present in patients with juvenile idiopathic arthritis who are in remission and will relapse upon anti-TNFα withdrawal. Ann. Rheum. Dis. 2019, 78, 1712–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spreafico, R.; Rossetti, M.; Whitaker, J.W.; Wang, W.; Lovell, D.J.; Albani, S. Epipolymorphisms associated with the clinical outcome of autoimmune arthritis affect CD4+T cell activation pathways. Proc. Natl. Acad. Sci. USA 2016, 113, 13845–13850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, J.G.; Vervoort, S.J.; Tan, S.; Mijnheer, G.; De Roock, S.; Vastert, S.J.; Nieuwenhuis, E.E.S.; Van Wijk, F.; Prakken, B.J.; Creyghton, M.P.; et al. Inhibition of Super-Enhancer Activity in Autoinflammatory Site-Derived T Cells Reduces Disease-Associated Gene Expression. Cell Rep. 2015, 12, 1986–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, C.H.; Ceuppens, J.L.; Stevens, E.A.M. Different circulating lymphocyte profiles in patients with different subtypes of juvenile idiopathic arthritis. Clin. Exp. Rheumatol. 2002, 20, 239–248. [Google Scholar] [PubMed]
- Stamatoyannopoulos, J. What does our genome encode? Genome Res. 2012, 22, 1602–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquier, A. The complex eukaryotic transcriptome: Unexpected pervasive transcription and novel small RNAs. Nat. Rev. Genet. 2009, 10, 833–844. [Google Scholar] [CrossRef]
- Kapranov, P.; Willingham, A.T.; Gingeras, T.R. Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet. 2007, 8, 413–423. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Zhu, L.; Buck, M.J.; Chen, Y.; Carrier, B.; Liu, T.; Jarvis, J.N. Disease-Associated Single-Nucleotide Polymorphisms From Noncoding Regions in Juvenile Idiopathic Arthritis Are Located Within or Adjacent to Functional Genomic Elements of Human Neutrophils and CD4+ T Cells. Arthritis Rheumatol. 2015, 67, 1966–1977. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Jiang, K.; Webber, K.; Wong, L.P.; Liu, T.; Chen, Y.; Jarvis, J.N. Chromatin landscapes and genetic risk for juvenile idiopathic arthritis. Arthritis Res. Ther. 2017, 19, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, J.A.; Munro, J.E.; Chavez, R.A.; Gordon, L.; Joo, J.E.; Akikusa, J.D.; Allen, R.C.; Ponsonby, A.-L.; Craig, J.M.; Saffery, R. Genome-scale case-control analysis of CD4+ T-cell DNA methylation in juvenile idiopathic arthritis reveals potential targets involved in disease. Clin. Epigenetics 2012, 4, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, R.; Whitaker, J.W.; Boyle, D.L.; Tak, P.P.; Gerlag, D.M.; Wang, W.; Firestein, G.S. DNA Methylome Signature in Synoviocytes from Patients with Early Rheumatoid Arthritis Compared to Synoviocytes from Patients with Longstanding Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 67, 1978–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijhuis, L.; Peeters, J.G.C.; Vastert, S.J.; Van Loosdregt, J. Restoring T Cell Tolerance, Exploring the Potential of Histone Deacetylase Inhibitors for the Treatment of Juvenile Idiopathic Arthritis. Front. Immunol. 2019, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, R.Y.; Clowney, E.J.; Agamy, O.; Kim, M.J.; Zhao, J.; Yamanaka, T.; Pappalardo, Z.; Clarke, S.L.; Wenger, A.M.; Nguyen, L.; et al. Coding exons function as tissue-specific enhancers of nearby genes. Genome Res. 2012, 22, 1059–1068. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Q.; Ding, Y.; Liu, Y.; Zhao, D.; Zhao, K.; Shen, Q.; Liu, X.; Zhu, X.; Li, N.; et al. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat. Immunol. 2016, 17, 806–815. [Google Scholar] [CrossRef] [Green Version]
- Vojinovic, J.; Damjanov, N. HDAC Inhibition in Rheumatoid Arthritis and Juvenile Idiopathic Arthritis. Mol. Med. 2011, 17, 397–403. [Google Scholar] [CrossRef]
- Vojinovic, J.; Damjanov, N.; D’Urzo, C.; Furlan, A.; Susic, G.; Pasic, S.; Iagaru, N.; Stefan, M.; Dinarello, C.A. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2011, 63, 1452–1458. [Google Scholar] [CrossRef]



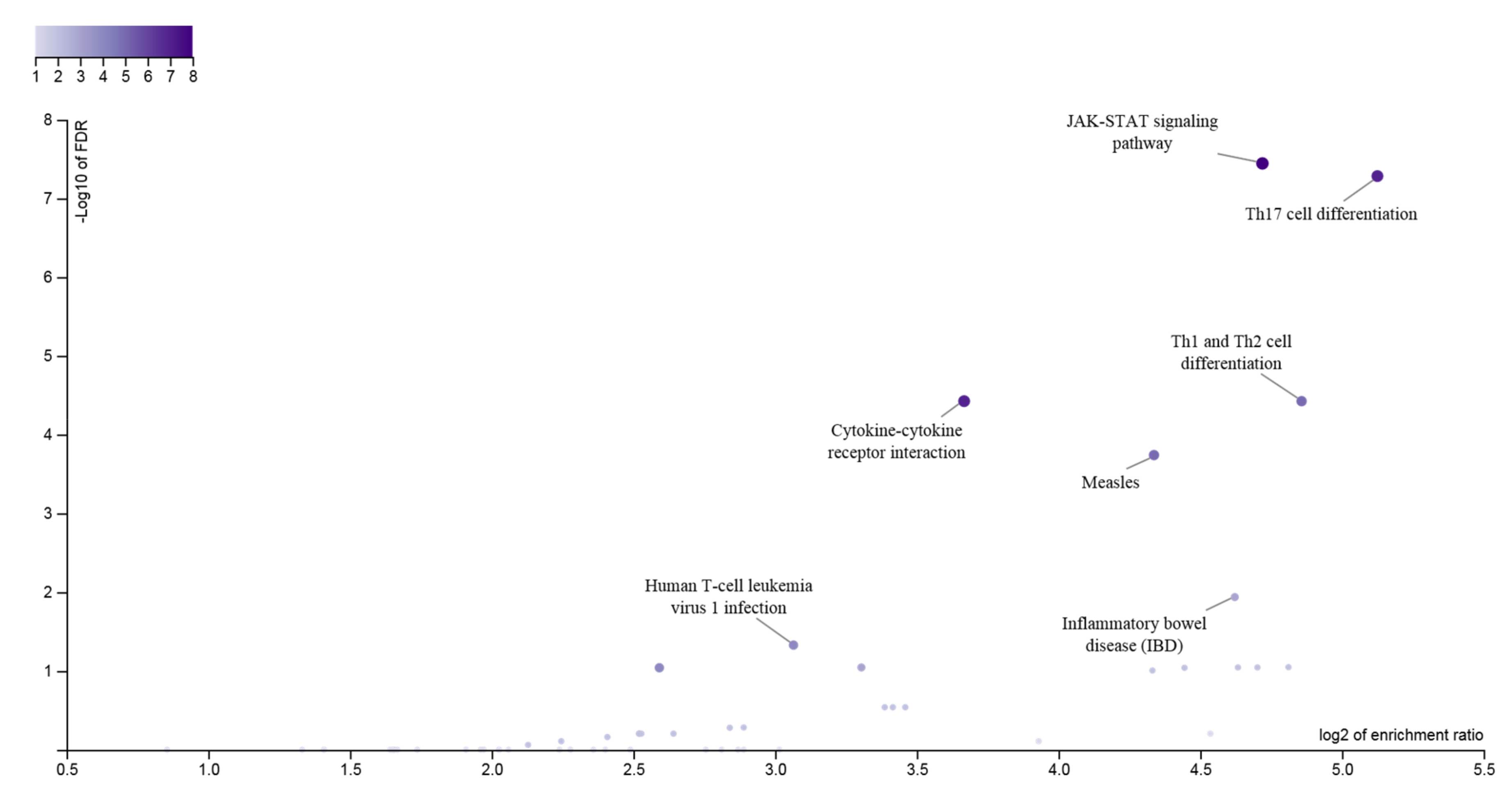{kind=link}
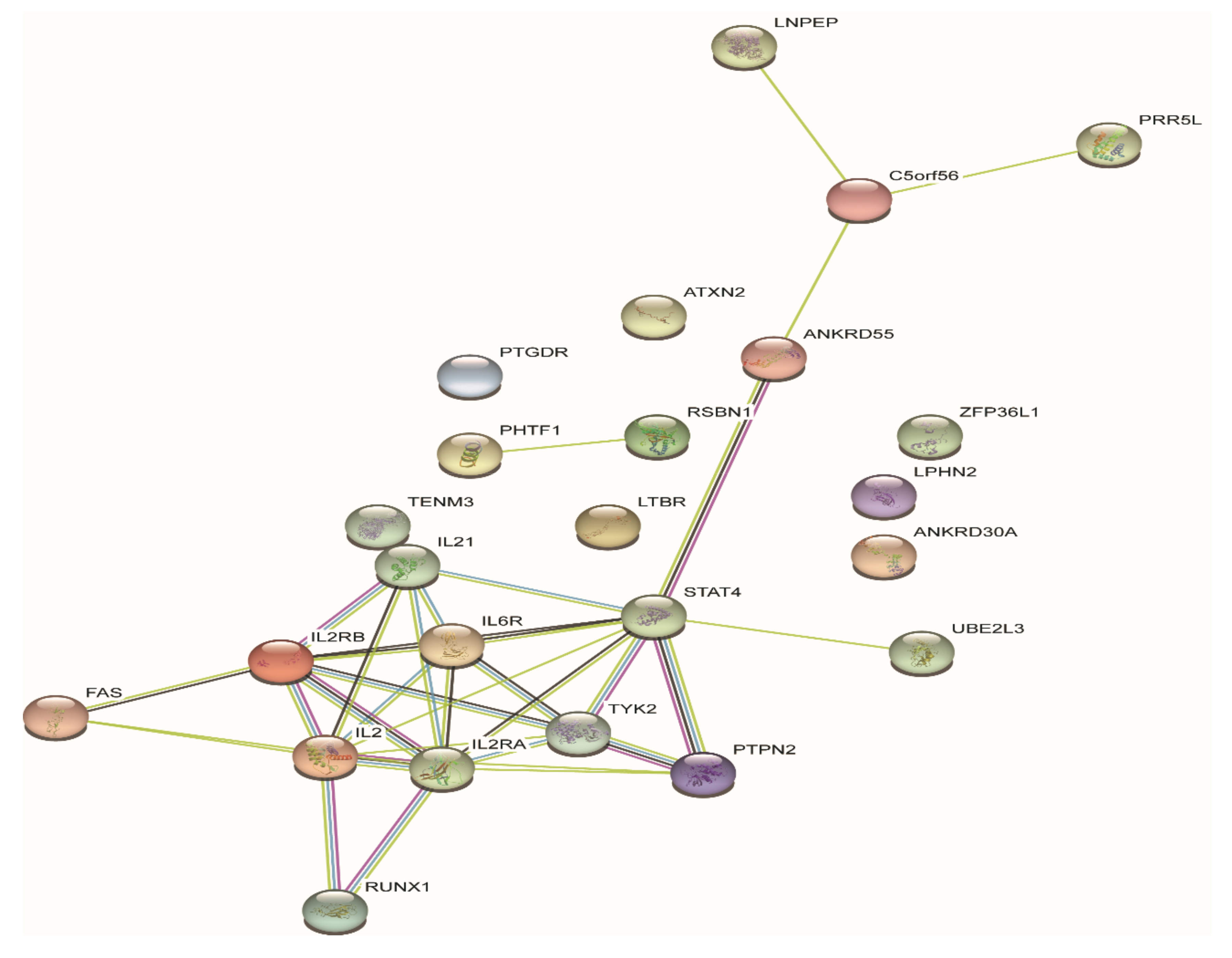{kind=link}
| Genes | Causal Mutations (PMID) | Related Subtype of JIA | Functional Evidence (PMID) | Mechanism |
|---|---|---|---|---|
| LRBA | Oligoarthritis | Lrba−/− mice produce high levels of serum and secretory IgA (28652580). | Defects in peripheral tolerance. | |
| NFIL3 | p.M170I (30552177) | systemic JIA | NFIL3 mutations drive elevated IL-1β (30552177) | Sensitizing for arthritis. Development and rewiring the innate immune system for IL-1 overproduction |
| LACC1 | p.Cys284Arg (27881174) p.Ile254Val (27881174) rs3816311 (27098602) p.Arg414Ter (2917096) p.Ile330del (2917096) p.Cys43Tyrfs*6 (30872671) | systemic JIA | TNF levels were increased in LACC1−/− mice. LACC1 transcripts and protein were upregulated by LPS and other TLR ligands in macrophages and dendritic cells (30510070). | Regulating inflammation. |
| UNCD13 | c.117 + 143A>G (29409136) 753 + 3 [G>A] (18240215) 1579 [C>T] R527W (18240215) | systemic JIA | Munc13-4 was highly expressed in differentiated human NK cells and effector CD8+ T lymphocytes. Munc13-4 expression levels were selectively upregulated upon cytotoxic lymphocyte differentiation (24842371). | Disrupting transcription factor binding. |
| Subtype of JIA | Predisposing Allele | Protective Allele |
|---|---|---|
| Oligoarthritis and polyarthritis RF-negative | A2, DRB1*01,DRB1*08, DRB1*11, DRB1*13,DPB1*02,DPB1*03, DQB1*04, | DRB1*04, DRB1*07,DRB1*15:01 |
| Polyarthritis RF-positive | DRB1*04:01,DRB1*04:05 | |
| Systemic JIA | HLA-DRB1*11 | |
| Enthesitis-related JIA | B*27:04,B*27:05 |
| Index SNPs | Chr | Position | Region | Ref Allele | Risk Allele | Mapped Gene | Risk Allele Frequency | p-Value | Odds Ratio | Associated JIA Subtypes | PMID |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs72632736 | 1 | 4389144 | 1p36.32 | A | G | EEF1DP6-LINC01777 | 3.00E–09 | 2.4 | SYS | 27927641 | |
| rs2066363 | 1 | 81771892 | 1p31.1 | C | T | ADGRL2 | 0.34 | 8.00E–11 | 26301688 | ||
| rs6679677 | 1 | 113761186 | 1p13.2 | C | A | PHTF1-RSBN1 | 0.10 | 3.00E–25 | 1.59 | OLG,PRFN | 23603761 |
| rs6679677 | 1 | 113761186 | 1p13.2 | C | A | PHTF1-RSBN1 | 0.09 | 8.00E–11 | 26301688 | ||
| rs72698115 | 1 | 154406893 | 1q21.3 | A | C | IL6R | 0.1 | 1.00E–08 | 1.36 | OLG,PRFN | 23603761 |
| rs10174238 | 2 | 191108308 | 2q32.3 | G | A | STAT4 | 0.23 | 1.00E–13 | 1.29 | OLG,PRFN | 23603761 |
| rs1479924 | 4 | 122466445 | 4q27 | G | A | IL2-IL21 | 0.71 | 6.00E–11 | 1.27 | OLG,PRFN | 23603761 |
| rs62324212 | 4 | 122639784 | 4q27 | C | A/G | IL21-AS1 | 0.42 | 3.00E–08 | 26301688 | ||
| rs7660520 | 4 | 182824168 | 4q35.1 | G | A/C | TENM3-AC114798.1 | 0.26 | 8.00E–11 | 26301688 | ||
| rs10213692 | 5 | 56146422 | 5q11.2 | T | C/T | ANKRD55 | 0.75 | 3.00E–11 | 1.27 | OLG,PRFN | 23603761 |
| rs7731626 | 5 | 56148856 | 5q11.2 | G | A | ANKRD55 | 0.39 | 1.00E–10 | 26301688 | ||
| rs27293 | 5 | 97021474 | 5q15 | A | G/T | LNPEP | 0.44 | 7.00E–09 | 1.31 | OLG,PRFN | 23603761 |
| rs6894249 | 5 | 132461855 | 5q31.1 | A | G | AC116366.3, C5orf56 | 0.61 | 1.00E–09 | 1.32 | OLG,PRFN | 23603761 |
| rs6946509 | 7 | 22769871 | 7p15.3 | T | A/C | MTCYBP42-AC073072.2 | 0.45 | 3.00E–08 | 1.19 | OLG,PRFN | 23603761 |
| rs7909519 | 10 | 6047878 | 10p15.1 | T | G | IL2RA | 0.89 | 8.00E–10 | 1.39 | OLG,PRFN | 23603761 |
| rs706778 | 10 | 6056986 | 10p15.1 | C | T | IL2RA | 0.41 | 6.00E–09 | 26301688 | ||
| rs7100025 | 10 | 37303610 | 10p11.21 | G | A | LINC00993, ANKRD30A | 0.34 | 8.00E–11 | 26301688 | ||
| rs7069750 | 10 | 89002619 | 10q23.31 | G | C/T | FAS | 0.44 | 3.00E–08 | 1.18 | OLG,PRFN | 23603761 |
| rs7127214 | 11 | 36322143 | 11p13 | C | G/T | PRR5L, AC087277.1 | 0.65 | 2.00E–08 | 1.28 | OLG,PRFN | 23603761 |
| rs10849448 | 12 | 6384185 | 12p13.31 | A | G | LTBR | 0.24 | 5.00E–09 | 1.24 | OLG,PRFN | 23603761 |
| rs7137828 | 12 | 111494996 | 12q24.12 | C | A/T | ATXN2 | 0.49 | 2.00E–09 | 1.20 | OLG,PRFN | 23603761 |
| rs3825568 | 14 | 68793871 | 14q24.1 | C | G/T | ZFP36L1 | 0.56 | 1.00E–08 | 1.30 | OLG,PRFN | 23603761 |
| rs2847293 | 18 | 12782449 | 18p11.21 | A | G/T | AP005482.1-PTPN2 | 0.17 | 1.00E–12 | 1.31 | OLG,PRFN | 23603761 |
| rs34536443 | 19 | 10352442 | 19p13.2 | G | C | TYK2 | 0.95 | 1.00E–10 | 1.79 | OLG,PRFN | 23603761 |
| rs8129030 | 21 | 35340290 | 21q22.12 | T | A/G | RUNX1 | 0.63 | 5.00E–09 | 1.28 | OLG,PRFN | 23603761 |
| rs2266959 | 22 | 21568615 | 22q11.21 | G | T | UBE2L3 | 0.19 | 6.00E–09 | 1.24 | OLG,PRFN | 23603761 |
| rs2284033 | 22 | 37137994 | 22q12.3 | G | A | IL2RB | 0.56 | 2.00E–08 | 1.19 | OLG,PRFN | 23603761 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, X.; Qu, H.; Zhang, S.; Qi, X.; Hakonarson, H.; Xia, Q.; Li, J. The Multi-Omics Architecture of Juvenile Idiopathic Arthritis. Cells 2020, 9, 2301. https://doi.org/10.3390/cells9102301
Hou X, Qu H, Zhang S, Qi X, Hakonarson H, Xia Q, Li J. The Multi-Omics Architecture of Juvenile Idiopathic Arthritis. Cells. 2020; 9(10):2301. https://doi.org/10.3390/cells9102301
Chicago/Turabian StyleHou, Xiaoyuan, Huiqi Qu, Sipeng Zhang, Xiaohui Qi, Hakon Hakonarson, Qianghua Xia, and Jin Li. 2020. "The Multi-Omics Architecture of Juvenile Idiopathic Arthritis" Cells 9, no. 10: 2301. https://doi.org/10.3390/cells9102301
APA StyleHou, X., Qu, H., Zhang, S., Qi, X., Hakonarson, H., Xia, Q., & Li, J. (2020). The Multi-Omics Architecture of Juvenile Idiopathic Arthritis. Cells, 9(10), 2301. https://doi.org/10.3390/cells9102301