Hypothermia Advocates Functional Mitochondria and Alleviates Oxidative Stress to Combat Acetaminophen-Induced Hepatotoxicity



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Hypothermic Conditioning
2.3. Immunofluorescence Imaging
2.4. Measurement of Mitochondrial Membrane Potential
2.5. Isolation of Mitochondrial and Cytosolic Fraction
2.6. Western Blotting
2.7. Real-Time PCR Analysis
2.8. Measurement of Hepatic GSH Content and GSH/GSSG Ratio
2.9. Cell Viability Assay
2.10. Cell Death Analysis
2.11. Statistical Analysis
3. Results
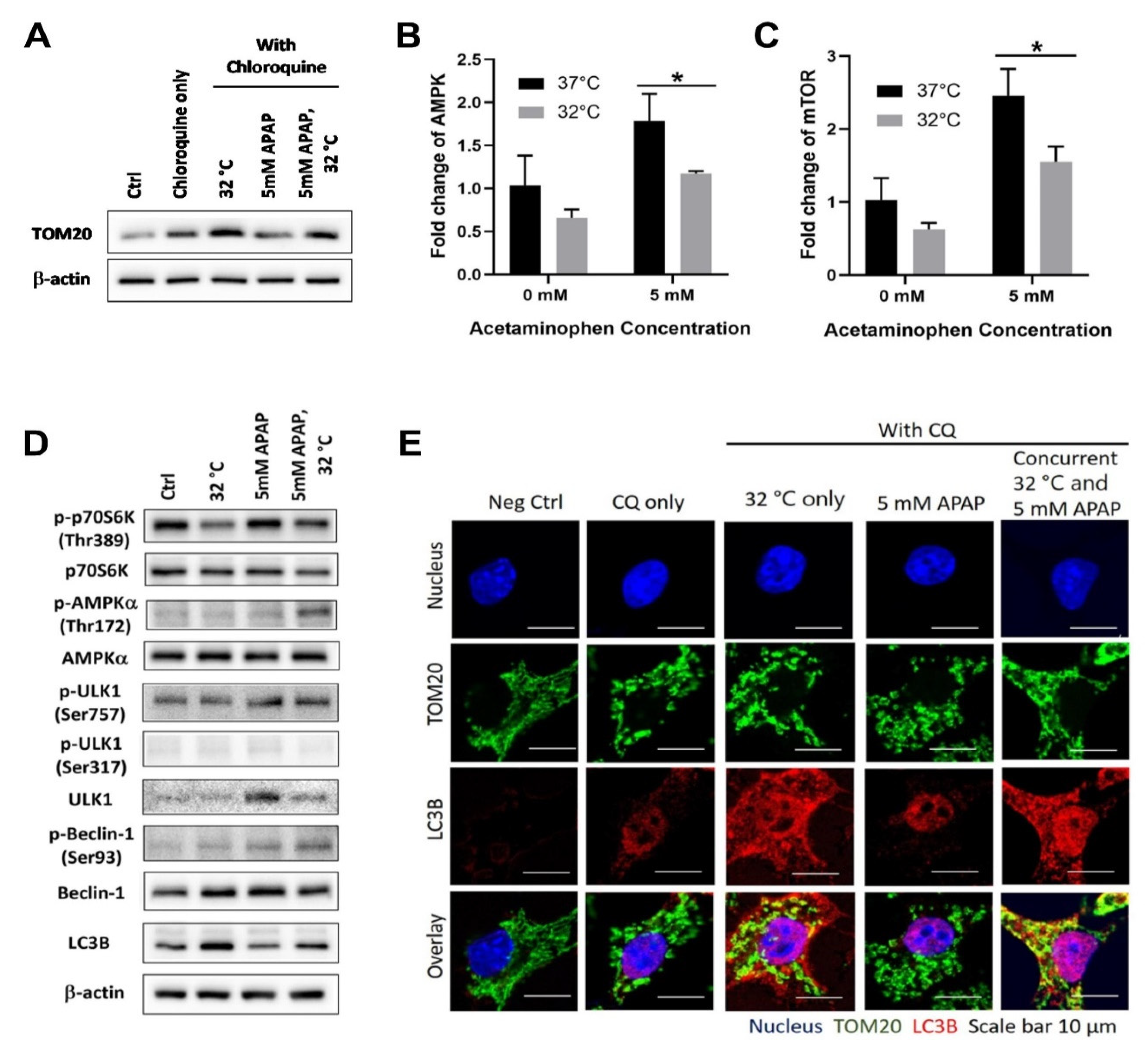
3.1. Moderate Hypothermic (32 °C) Conditioning Promotes ULK1-Independent Mitophagy via AMPKα Activation in the Presence of AIHI
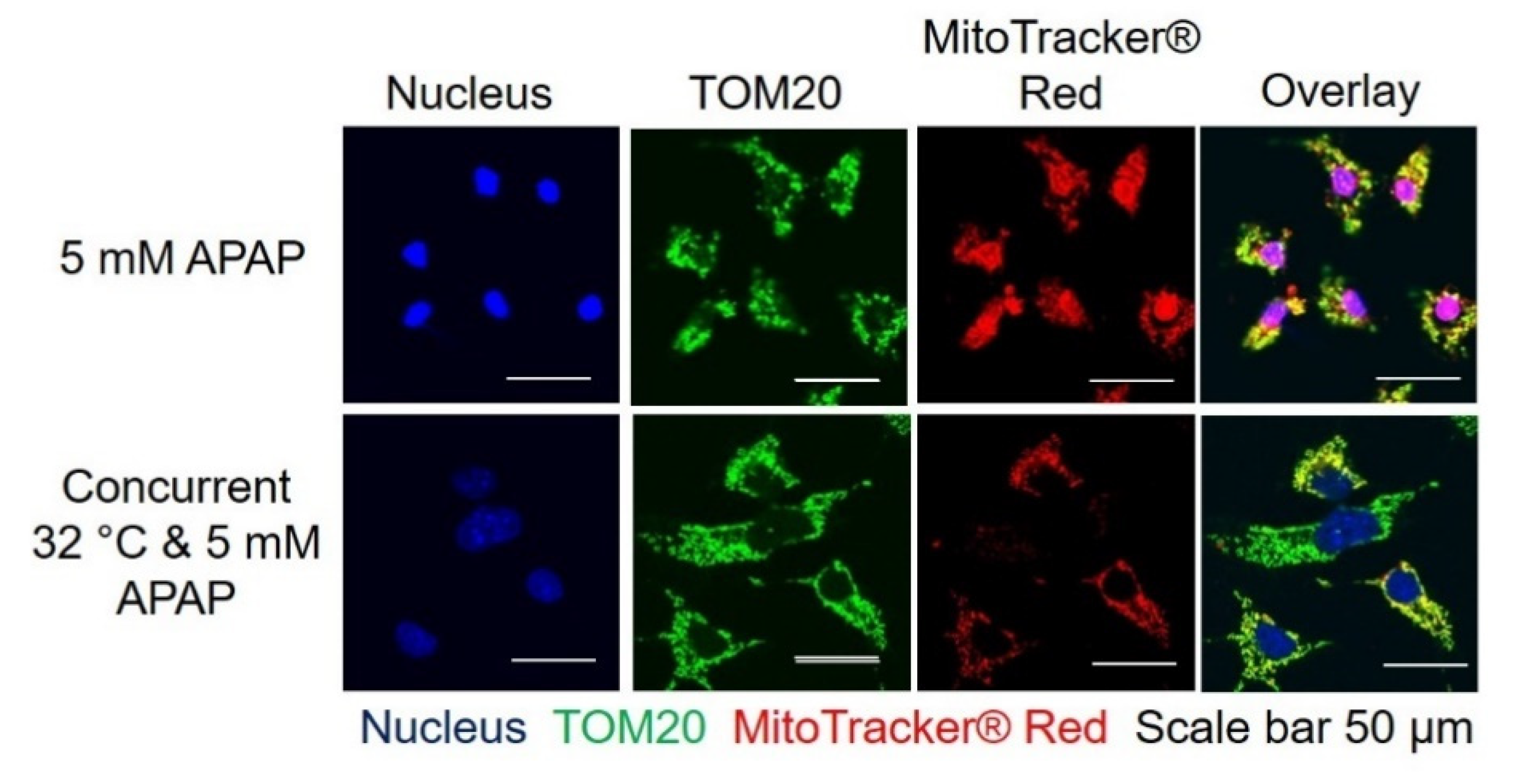
3.2. Moderate Hypothermic (32 °C) Conditioning Fosters Mitochondrial Biogenesis in the Presence of AIHI
3.3. Moderate Hypothermic (32 °C) Conditioning Renders a Concomitant Interplay of Mitophagy and Mitochondrial Biogenesis During AIHI
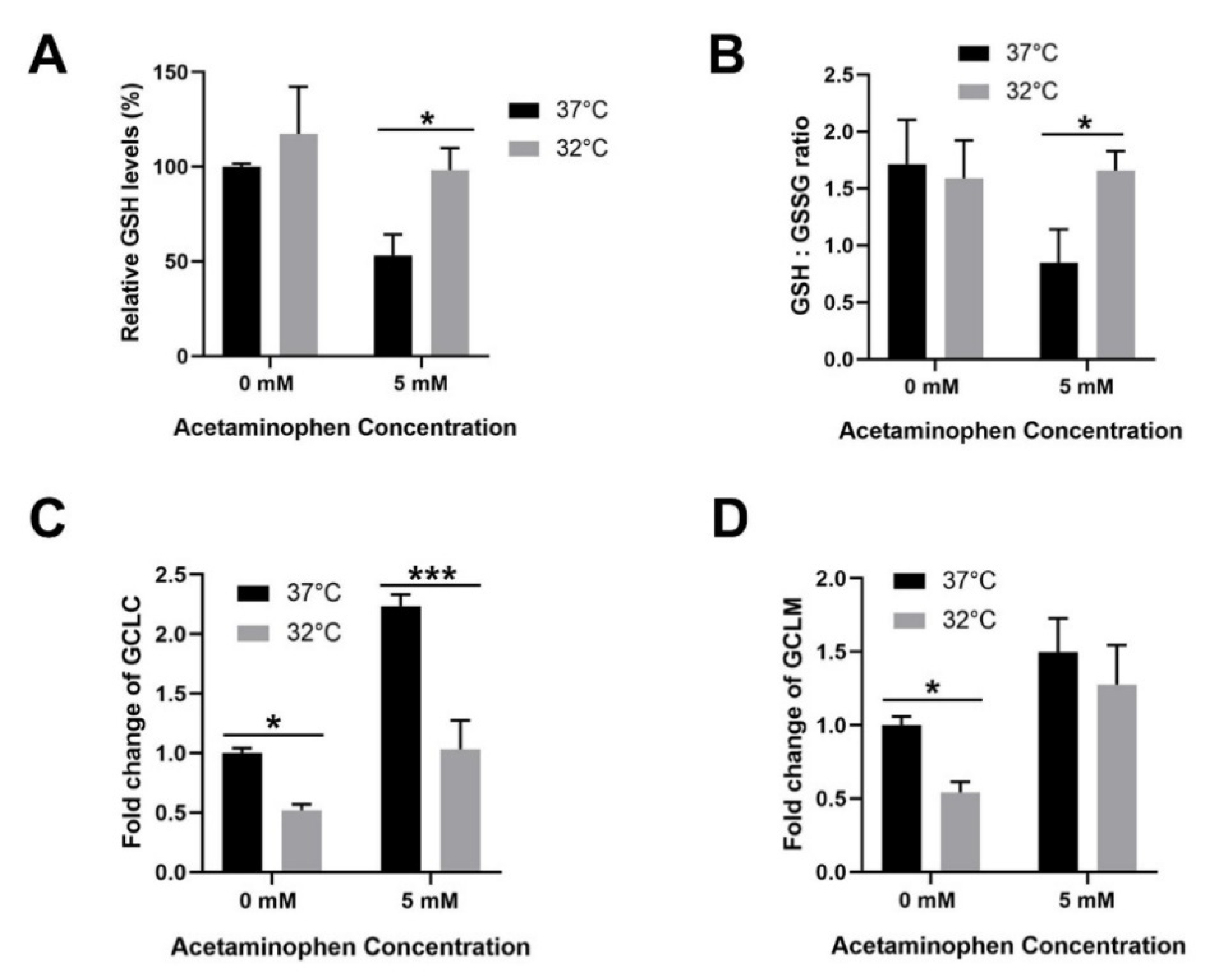
3.4. Moderate Hypothermic (32 °C) Conditioning Facilitates Effective Hepatic GSH Recycling to Evade a Depletion of ROS Scavenger in AIHI
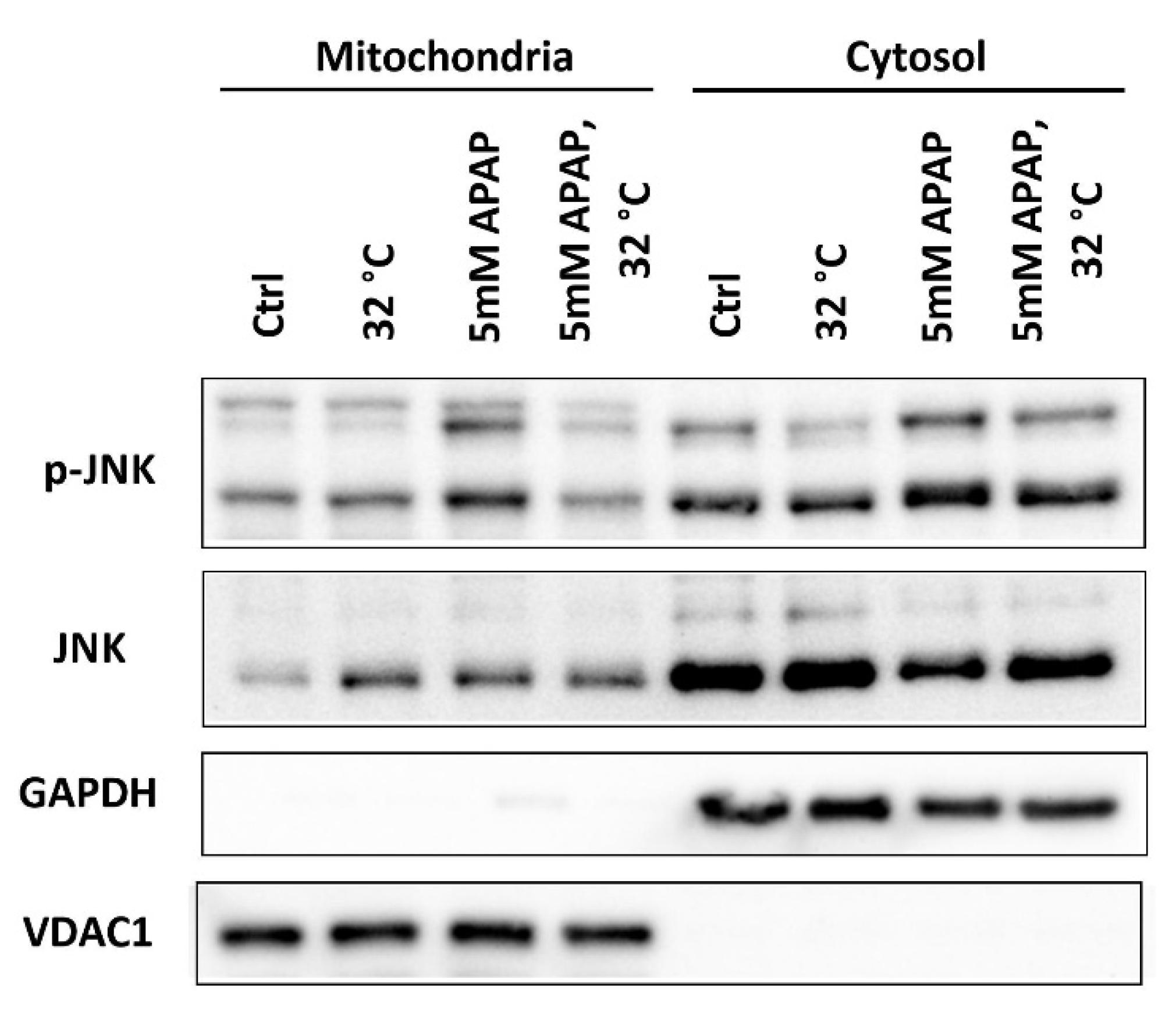
3.5. Moderate Hypothermic (32 °C) Conditioning Suppressed JNK Activation and Subsequent Mitochondrial Translocation to Abate the Amplification of Oxidative Stress in AIHI
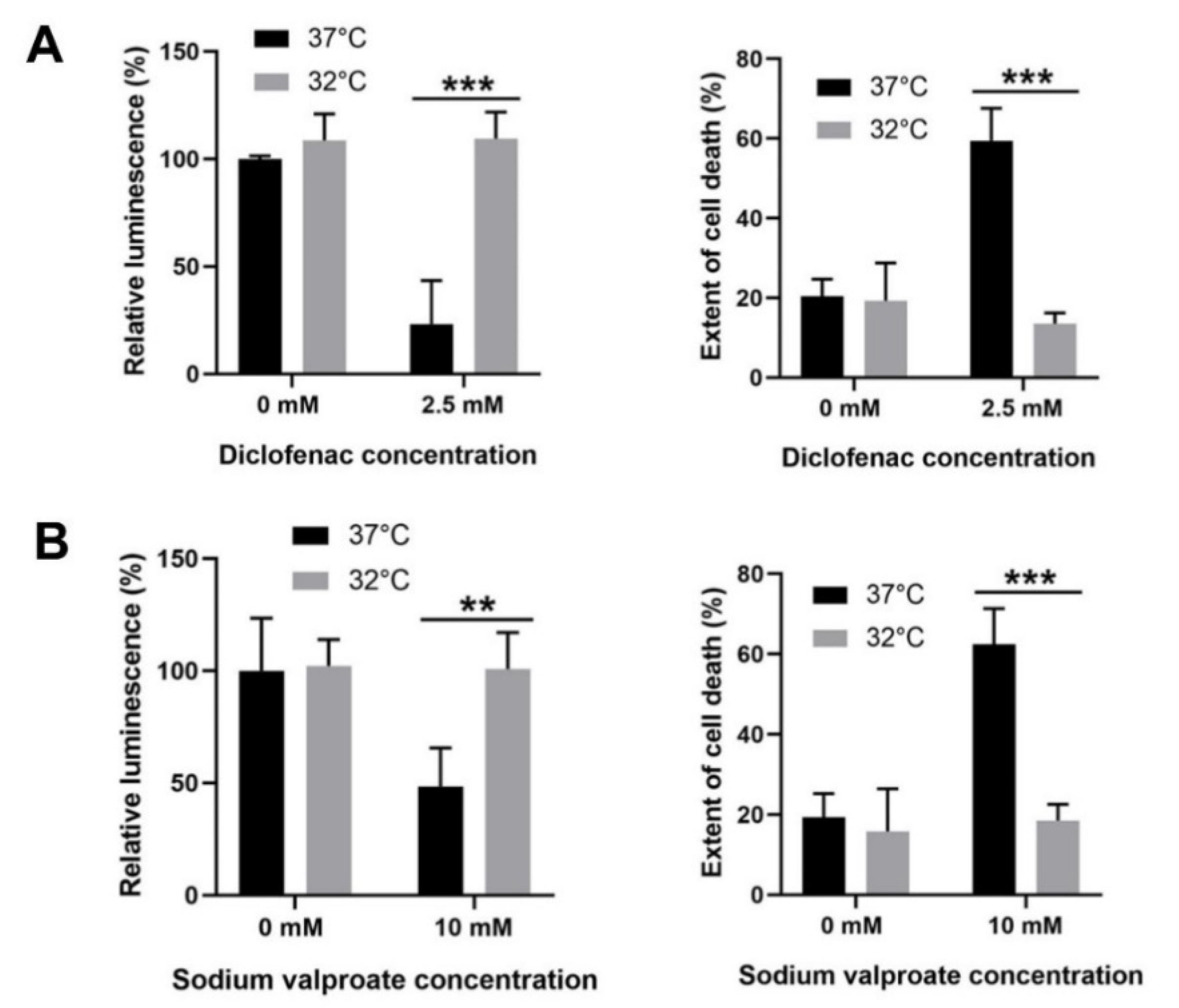
3.6. Moderate Hypothermic (32 °C) Conditioning Displayed Cytoprotective Potential in Other DILI with Similar Pathophysiology as APAP Toxicity
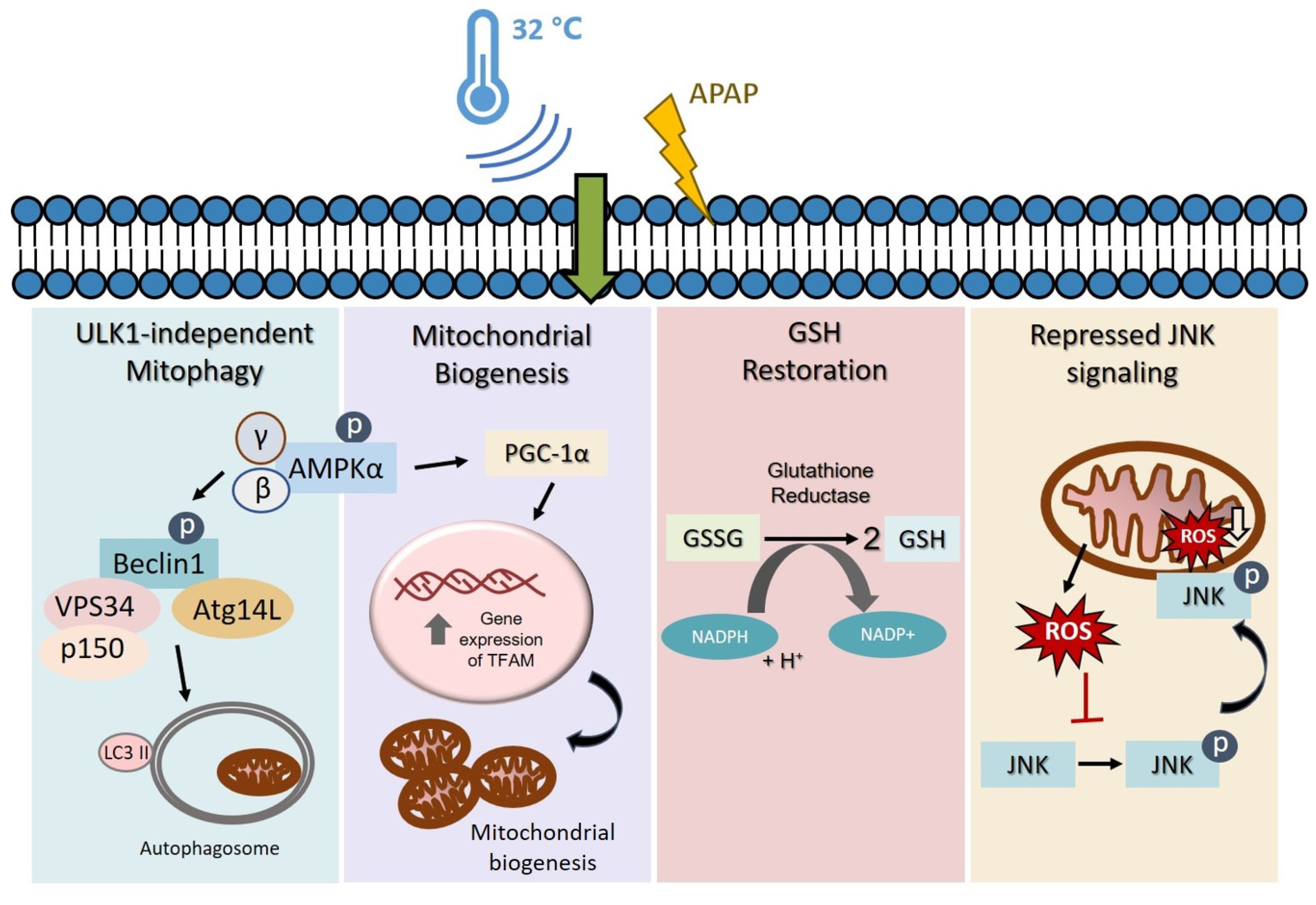
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APAP | Acetaminophen |
| AIHI | APAP-induced hepatocellularinjury |
| ALF | Acute liver failure |
| NAPQI | N-acetyl-p-benzoquinone imine |
| GSH | Reduced glutathione |
| JNK | c-Jun N-terminal kinase |
| NAC | N-acetyl cysteine |
| ROS | Reactive oxygen species |
| TAMH | Transforming growth factor-α transgenic mouse hepatocytes |
| CQ | Chloroquine |
| BSA | Bovine serum albumin |
| TMRM | Tetramethylrhodamine methyl ester |
| FCCP | Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone |
| SDS | Sodium dodecyl sulfate |
| TBS-T | Tris-buffered saline-tween 20 |
| GSSG | Oxidised glutathione |
| LC3B | Microtubule-associated proteins 1A/1B light chain 3B |
| AMPK | AMP-activated protein kinase |
| ULK1 | Unc-51-like autophagy activating kinase 1 |
| mTORC1 | Mechanistic target of rapamycin complex 1 |
| TOM | Translocases of outer membrane |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| TFAM | Transcription of transcription factor A, mitochondrial |
| DILI | Drug-induced liver injury |
| TGF-α | Transforming growth factor alpha |
| Vps34 | Phosphatidylinositol 3-kinase |
| NRF1 | Nuclear respiratory factor 1 |
| NRF2 | Nuclear respiratory factor 2 |
| Tim23 | Presequence translocase of the inner membrane |
| GPx | Glutathione peroxidase |
| MPT | Membrane permeability transition |
| MPK-1 | Mitogen-activated protein kinase phosphatase-1 |
| GCLC | Glutamate-cysteine ligase, catalytic subunit |
| GCLM | Glutamate-cysteine ligase, modifier subunit |
| ND1 | Mitochondrially encoded NADH dehydrogenase 1 |
| HK2 | Hexokinase 2 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| VDAC1 | Voltage dependent anion channel 1 |
| p70S6K | Ribosomal protein S6 kinase B1 |
| RT-PCR | Reverse transcription-polymerase chain reaction |
References
- Lee, W.M. Acetaminophen (APAP) hepatotoxicity—Isn’t it time for APAP to go away? J. Hepatol. 2017, 67, 1324–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIH. Acetaminophen. 2019. Available online: https://livertox.nih.gov/Acetaminophen.htm (accessed on 24 October 2020).
- Ostapowicz, G.; Fontana, R.J.; Schiodt, F.V.; Larson, A.; Davern, T.J.; Han, S.H.; McCashland, T.M.; Shakil, A.O.; Hay, J.E.; Hynan, L.; et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann. Intern. Med. 2002, 137, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Acute liver failure. Semin. Respir. Crit. Care Med. 2012, 33, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalimar, S.K.A. Management in acute liver failure. J. Clin. Exp. Hepatol. 2015, 5, S104–S115. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Huo, Y.; Yin, S.; Hu, H. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol. 2018, 17, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.D. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin. Liver Dis. 1990, 10, 267–278. [Google Scholar] [CrossRef]
- Gunawan, B.K.; Liu, Z.X.; Han, D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 2006, 131, 165–178. [Google Scholar] [CrossRef]
- Han, D.; Dara, L.; Win, S.; Than, T.A.; Yuan, L.; Abbasi, S.Q.; Liu, Z.-X.; Kaplowitz, N. Regulation of drug-induced liver injury by signal transduction pathways: Critical role of mitochondria. Trends Pharmacol. Sci. 2013, 34, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, G.B.; Todd, E.L.; Racz, W.J.; Hughes, H.; Smith, C.V.; Mitchell, J.R. Effects of N-acetylcysteine on the disposition and metabolism of acetaminophen in mice. J. Pharmacol. Exp. Ther. 1985, 232, 857–863. [Google Scholar] [PubMed]
- Wang, L.; Wei, W.; Xiao, Q.; Yang, H.; Ci, X. Farrerol Ameliorates APAP-induced Hepatotoxicity via Activation of Nrf2 and Autophagy. Int. J. Biol. Sci. 2019, 15, 788–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaquero, J.; Belanger, M.; James, L.; Herrero, R.; Desjardins, P.; Côté, J.; Blei, A.T.; Butterworth, R.F. Mild Hypothermia Attenuates Liver Injury and Improves Survival in Mice with Acetaminophen Toxicity. Gastroenterology 2007, 132, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Bemeur, C.; Desjardins, P.; Butterworth, R.F. Antioxidant and anti-inflammatory effects of mild hypothermia in the attenuation of liver injury due to azoxymethane toxicity in the mouse. Metab. Brain Dis. 2010, 25, 23–29. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, K.; Jo, Y.H.; Kim, M.A.; Rim, K.P.; Kang, K.W.; Rhee, J.E.; Lee, M.J.; Lee, H.S.; Kwon, W.Y.; et al. Therapeutic hypothermia attenuates liver injury in polymicrobial sepsis model of rats via Akt survival pathway. J. Surg. Res. 2013, 181, 114–120. [Google Scholar] [CrossRef]
- Zhu, Y.; Xiang, T.; Hu, D.; Shi, B.; Zhang, W.; Zhang, S.; Wu, X. Protective effects of mild hypothermia against hepatic injury in rats with acute liver failure. Ann. Hepatol. 2019, 18, 770–776. [Google Scholar] [CrossRef]
- Gong, P.; Li, C.-S.; Hua, R.; Zhao, H.; Tang, Z.-R.; Mei, X.; Zhang, M.-Y.; Cui, J. Mild Hypothermia Attenuates Mitochondrial Oxidative Stress by Protecting Respiratory Enzymes and Upregulating MnSOD in a Pig Model of Cardiac Arrest. PLoS ONE 2012, 7, e35313. [Google Scholar] [CrossRef] [Green Version]
- Chisholm, K.I.; Ida, K.K.; Davies, A.L.; Tachtsidis, I.; Papkovsky, D.B.; Dyson, A.; Singer, M.; Duchen, M.R.; Smith, K.J. Hypothermia protects brain mitochondrial function from hypoxemia in a murine model of sepsis. J. Cereb. Blood Flow Metab. 2016, 36, 1955–1964. [Google Scholar] [CrossRef] [Green Version]
- Hackenhaar, F.S.; Medeiros, T.M.; Heemann, F.M.; Behling, C.S.; Putti, J.S.; Mahl, C.D.; Verona, C.; Da Silva, A.C.A.; Guerra, M.C.; Gonçalves, C.A.S.; et al. Therapeutic Hypothermia Reduces Oxidative Damage and Alters Antioxidant Defenses after Cardiac Arrest. Oxidative Med. Cell. Longev. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Huang, C.-H.; Tsai, M.-S.; Chiang, C.-Y.; Su, Y.-J.; Wang, T.-D.; Chang, W.-T.; Chen, H.-W.; Chen, W.-J. Activation of mitochondrial STAT-3 and reduced mitochondria damage during hypothermia treatment for post-cardiac arrest myocardial dysfunction. Basic Res. Cardiol. 2015, 110, 1–13. [Google Scholar] [CrossRef]
- Tan, Y.L.; Tey, S.M.; Ho, H.K. Moderate hypothermia effectively alleviates acetaminophen-induced liver injury with prolonged action beyond cooling. Dose Response 2020, in press. [Google Scholar]
- Wu, J.C.; Merlino, G.; Cveklova, K.; Mosinger, B., Jr.; Fausto, N. Autonomous growth in serum-free medium and production of hepatocellular carcinomas by differentiated hepatocyte lines that overexpress transforming growth factor alpha 1. Cancer Res. 1994, 54, 5964–5973. [Google Scholar] [PubMed]
- Tan, C.T.; Zhou, Q.-L.; Su, Y.-C.; Fu, N.; Chang, H.-C.; Tao, R.N.; Sukumaran, S.K.; Baksh, S.; Tan, Y.-J.; Sabapathy, K.; et al. MOAP-1 Mediates Fas-Induced Apoptosis in Liver by Facilitating tBid Recruitment to Mitochondria. Cell Rep. 2016, 16, 174–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, H.-M.; Bockus, A.; Boggess, N.; Jaeschke, H.; Ding, W.-X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012, 55, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, H.-M.; Jaeschke, H.; Ding, W.-X. Targeting autophagy for drug-induced hepatotoxicity. Autophagy 2012, 8, 709–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marek-Iannucci, S.; Thomas, A.; Hou, J.; Crupi, A.; Sin, J.; Taylor, D.J.; Czer, L.S.; Esmailian, F.; Mentzer, R.M., Jr.; Andres, A.M.; et al. Myocardial hypothermia increases autophagic flux, mitochondrial mass and myocardial function after ischemia-reperfusion injury. Sci. Rep. 2019, 9, 10001. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- McGill, M.R.; Sharpe, M.R.; Williams, C.D.; Taha, M.; Curry, S.C.; Jaeschke, H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Investig. 2012, 122, 1574–1583. [Google Scholar] [CrossRef] [Green Version]
- Latchoumycandane, C.; Goh, C.W.; Ong, M.M.; Boelsterli, U.A. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen-induced liver injury. Hepatology 2007, 45, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Pollock, K.J.; Frew, J.; MacKinnon, A.C.; A Flavell, R.; Davis, R.J.; Sethi, T.; Simpson, K.J. Critical role of c-jun (NH2) terminal kinase in paracetamol- induced acute liver failure. Gut 2007, 56, 982–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aithal, G.P. Diclofenac-induced liver injury: A paradigm of idiosyncratic drug toxicity. Expert Opin. Drug Saf. 2004, 3, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Visschers, R.G.J.; Duan, L.; Akakpo, J.Y.; Jaeschke, H. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: Current understanding and future perspectives. J. Clin. Transl. Res. 2018, 4, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Varon, J.; Acosta, P. Therapeutic hypothermia: Past, present, and future. Chest 2008, 133, 1267–1274. [Google Scholar] [CrossRef]
- Davis, M.; Stamper, B. TAMH: A Useful In Vitro Model for Assessing Hepatotoxic Mechanisms. BioMed Res. Int. 2016, 2016, 1–9. [Google Scholar] [CrossRef]
- Donato, M.T.; Klocke, R.; Castell, J.V.; Stenzel, K.; Paul, D.; Gómez-Lechón, M.J. Constitutive and inducible expression of CYP enzymes in immortal hepatocytes derived from SV40 transgenic mice. Xenobiotica 2003, 33, 459–473. [Google Scholar] [CrossRef]
- Pierce, R.H.; Franklin, C.C.; Campbell, J.S.; Tonge, R.P.; Chen, W.; Fausto, N.; Nelson, S.D.; Bruschi, A.S. Cell culture model for acetaminophen-induced hepatocyte death in vivo. Biochem. Pharmacol. 2002, 64, 413–424. [Google Scholar] [CrossRef]
- Jin, Y.; Lin, Y.; Feng, J.-F.; Jia, F.; Gao, G.-Y.; Jiang, J.-Y. Moderate Hypothermia Significantly Decreases Hippocampal Cell Death Involving Autophagy Pathway after Moderate Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1090–1100. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Yin, M.; Lu, Y.; Yang, Y.; Li, B.; Liao, X.X.; Dai, G.; Jing, X.; Xiong, Y.; Hu, C. Mild hypothermia improves neurological outcome in mice after cardiopulmonary resuscitation through Silent Information Regulator 1-actviated autophagy. Cell Death Discov. 2019, 5, 129. [Google Scholar] [CrossRef] [Green Version]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, N.; Kageyama, S.; Komatsu, M.; Waguri, S. Hyperosmotic Stress Induces Unconventional Autophagy Independent of the Ulk1 Complex. Mol. Cell. Biol. 2019, 39, e00024-19. [Google Scholar] [PubMed] [Green Version]
- Cheong, H.; Lindsten, T.; Wu, J.; Lu, C.; Thompson, C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Nat. Acad. Sci. USA 2011, 108, 11121–11126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim Young, C.; Fang, C.; Russell Ryan, C.; Kim Jeong, H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.-L. Differential Regulation of Distinct Vps34 Complexes by AMPK in Nutrient Stress and Autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.B.; Zhou, G.; Li, C. AMPK: An emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009, 9, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Nat. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef]
- Truscott, K.N.; Kovermann, P.; Geissler, A.; Merlin, A.; Meijer, M.; Driessen, A.J.M.; Rassow, J.; Pfanner, N.; Wagner, R. A presequence- and voltage-sensitive channel of the mitochondrial preprotein translocase formed by Tim23. Nat. Struct. Biol. 2001, 8, 1074–1082. [Google Scholar] [CrossRef] [Green Version]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Moon, K.-H.; Chen, C.; Gonzalez, F.J.; Song, B.-J. Role of cytochrome P450 2E1 in protein nitration and ubiquitin-mediated degradation during acetaminophen toxicity. Biochem. Pharmacol. 2010, 79, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2017, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.-S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskla, K.-L.; Porosk, R.; Reimets, R.; Visnapuu, T.; Vasar, E.; Hundahl, C.A.; Luuk, H. Hypothermia augments stress response in mammalian cells. Free Radic. Biol. Med. 2018, 121, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Hackenhaar, F.S.; Medeiros, T.M.; Heemann, F.M.; Behling, C.S.; Mahl, C.D.; Verona, C.; Silva, A.C.A.; Oliveira, V.M.; Riveiro, D.F.M.; Vieira, S.R.R.; et al. Mild Therapeutic Hypothermia Increases Glutathione Levels in Postcardiac Arrest Patients. Ther. Hypothermia Temp. Manag. 2019, 9, 63–69. [Google Scholar] [CrossRef]
- Alva, N.; Carbonell, T.; Palomeque, J. Hypothermic protection in an acute hypoxia model in rats: Acid-base and oxidant/antioxidant profiles. Resuscitation 2010, 81, 609–616. [Google Scholar] [CrossRef]
- Han, X.; Song, X.; Yu, F.; Chen, L. A ratiometric fluorescent probe for imaging and quantifying anti-apoptotic effects of GSH under temperature stress. Chem. Sci. 2017, 8, 6991–7002. [Google Scholar] [CrossRef] [Green Version]
- Slikker, W.; Desai, V.G.; Duhart, H.; Feuers, R.; Imam, S.Z. Hypothermia enhances bcl-2 expression and protects against oxidative stress-induced cell death in chinese hamster ovary cells. Free Radic. Biol. Med. 2001, 31, 405–411. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 26. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinson, J.A.; Reid, A.B.; McCullough, S.S.; James, L.P. Acetaminophen-induced hepatotoxicity: Role of metabolic activation, reactive oxygen/nitrogen species, and mitochondrial permeability transition. Drug Metab. Rev. 2004, 36, 805–822. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Guo, S.; Zhang, T.; Li, H. Hypothermia attenuates ischemia/reperfusion-induced endothelial cell apoptosis via alterations in apoptotic pathways and JNK signaling. FEBS Lett. 2009, 583, 2500–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Xie, P.; Guo, S.; Li, H. Induction of MAPK phosphatase-1 by hypothermia inhibits TNF-alpha-induced endothelial barrier dysfunction and apoptosis. Cardiovasc. Res. 2010, 85, 520–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wancket, L.M.; Meng, X.; Rogers, L.K.; Liu, Y. Mitogen-activated protein kinase phosphatase (Mkp)-1 protects mice against acetaminophen-induced hepatic injury. Toxicol. Pathol. 2012, 40, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gorospe, M.; Yang, C.; Holbrook, N.J. Role of mitogen-activated protein kinase phosphatase during the cellular response to genotoxic stress. Inhibition of c-Jun N-terminal kinase activity and AP-1-dependent gene activation. J. Biol. Chem. 1995, 270, 8377–8380. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, Y. Regulation of innate immune response by MAP kinase phosphatase-1. Cell Signal 2007, 19, 1372–1382. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wang, L.; Dong, X.; Hu, X.; Zhou, L.; Liu, Q.; Song, B.; Wu, Q.; Li, L. The c-jun n-terminal kinase (jnk) pathway is activated in human interstitial cystitis (ic) and rat protamine sulfate induced cystitis. Sci. Rep. 2016, 6, 19670. [Google Scholar] [CrossRef] [Green Version]
- Donnino, M.W.; Andersen, L.W.; Berg, K.M.; Reynolds, J.C.; Nolan, J.P.; Morley, P.T.; Lang, E.; Cocchi, M.N.; Xanthos, T.; Callaway, C.W.; et al. Temperature Management After Cardiac Arrest: An Advisory Statement by the Advanced Life Support Task Force of the International Liaison Committee on Resuscitation and the American Heart Association Emergency Cardiovascular Care Committee and the Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation. Resuscitation 2016, 98, 97–104. [Google Scholar]
- Perlman, J.M.; Wyllie, J.; Kattwinkel, J.; Atkins, D.L.; Chameides, L.; Goldsmith, J.P.; Guinsburg, R.; Hazinski, M.F.; Morley, C.; Richmond, S.; et al. Part 11: Neonatal Resuscitation. Circulation 2010, 122, S516–S538. [Google Scholar] [CrossRef] [Green Version]
- Dubay, D.; Gallinger, S.; Hawryluck, L.; Swallow, C.; McCluskey, S.; McGilvray, I. In situ hypothermic liver preservation during radical liver resection with major vascular reconstruction. Br. J. Surg. 2009, 96, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, G.H.; Abelev, N.; Tevez-Ortiz, A. Noninvasive Selective Cryolipolysis and Reperfusion Recovery for Localized Natural Fat Reduction and Contouring. Aesthetic Surg. J. 2014, 34, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Bonkovsky, H.L.; Fontana, R.J.; Lee, W.; Stolz, A.; A Talwalkar, J.; Reddy, K.R.; Watkins, P.B.; Navarro, V.J.; Barnhart, H.X.; et al. Features and Outcomes of 899 Patients with Drug-Induced Liver Injury: The DILIN Prospective Study. Gastroenterology 2015, 148, 1340–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Primer Sequence (5′ → 3′) |
|---|---|---|
| Mechanistic target of rapamycin kinase (mTOR) - homologous to human mTOR | NM_020009.2 | F: ATGTGTCCCCCAAACTTCTG R: ATCTTCATGGCCTTTCAGGA |
| Protein kinase, AMP-activated catalytic subunit alpha 1 (Prkaa1) - homologous to human AMPK | NM_001013367.3 | F: AGAGGGCCGCAATAAAAGAT R: TCCTCCGAACACTCGAACTT |
| Glutamate-cysteine ligase, catalytic subunit (GCLC) -homologous to human GCLC | NM_010295.2 | F: TCCATTTTACCGAGGCTACG R: CGATGGTCAGGTCGATGTC |
| Glutamate-cysteine ligase, modifier subunit (GCLM) - homologous to human GCLM | NM_008129.4 | F: CCAGATTTGACTGCCTTTGC R: TGATGATTCCCCTGCTCTTC |
| Mitochondrially encoded NADH dehydrogenase 1 (ND1) [25] - homologous to human ND1 | KY018919.1 | F: CTAGCAGAAACAAACCGGGC R: CCGGCTGCGTATTCTACGTT |
| Hexokinase 2 (HK2) [25] - homologous to human HK2 | Y11668.1 | F: GCCAGCCTCTCCTGATTTTAGTGT R: GGGAACACAAAAGACCTCTTCTGG |
| Beta-actin (β-actin) | NM_007393.5 | F: TGTTACCAACTGGGACGACA R: GGGGTGTTGAAGGTCTCAAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, Y.L.; Ho, H.K. Hypothermia Advocates Functional Mitochondria and Alleviates Oxidative Stress to Combat Acetaminophen-Induced Hepatotoxicity. Cells 2020, 9, 2354. https://doi.org/10.3390/cells9112354
Tan YL, Ho HK. Hypothermia Advocates Functional Mitochondria and Alleviates Oxidative Stress to Combat Acetaminophen-Induced Hepatotoxicity. Cells. 2020; 9(11):2354. https://doi.org/10.3390/cells9112354
Chicago/Turabian StyleTan, Yeong Lan, and Han Kiat Ho. 2020. "Hypothermia Advocates Functional Mitochondria and Alleviates Oxidative Stress to Combat Acetaminophen-Induced Hepatotoxicity" Cells 9, no. 11: 2354. https://doi.org/10.3390/cells9112354
APA StyleTan, Y. L., & Ho, H. K. (2020). Hypothermia Advocates Functional Mitochondria and Alleviates Oxidative Stress to Combat Acetaminophen-Induced Hepatotoxicity. Cells, 9(11), 2354. https://doi.org/10.3390/cells9112354