Quick, Coordinated and Authentic Reprogramming of Ribosome Biogenesis during iPSC Reprogramming


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
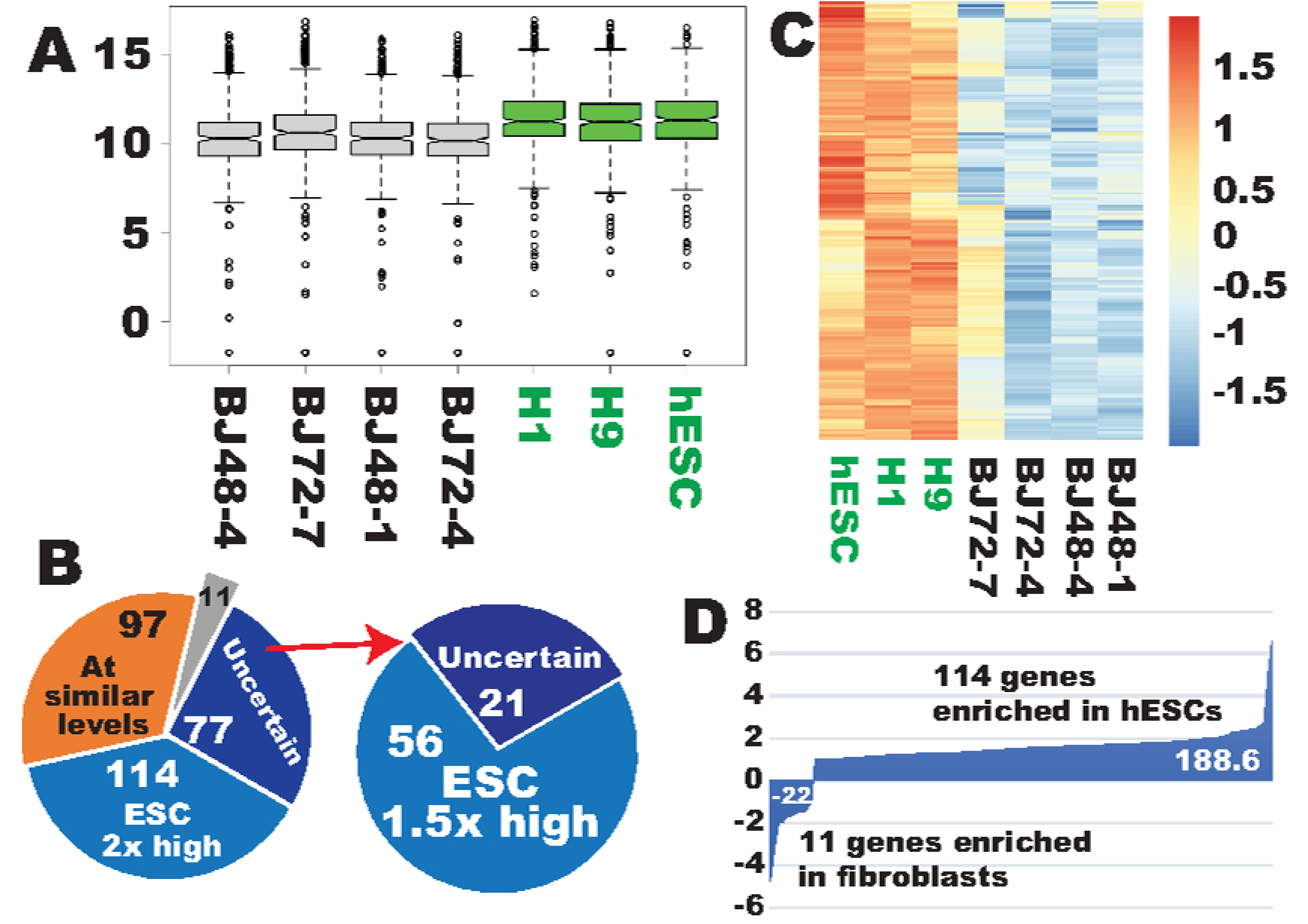
2.1. More Robust Ribosome Biogenesis (RB) in hESCs than in the Reprogramming Starting Fibroblasts
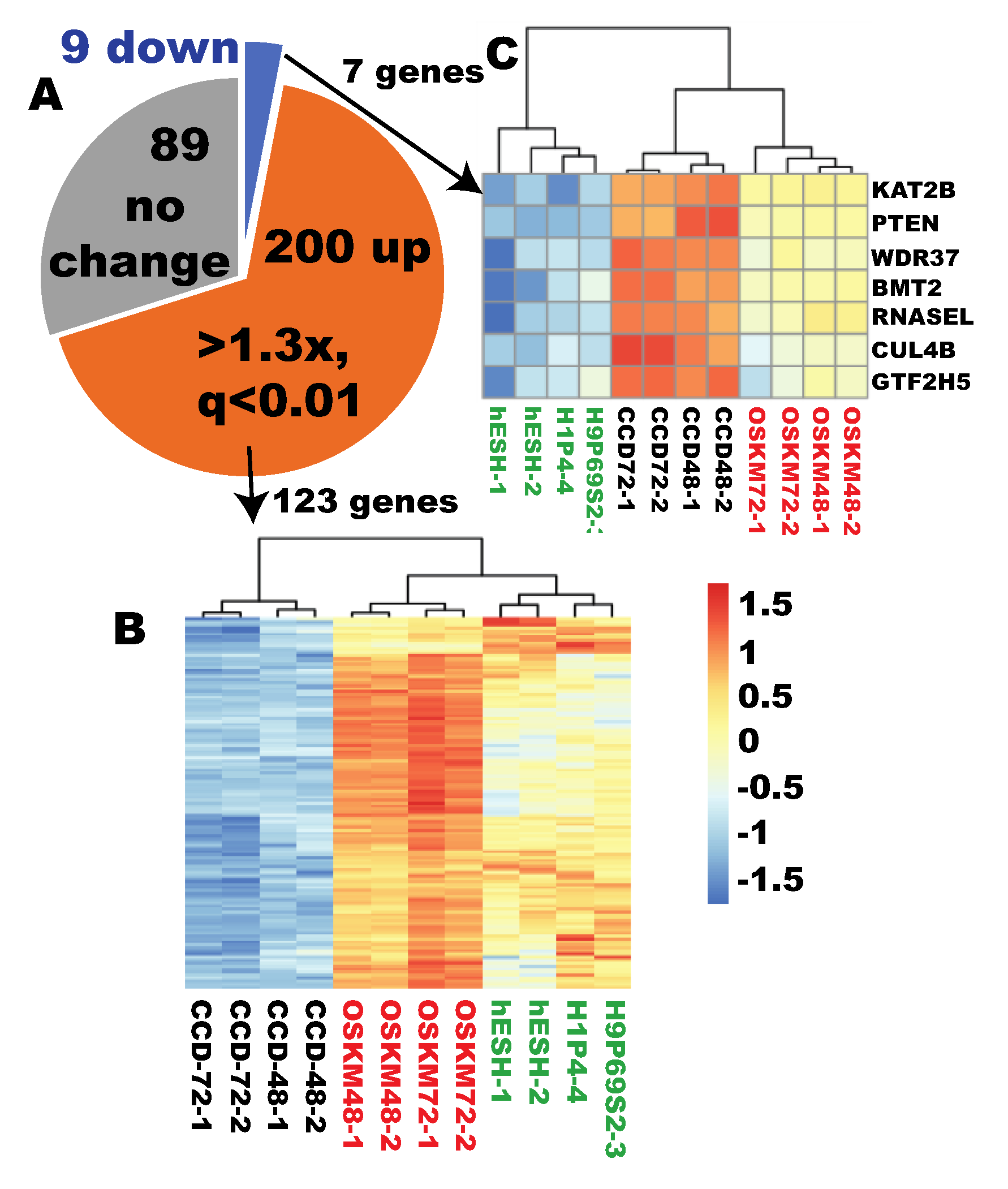
2.2. Ribosome Biogenesis Was Properly Reprogrammed within 48 Hours
2.3. Accurate Reprogramming of the Ribosome Biogenesis
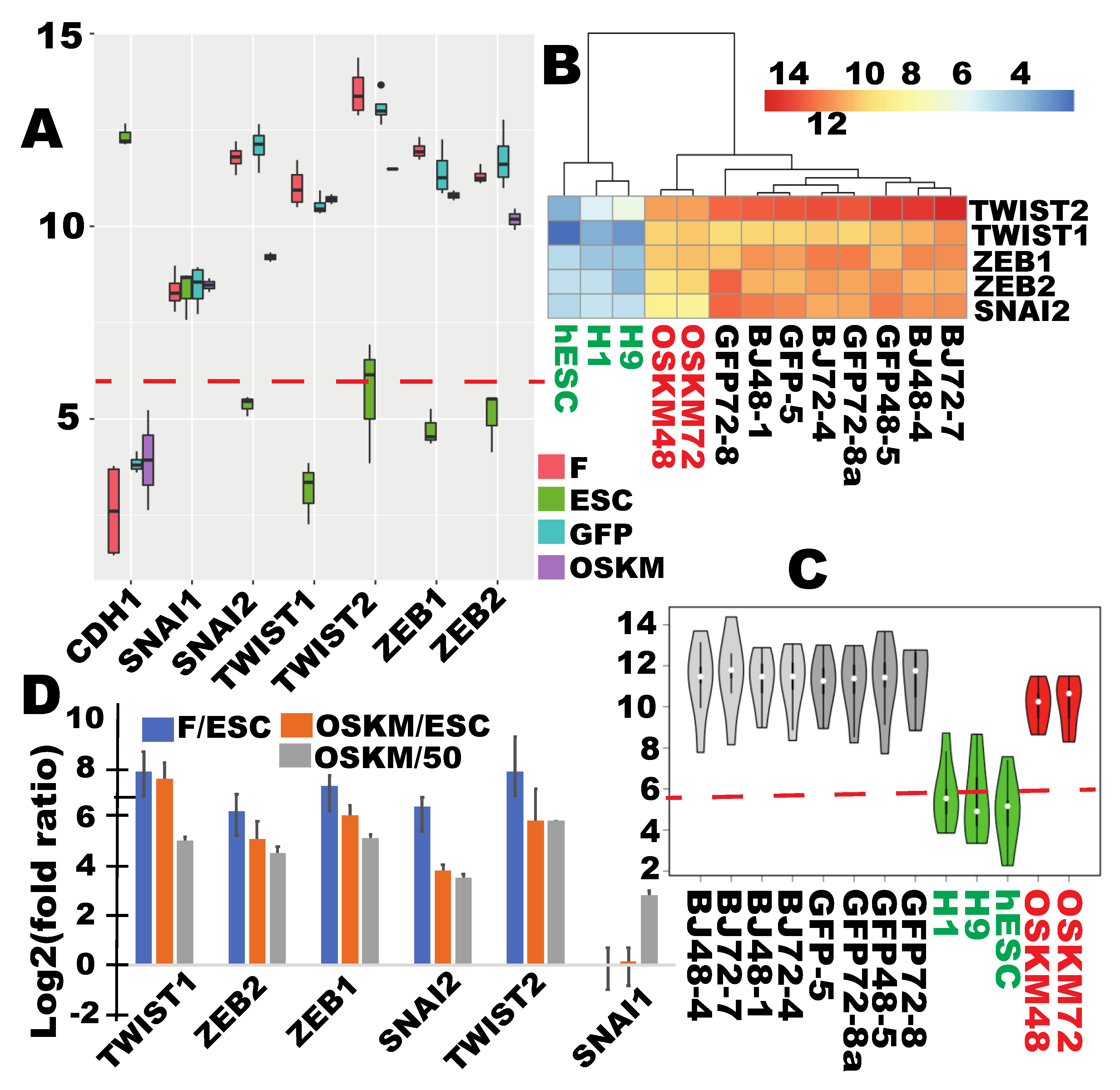
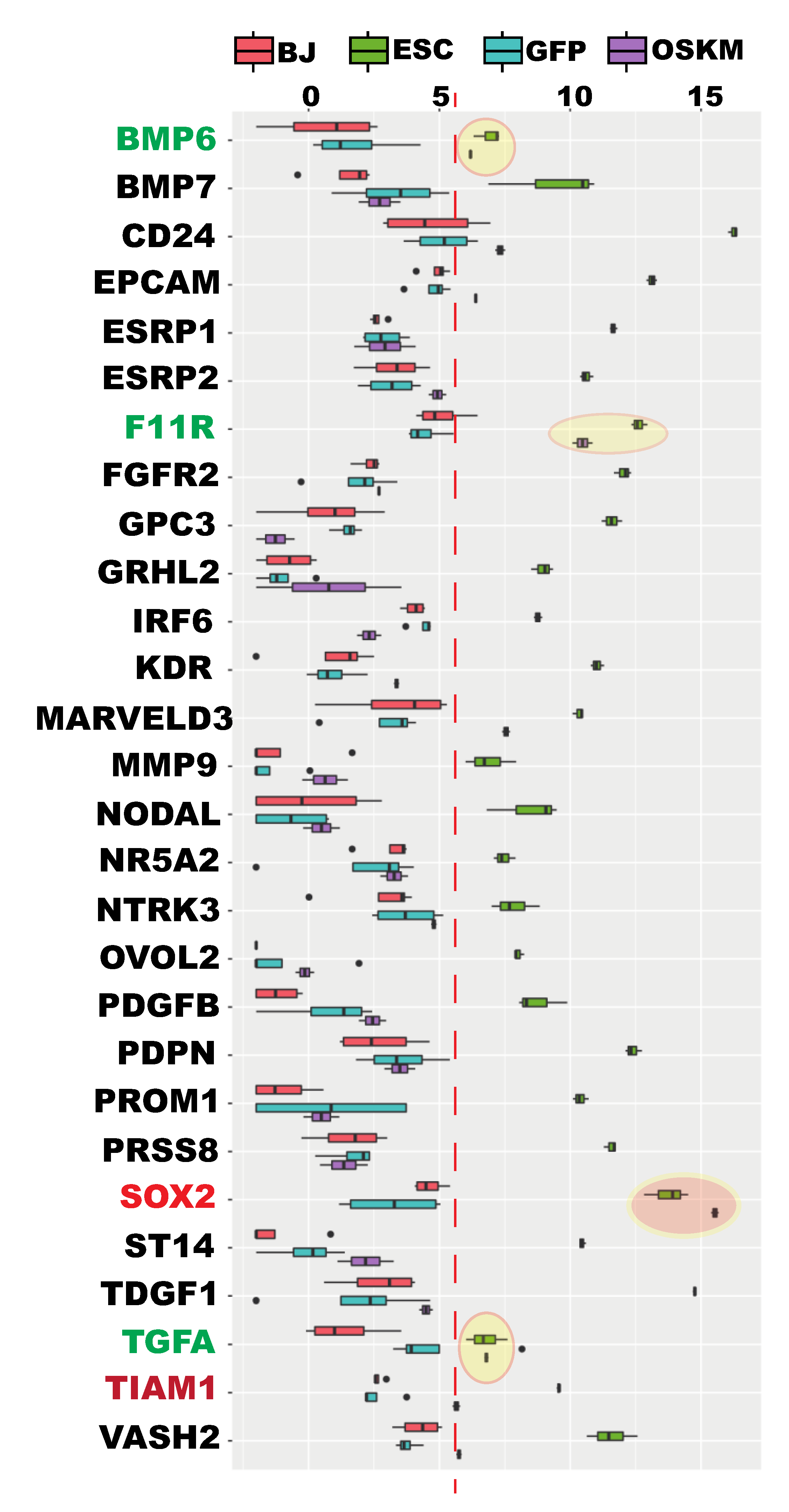
2.4. Mesenchymal-to-Epithelial (MET) Transition Was not Achieved at the Time of Proper RB Reprogramming
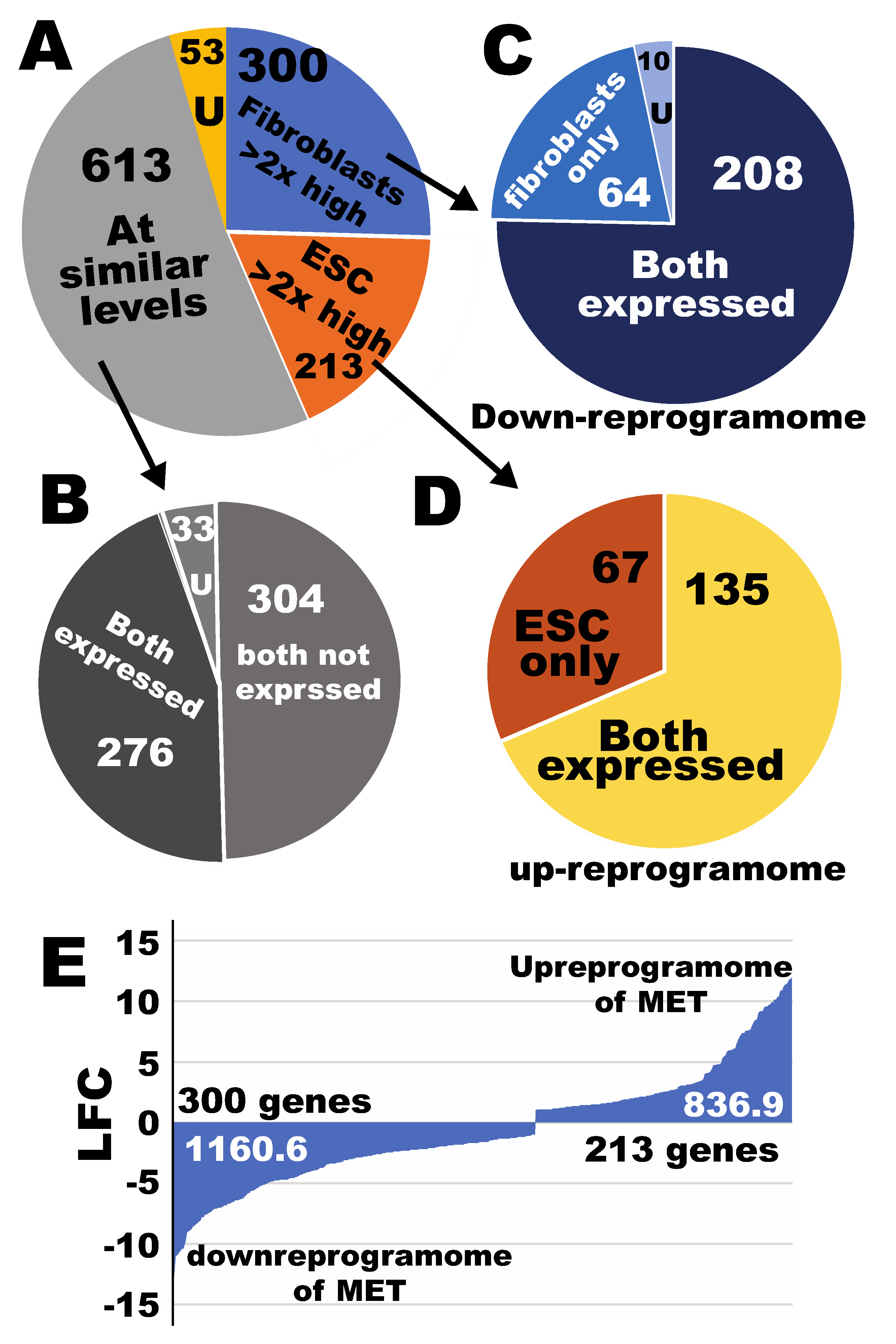
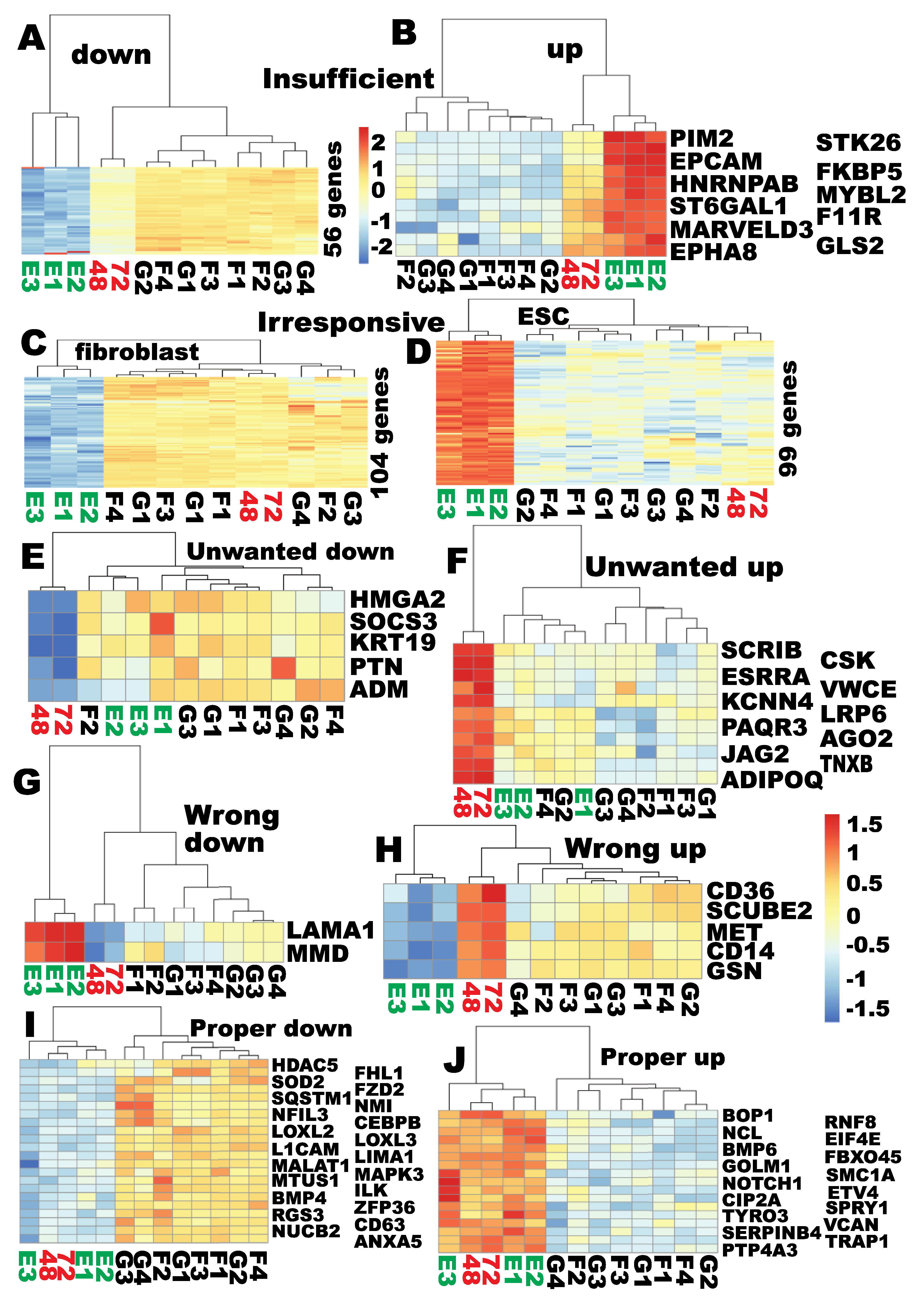
2.5. Defining the Sub-Reprogramomes of MET for Human Fibroblast Reprogramming to Pluripotency
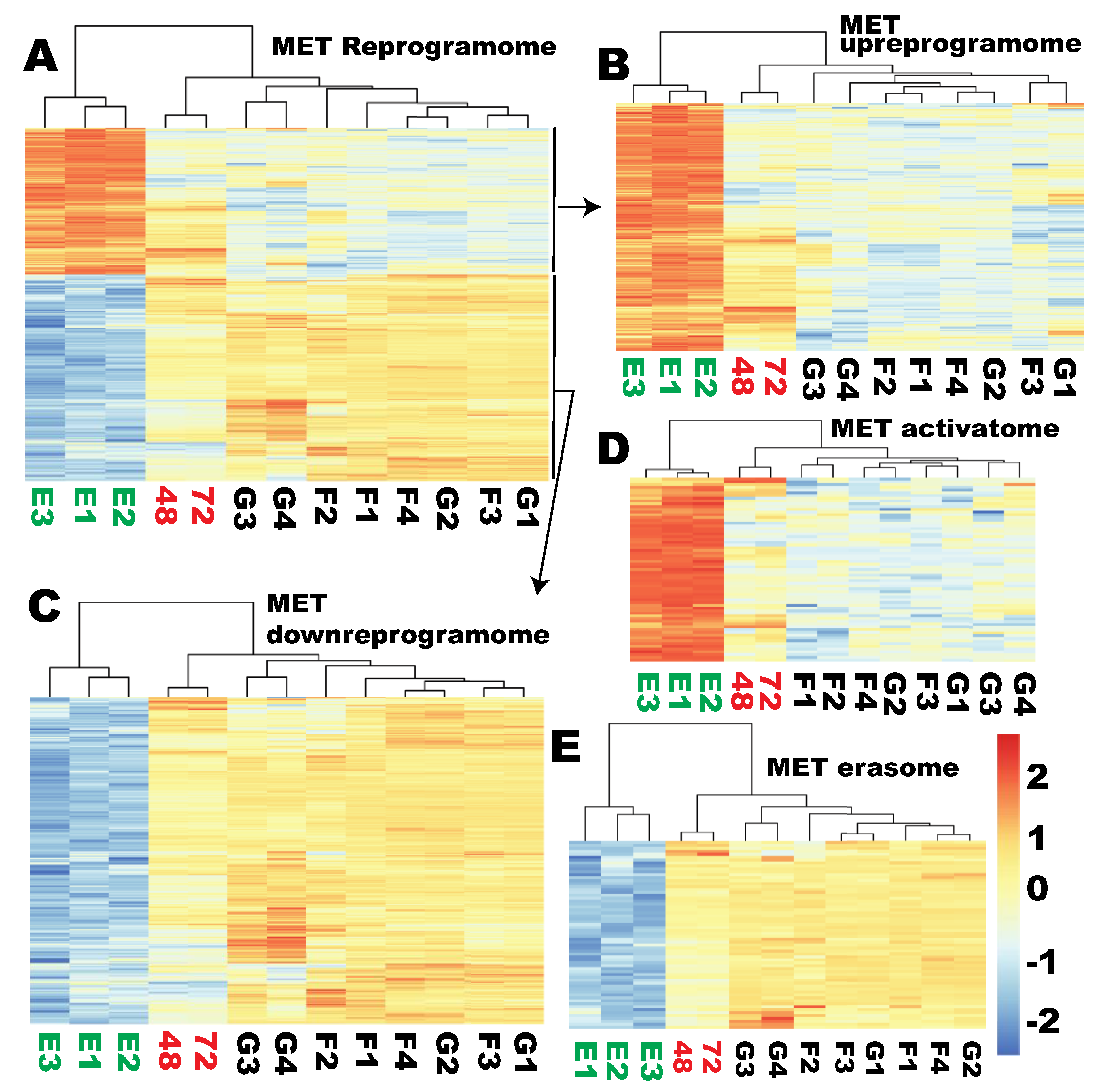
2.6. None of the Sub-Reprogramome of the MET Gene Set Was Reprogrammed at the Time of Proper Reprogramming of Ribosome Biogenesis
2.7. PIANO Responses of the MET/EMT Genes to the Yamanaka Factors at the Time of Proper RB Reprogramming
2.8. Additional Epithelial Signatures Were Not Reprogrammed at the Time of Proper Reprogramming of Ribosome Biogenesis
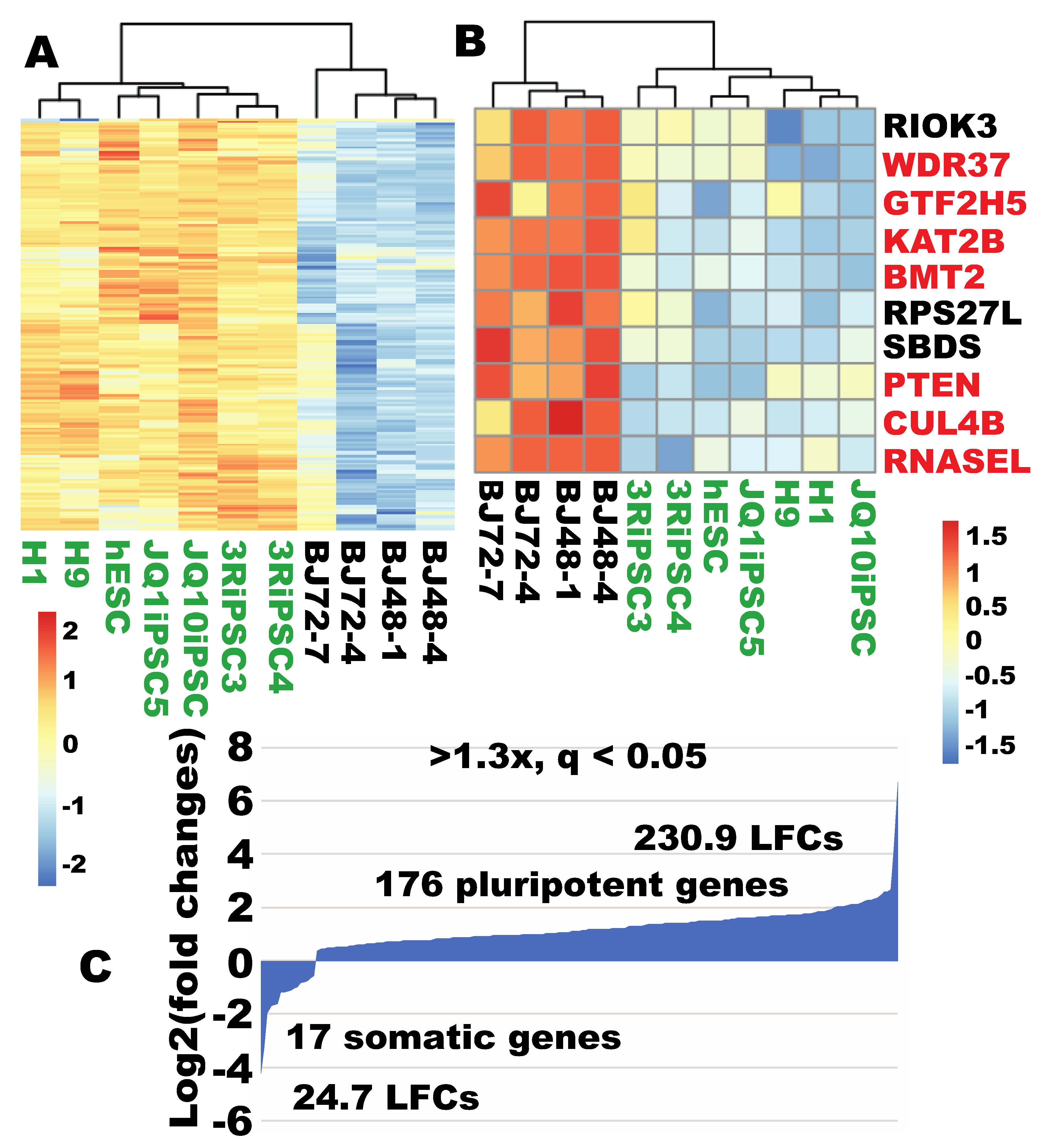
2.9. More Robust Ribosome Biogenesis was Observed in iPSCs
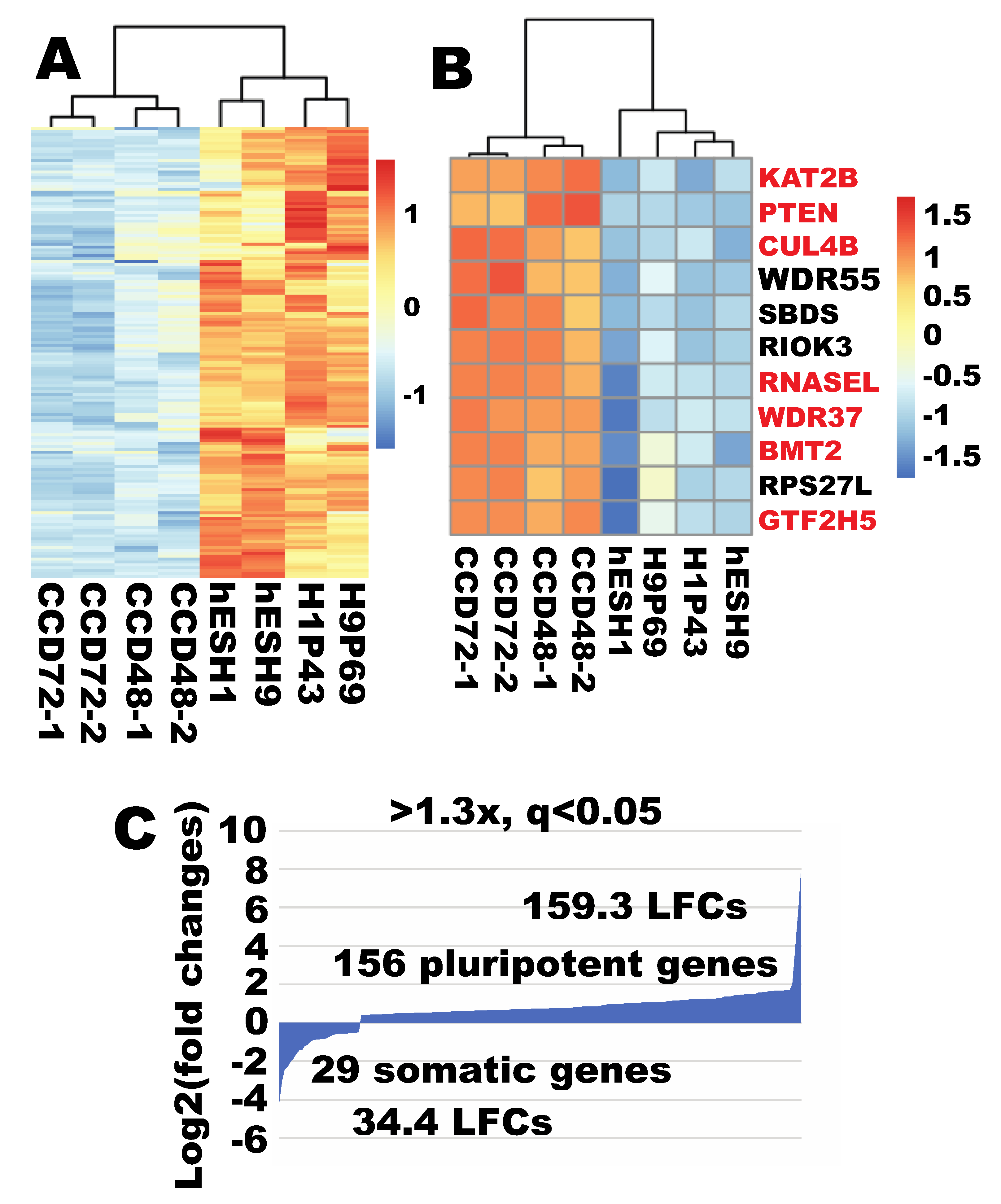
2.10. Robust Ribosome Biogenesis Remained a Pluripotent Feature When Compared to an Independent Human Fibroblast Line
2.11. OSKM Quickly Upregulated Ribosome Biogenesis to Pluripotent State in An Independent Human Fibroblast Line
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cultures
4.2. iPSC Reprogramming from Human Fibroblasts
4.3. RNA-seq
4.4. Bioinformatics
4.5. Gene set of Human Ribosome Biogenesis
4.6. Gene Set of Human MET/EMT-Related Genes
4.7. Criteria for the Expressed Genes and Differentially Expressed Genes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hu, K. All roads lead to induced pluripotent stem cells: The technologies of iPSC generation. Stem Cells Dev. 2014, 23, 1285–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Yu, J.; Suknuntha, K.; Tian, S.; Montgomery, K.; Choi, K.D.; Stewart, R.; Thomson, J.A.; Slukvin, I.I. Efficient generation of transgene-free induced pluripotent stem cells from normal and neoplastic bone marrow and cord blood mononuclear cells. Blood 2011, 117, e109–e119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Z.; Zhang, R.; Khodadadi-Jamayran, A.; Chen, B.; Crowley, M.R.; Festok, M.A.; Crossman, D.K.; Townes, T.M.; Hu, K. The acetyllysine reader BRD3R promotes human nuclear reprogramming and regulates mitosis. Nat. Commun. 2016, 7, 10869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Z.; Yao, C.; Khodadadi-Jamayran, A.; Xu, W.; Townes, T.M.; Crowley, M.R.; Hu, K. Reprogramming by De-bookmarking the Somatic Transcriptional Program through Targeting of BET Bromodomains. Cell Rep. 2016, 16, 3138–3145. [Google Scholar] [CrossRef] [Green Version]
- Hu, K. Vectorology and factor delivery in induced pluripotent stem cell reprogramming. Stem Cells Dev. 2014, 23, 1301–1315. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [Green Version]
- Hu, K. A PIANO (Proper, Insufficient, Aberrant, and NO Reprogramming) Response to the Yamanaka Factors in the Initial Stages of Human iPSC Reprogramming. Int. J. Mol. Sci. 2020, 21, 3229. [Google Scholar] [CrossRef]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.K.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef] [Green Version]
- Silverman, R.H. A scientific journey through the 2-5A/RNase L system. Cytokine Growth Factor Rev. 2007, 18, 381–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, T.S.; Hanna, J.; Zhang, X.; Ku, M.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Ianov, L.; Crossman, D. Profiling and quantification of pluripotency reprogramming reveal that WNT pathways and cell morphology have to be reprogramed extensively. Heliyon 2020, 6, e04035. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Nerurkar, P.; Altvater, M.; Gerhardy, S.; Schutz, S.; Fischer, U.; Weirich, C.; Panse, V.G. Eukaryotic Ribosome Assembly and Nuclear Export. Int. Rev. Cell Mol. Biol. 2015, 319, 107–140. [Google Scholar] [CrossRef]
- Mayer, C.; Grummt, I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006, 25, 6384–6391. [Google Scholar] [CrossRef] [Green Version]
- de la Cruz, J.; Gomez-Herreros, F.; Rodriguez-Galan, O.; Begley, V.; de la Cruz Munoz-Centeno, M.; Chavez, S. Feedback regulation of ribosome assembly. Curr. Genet. 2018, 64, 393–404. [Google Scholar] [CrossRef]
- McStay, B. Nucleolar organizer regions: Genomic ‘dark matter’ requiring illumination. Genes Dev. 2016, 30, 1598–1610. [Google Scholar] [CrossRef] [Green Version]
- Henras, A.K.; Plisson-Chastang, C.; O’Donohue, M.F.; Chakraborty, A.; Gleizes, P.E. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev. RNA 2015, 6, 225–242. [Google Scholar] [CrossRef]
- Nazar, R.N. Ribosomal RNA processing and ribosome biogenesis in eukaryotes. IUBMB Life 2004, 56, 457–465. [Google Scholar] [CrossRef]
- Klinge, S.; Woolford, J.L., Jr. Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol. 2019, 20, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Armistead, J.; Triggs-Raine, B. Diverse diseases from a ubiquitous process: The ribosomopathy paradox. FEBS Lett. 2014, 588, 1491–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe-Susaki, K.; Takada, H.; Enomoto, K.; Miwata, K.; Ishimine, H.; Intoh, A.; Ohtaka, M.; Nakanishi, M.; Sugino, H.; Asashima, M.; et al. Biosynthesis of ribosomal RNA in nucleoli regulates pluripotency and differentiation ability of pluripotent stem cells. Stem Cells 2014, 32, 3099–3111. [Google Scholar] [CrossRef] [PubMed]
- Corsini, N.S.; Peer, A.M.; Moeseneder, P.; Roiuk, M.; Burkard, T.R.; Theussl, H.C.; Moll, I.; Knoblich, J.A. Coordinated Control of mRNA and rRNA Processing Controls Embryonic Stem Cell Pluripotency and Differentiation. Cell Stem Cell 2018, 22, 543–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, K.T.; Park, J.; Kim, V.N. Role of the small subunit processome in the maintenance of pluripotent stem cells. Genes Dev. 2015, 29, 2004–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wang, B.; Yang, A.; Lu, R.; Wang, W.; Zhou, Y.; Shi, G.; Kwon, S.W.; Zhao, Y.; Jin, Y. Ly-1 antibody reactive clone is an important nucleolar protein for control of self-renewal and differentiation in embryonic stem cells. Stem Cells 2009, 27, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Gabut, M.; Bourdelais, F.; Durand, S. Ribosome and Translational Control in Stem Cells. Cells 2020, 9, 497. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Thiery, J.P. Epithelial-mesenchymal transitions: Insights from development. Development 2012, 139, 3471–3486. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Pei, D.; Shu, X.; Gassama-Diagne, A.; Thiery, J.P. Mesenchymal-epithelial transition in development and reprogramming. Nat. Cell Biol. 2019, 21, 44–53. [Google Scholar] [CrossRef]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zhang, L.; Mao, S.Q.; Li, Z.; Chen, J.; Zhang, R.R.; Wu, H.P.; Gao, J.; Guo, F.; Liu, W.; et al. Tet and TDG mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell 2014, 14, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Hu, K. Intron exclusion and the mystery of intron loss. FEBS Lett. 2006, 580, 6361–6365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Raay, T.J.; Connors, T.D.; Klinger, K.W.; Landes, G.M.; Burn, T.C. A novel ribosomal protein L3-like gene (RPL3L) maps to the autosomal dominant polycystic kidney disease gene region. Genomics 1996, 37, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Hu, K. Homologous recombination and innocuous intron elimination. In Genetic Recombination Research Progress; Schulz, J., Ed.; Nova Biomedical: Mörfelden-Walldorf, Germany, 2008; pp. 315–325. [Google Scholar]
- Uechi, T.; Maeda, N.; Tanaka, T.; Kenmochi, N. Functional second genes generated by retrotransposition of the X-linked ribosomal protein genes. Nucleic Acids Res. 2002, 30, 5369–5375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahn, B.T.; Page, D.C. A human sex-chromosomal gene family expressed in male germ cells and encoding variably charged proteins. Hum. Mol. Genet. 2000, 9, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Kakihara, Y.; Houry, W.A. The R2TP complex: Discovery and functions. Biochim. Biophys. Acta 2012, 1823, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Liu, Y.; Zheng, C.; Qu, H. dbEMT 2.0: An updated database for epithelial-mesenchymal transition genes with experimentally verified information and precalculated regulation information for cancer metastasis. J. Genet. Genom. 2019, 46, 595–597. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Malliri, A.; van Es, S.; Huveneers, S.; Collard, J.G. The Rac exchange factor Tiam1 is required for the establishment and maintenance of cadherin-based adhesions. J. Biol. Chem. 2004, 279, 30092–30098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, R.; Tchieu, J.; Mason, M.J.; Yachechko, R.; Kuoy, E.; Horvath, S.; Zhou, Q.; Plath, K. Role of the murine reprogramming factors in the induction of pluripotency. Cell 2009, 136, 364–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, J.; Rafiee, M.R.; Reiland, S.; Polo, J.M.; Gehring, J.; Okawa, S.; Huber, W.; Hochedlinger, K.; Krijgsveld, J. Highly coordinated proteome dynamics during reprogramming of somatic cells to pluripotency. Cell Rep. 2012, 2, 1579–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, E.W.; Green, R. Ribosomopathies: There’s strength in numbers. Science 2017, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Shalaby, N.A.; Buszczak, M. Changes in rRNA transcription influence proliferation and cell fate within a stem cell lineage. Science 2014, 343, 298–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolnough, J.L.; Atwood, B.L.; Liu, Z.; Zhao, R.; Giles, K.E. The Regulation of rRNA Gene Transcription during Directed Differentiation of Human Embryonic Stem Cells. PLoS ONE 2016, 11, e0157276. [Google Scholar] [CrossRef] [PubMed]
- Ginisty, H.; Amalric, F.; Bouvet, P. Nucleolin functions in the first step of ribosomal RNA processing. EMBO J. 1998, 17, 1476–1486. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Shi, G.; Zhou, C.; Lu, R.; Li, H.; Sun, L.; Jin, Y. Nucleolin maintains embryonic stem cell self-renewal by suppression of p53 protein-dependent pathway. J. Biol. Chem. 2011, 286, 43370–43382. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa, N.; Yoshikawa, H.; Magae, S.; Ishikawa, H.; Izumikawa, K.; Terukina, G.; Suzuki, A.; Nakamura-Fujiyama, S.; Miura, Y.; Hayano, T.; et al. Human cell growth regulator Ly-1 antibody reactive homologue accelerates processing of preribosomal RNA. Genes Cells 2014, 19, 273–286. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Wang, D.; Li, H.; Yu, Z.; Chen, X.; Fang, J. Identification of nucleolus-localized PTEN and its function in regulating ribosome biogenesis. Mol. Biol. Rep. 2014, 41, 6383–6390. [Google Scholar] [CrossRef]
- Okumura, K.; Mendoza, M.; Bachoo, R.M.; DePinho, R.A.; Cavenee, W.K.; Furnari, F.B. PCAF modulates PTEN activity. J. Biol. Chem. 2006, 281, 26562–26568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Blank, M.F.; Iyer, A.; Huang, B.; Wang, L.; Grummt, I.; Voit, R. SIRT7-dependent deacetylation of the U3-55k protein controls pre-rRNA processing. Nat. Commun. 2016, 7, 10734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S. RNase L and the NLRP3-inflammasome: An old merchant in a new trade. Cytokine Growth Factor Rev. 2016, 29, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Brennan-Laun, S.E.; Ezelle, H.J.; Li, X.L.; Hassel, B.A. RNase-L control of cellular mRNAs: Roles in biologic functions and mechanisms of substrate targeting. J. Interferon Cytokine Res. 2014, 34, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grummt, I. Life on a planet of its own: Regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003, 17, 1691–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theil, A.F.; Hoeijmakers, J.H.; Vermeulen, W. TTDA: Big impact of a small protein. Exp. Cell Res. 2014, 329, 61–68. [Google Scholar] [CrossRef]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Cevallos, R.; Hu, K. Reprogramming Human Fibroblasts to Induced Pluripotent Stem Cells Using the GFP-marked Lentiviral Vectors in the Chemically Defined Medium. In Nuclear Reprogramming; Hu, K., Ed.; Springer Science + Business Media: New York, NY, USA, 2020. [Google Scholar] [CrossRef]
- Koster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, K. Become competent within one day in generating boxplots and violin plots for a novice without prior R experience. Methods Protoc. 2020, 3, 64. [Google Scholar] [CrossRef]
- Hu, K. Become competent in generating RNA-seq heat maps in one day for novices without prior R experience. In Methods in Molecular Biology; Nuclear Reprogramming; Springer Science + Business Media: New York, NY, USA, 2020; Volume 2239. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, K. Quick, Coordinated and Authentic Reprogramming of Ribosome Biogenesis during iPSC Reprogramming. Cells 2020, 9, 2484. https://doi.org/10.3390/cells9112484
Hu K. Quick, Coordinated and Authentic Reprogramming of Ribosome Biogenesis during iPSC Reprogramming. Cells. 2020; 9(11):2484. https://doi.org/10.3390/cells9112484
Chicago/Turabian StyleHu, Kejin. 2020. "Quick, Coordinated and Authentic Reprogramming of Ribosome Biogenesis during iPSC Reprogramming" Cells 9, no. 11: 2484. https://doi.org/10.3390/cells9112484
APA StyleHu, K. (2020). Quick, Coordinated and Authentic Reprogramming of Ribosome Biogenesis during iPSC Reprogramming. Cells, 9(11), 2484. https://doi.org/10.3390/cells9112484