The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle

 ,
,  ,
,  ,
,
Abstract
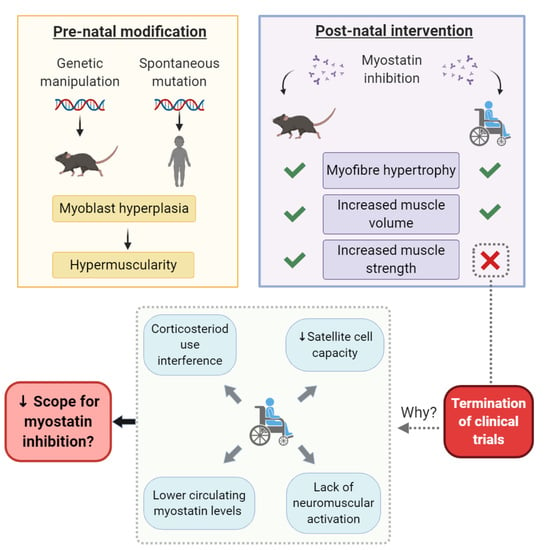
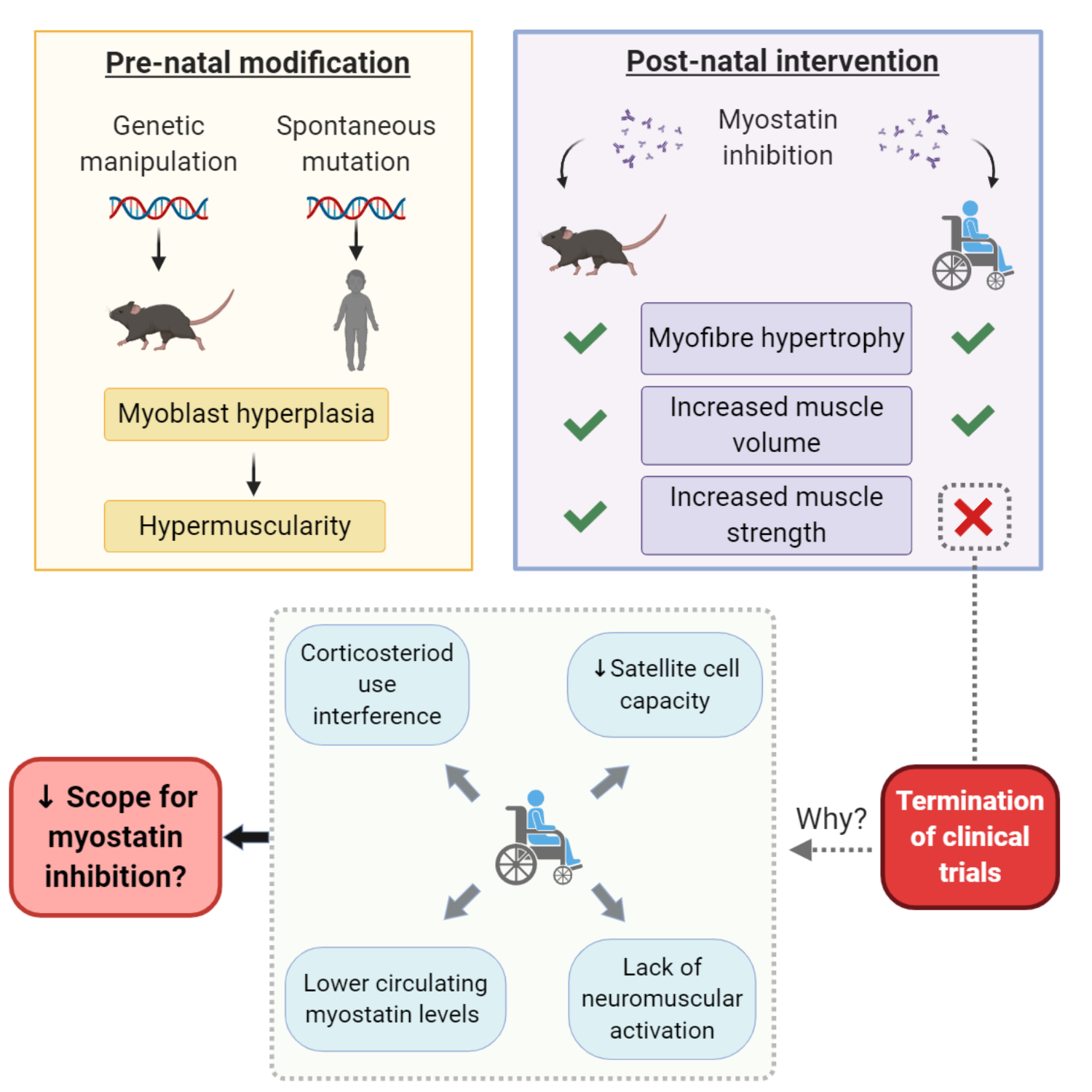
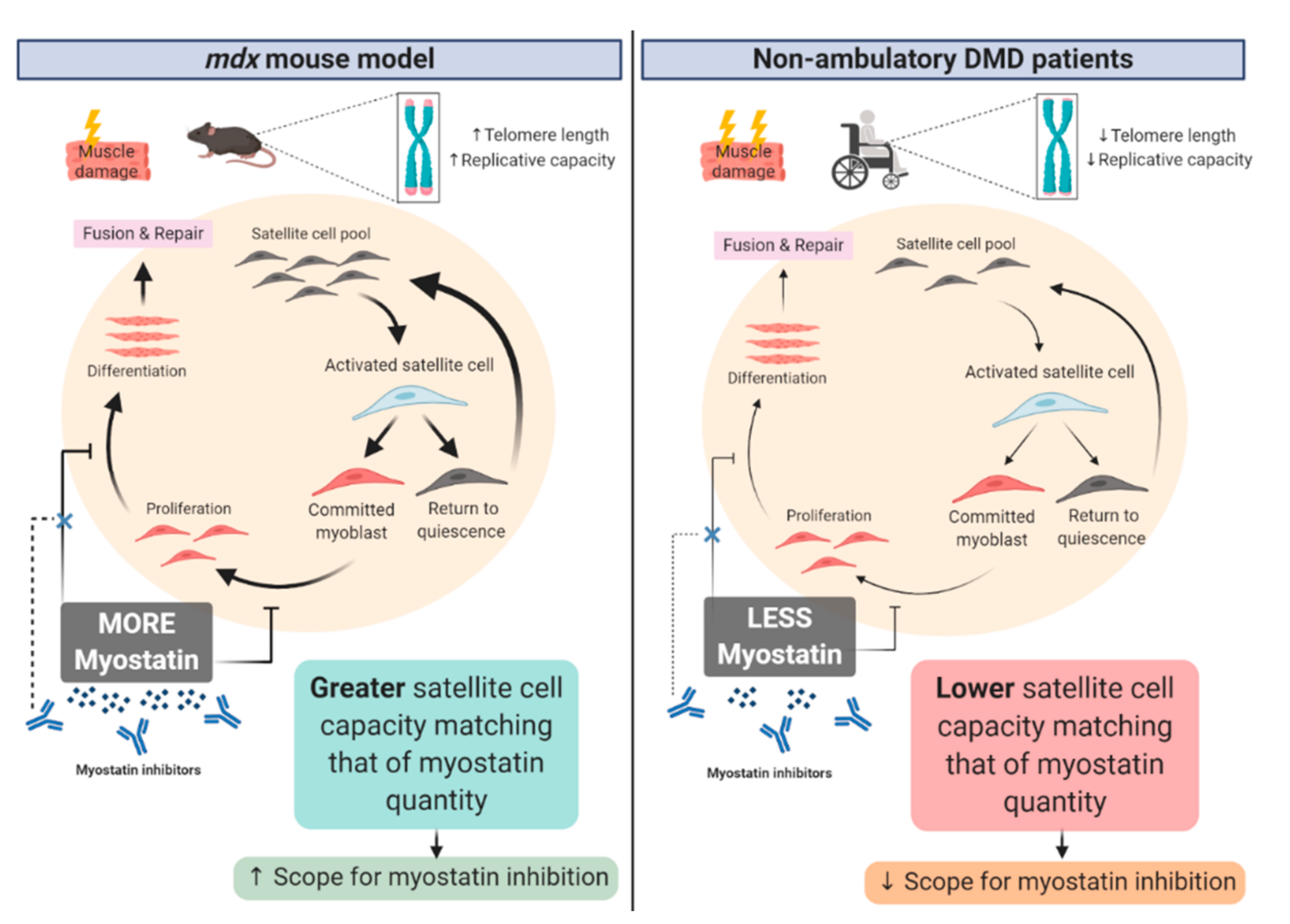
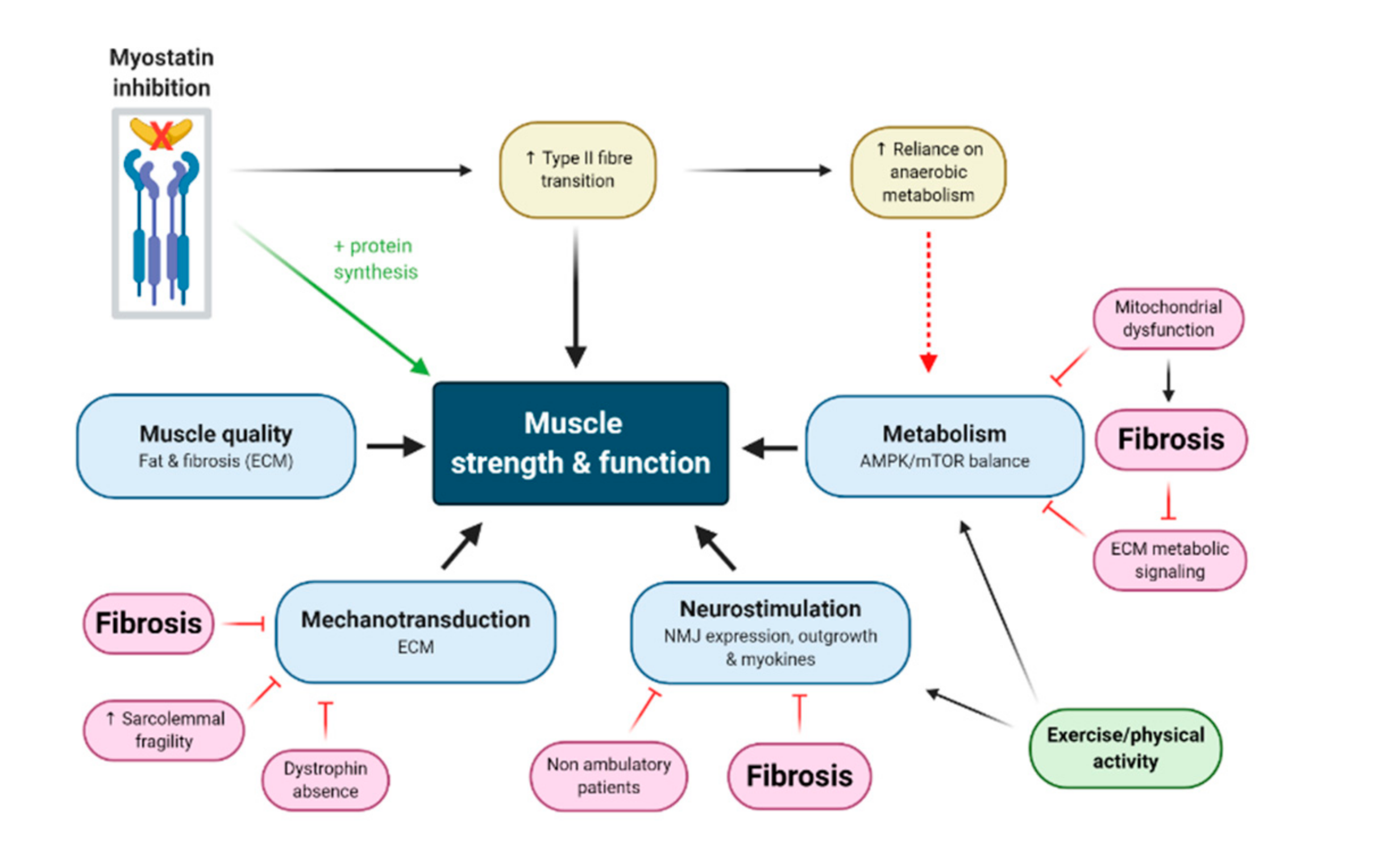
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Myostatin Is Differentially Expressed in Mice and Humans
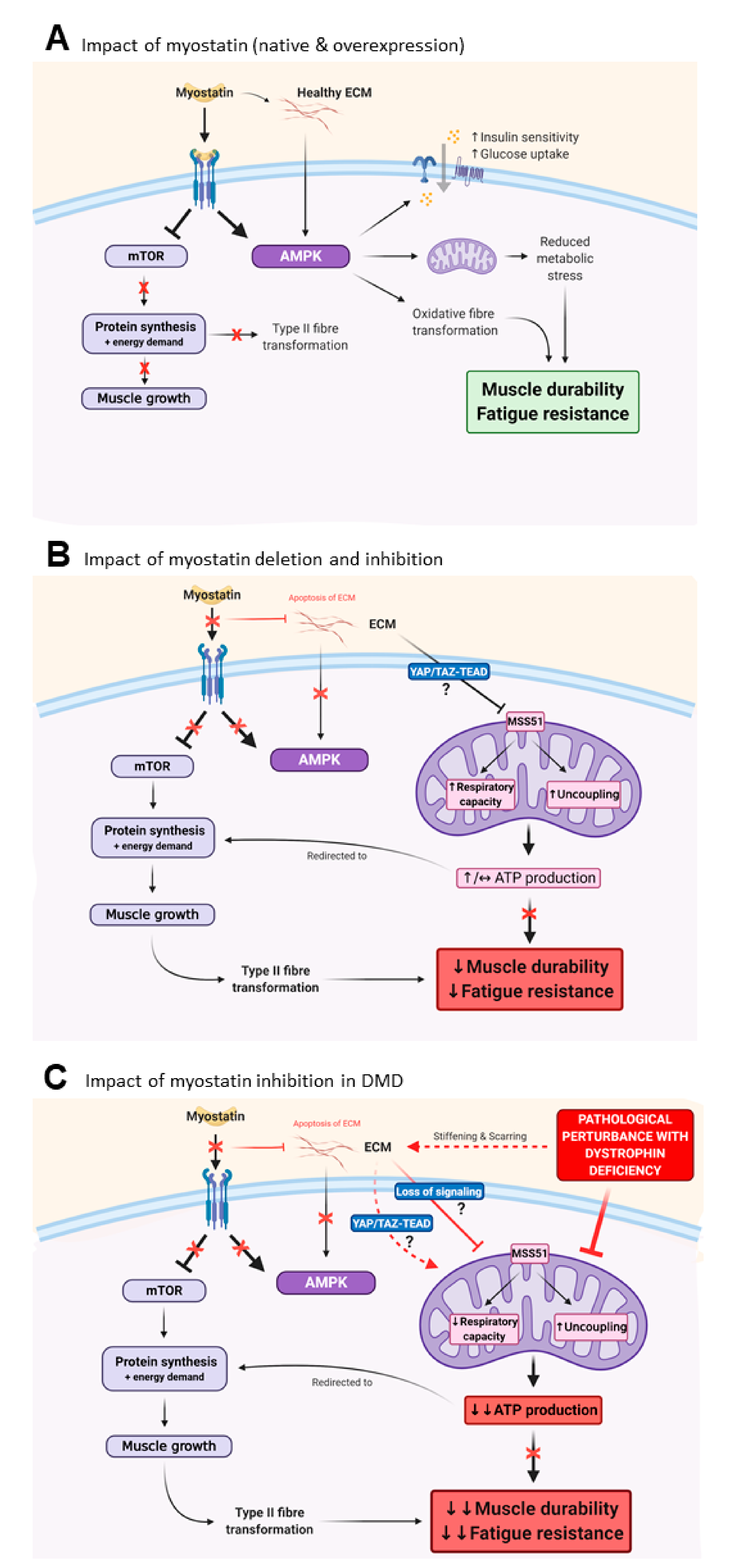
3. Myostatin Inhibition Enhances Muscle Mass but Not Function
3.1. Myostatin Induces Type II Fibre Type Transformations which Are More Susceptible to MD
3.2. Neuromotor Signaling Might Be Fundamentally Required to Convert Mass Gains to Strength Gains
3.3. Myostatin Inhibition Interferes with Muscle Metabolism and Endurance
3.4. Fibrosis of the Extracellular Matrix (ECM) May Drive Unmodifiable Loss of Function
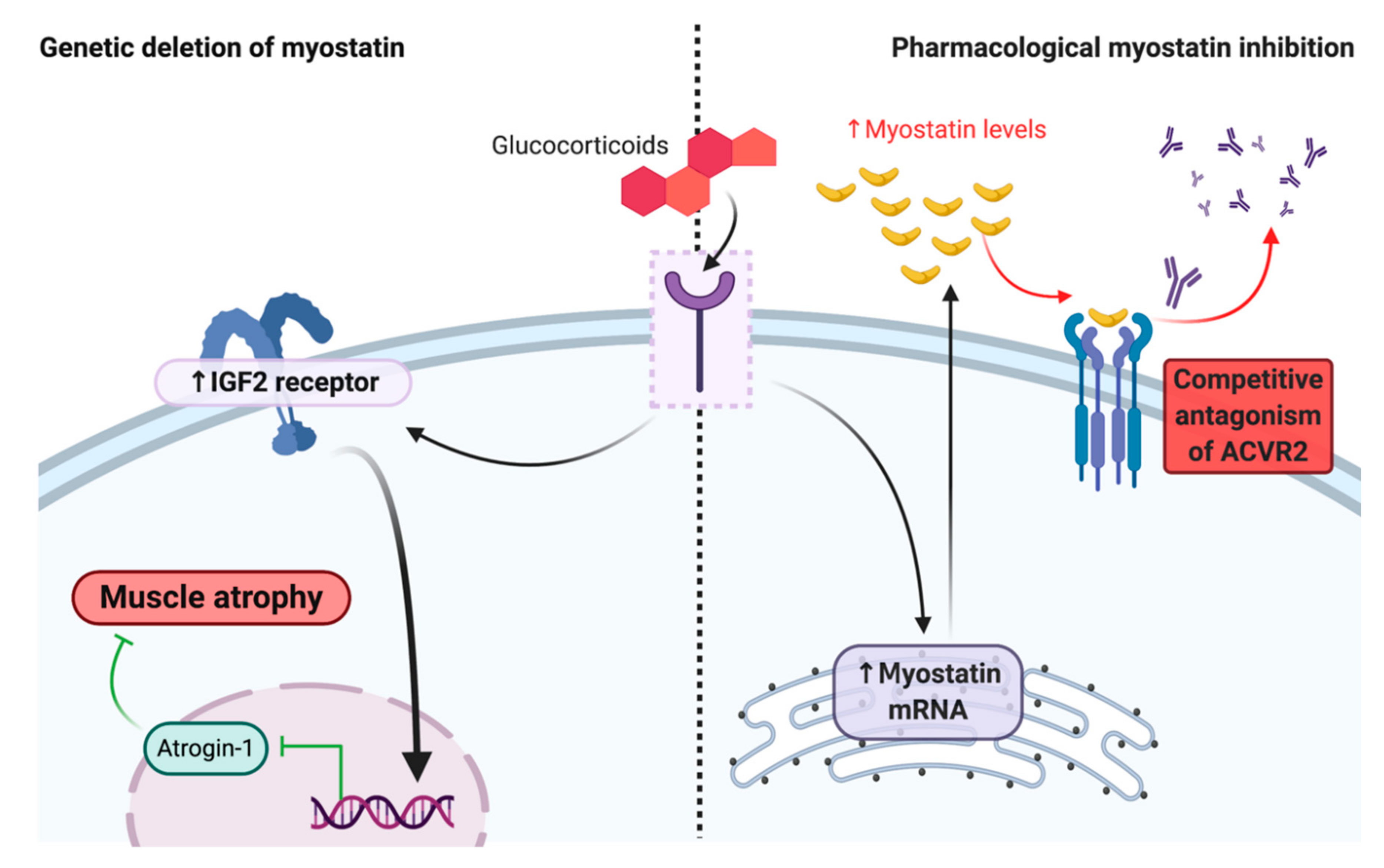
4. Corticosteroids Interfere with Myostatin Inhibition
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McPherron, A.C.; Lawler, A.M.; Lee, S.-J. Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Schuelke, M.; Wagner, K.R.; Stolz, L.E.; Hübner, C.; Riebel, T.; Kömen, W.; Braun, T.; Tobin, J.F.; Lee, S.-J. Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 2004, 350, 2682–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, M.; Ishida, J.; Ebner, N.; Anker, S.D.; Springer, J.; von Haehling, S. Myostatin inhibitors as pharmacological treatment for muscle wasting and muscular dystrophy. JCSM Clin. Rep. 2017, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R. The elusive promise of myostatin inhibition for muscular dystrophy. Curr. Opin. Neurol. 2020, 33, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Lee, Y.-S. Myostatin Inhibitors: Panacea or Predicament for Musculoskeletal Disorders? J. Bone Metab. 2020, 27, 151–165. [Google Scholar] [CrossRef]
- Taylor, W.E.; Bhasin, S.; Artaza, J.; Byhower, F.; Azam, M.; Darril, H.; Willard, J.; Frederick, C.; Kull, J.; Gonzalez-Cadavid, N. Myostatin inhibits cell proliferation and protein synthesis in C2C12 muscle cells. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E221–E228. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.K.; Matthews, K.G.; Oldham, J.M.; Jeanplong, F.; Falconer, S.J.; Bass, J.J.; Senna-Salerno, M.; Bracegirdle, J.W.; McMahon, C.D. Translational Signalling, Atrogenic and Myogenic Gene Expression during Unloading and Reloading of Skeletal Muscle in Myostatin-Deficient Mice. PLoS ONE 2014, 9, e94356. [Google Scholar] [CrossRef]
- Rodriguez, J.; Vernus, B.; Chelh, I.; Cassar-Malek, I.; Gabillard, J.C.; Hadj Sassi, A.; Seiliez, I.; Picard, B.; Bonnieu, A. Myostatin and the skeletal muscle atrophy and hypertrophy signaling pathways. Cell. Mol. Life Sci. 2014, 71, 4361–4371. [Google Scholar] [CrossRef]
- Sartori, R.; Milan, G.; Patron, M.; Mammucari, C.; Blaauw, B.; Abraham, R.; Sandri, M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 2009, 296, C1248–C1257. [Google Scholar] [CrossRef] [Green Version]
- Allen, D.L.; Cleary, A.S.; Lindsay, S.F.; Loh, A.S.; Reed, J.M. Myostatin expression is increased by food deprivation in a muscle-specific manner and contributes to muscle atrophy during prolonged food deprivation in mice. J. Appl. Physiol. 2010, 109, 692–701. [Google Scholar] [CrossRef] [Green Version]
- Thomson, D.M. The role of AMPK in the regulation of skeletal muscle size, hypertrophy, and regeneration. Int. J. Mol. Sci. 2018, 19, 3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Z.; Luo, P.; Lai, W.; Song, T.; Peng, J.; Wei, H.-K. Myostatin inhibits eEF2K-eEF2 by regulating AMPK to suppress protein synthesis. Biochem. Biophys. Res. Commun. 2017, 494, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A. Role of mTORC1 in mechanically induced increases in translation and skeletal muscle mass. J. Appl. Physiol. 2019, 127, 581–590. [Google Scholar] [CrossRef] [PubMed]
- von Walden, F. Ribosome biogenesis in skeletal muscle: Coordination of transcription and translation. J. Appl. Physiol. 2019, 127, 591–598. [Google Scholar] [CrossRef] [PubMed]
- McCroskery, S.; Thomas, M.; Maxwell, L.; Sharma, M.; Kambadur, R. Myostatin negatively regulates satellite cell activation and self-renewal. J. Cell Biol. 2003, 162, 1135–1147. [Google Scholar] [CrossRef] [Green Version]
- Merrell, A.J.; Kardon, G. Development of the diaphragm—A skeletal muscle essential for mammalian respiration. FEBS J. 2013, 280, 4026–4035. [Google Scholar] [CrossRef] [Green Version]
- Malina, R.M. Growth of Muscle Tissue and Muscle Mass. In Human Growth: 2 Postnatal Growth; Falkner, F., Tanner, J.M., Eds.; Springer US: Boston, MA, USA, 1978; pp. 273–294. [Google Scholar] [CrossRef]
- Jones, D.A.; Round, J.M. Muscle development during childhood and adolescence. In The Young Athlete; Hebestreit, H., Bar-Or, O., Eds.; Wiley Online Library: Hoboken, NJ, USA, 2007; p. 18. [Google Scholar]
- Manceau, M.; Gros, J.; Savage, K.; Thomé, V.; McPherron, A.; Paterson, B.; Marcelle, C. Myostatin promotes the terminal differentiation of embryonic muscle progenitors. Genes Dev. 2008, 22, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Schuster-Gossler, K.; Cordes, R.; Gossler, A. Premature myogenic differentiation and depletion of progenitor cells cause severe muscle hypotrophy in Delta1 mutants. Proc. Natl. Acad. Sci. USA 2007, 104, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Vasyutina, E.; Lenhard, D.C.; Wende, H.; Erdmann, B.; Epstein, J.A.; Birchmeier, C. RBP-J (Rbpsuh) is essential to maintain muscle progenitor cells and to generate satellite cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4443–4448. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R.; McPherron, A.C.; Winik, N.; Lee, S.J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol. 2002, 52, 832–836. [Google Scholar] [CrossRef]
- Mosher, D.S.; Quignon, P.; Bustamante, C.D.; Sutter, N.B.; Mellersh, C.S.; Parker, H.G.; Ostrander, E.A. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007, 3, e79. [Google Scholar] [CrossRef] [PubMed]
- Clop, A.; Marcq, F.; Takeda, H.; Pirottin, D.; Tordoir, X.; Bibé, B.; Bouix, J.; Caiment, F.; Elsen, J.-M.; Eychenne, F. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 2006, 38, 813–818. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lee, S.-J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall’Olio, S.; Fontanesi, L.; Nanni Costa, L.; Tassinari, M.; Minieri, L.; Falaschini, A. Analysis of horse myostatin gene and identification of single nucleotide polymorphisms in breeds of different morphological types. J. Biomed. Biotechnol. 2010, 2010, 542945. [Google Scholar] [PubMed] [Green Version]
- Hill, E.W.; Gu, J.; Eivers, S.S.; Fonseca, R.G.; McGivney, B.A.; Govindarajan, P.; Orr, N.; Katz, L.M.; MacHugh, D. A Sequence Polymorphism in MSTN Predicts Sprinting Ability and Racing Stamina in Thoroughbred Horses. PLoS ONE 2010, 5, e8645. [Google Scholar] [CrossRef]
- Gilson, H.; Schakman, O.; Combaret, L.; Lause, P.; Grobet, L.; Attaix, D.; Ketelslegers, J.-M.; Thissen, J.-P. Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology 2007, 148, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Andre, M.S.; Johnson, M.; Bansal, P.N.; Wellen, J.; Robertson, A.; Opsahl, A.; Burch, P.M.; Bialek, P.; Morris, C.; Owens, J. A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys. Skelet. Muscle 2017, 7, 25. [Google Scholar]
- Bogdanovich, S.; Krag, T.O.; Barton, E.R.; Morris, L.D.; Whittemore, L.-A.; Ahima, R.S.; Khurana, T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002, 420, 418–421. [Google Scholar] [CrossRef]
- Bogdanovich, S.; Perkins, K.J.; Krag, T.O.B.; Whittemore, L.-A.; Khurana, T.S. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. 2005, 19, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Iskenderian, A.; Liu, N.; Deng, Q.; Huang, Y.; Shen, C.; Palmieri, K.; Crooker, R.; Lundberg, D.; Kastrapeli, N.; Pescatore, B.; et al. Myostatin and activin blockade by engineered follistatin results in hypertrophy and improves dystrophic pathology in mdx mouse more than myostatin blockade alone. Skelet. Muscle 2018, 8, 34. [Google Scholar] [CrossRef]
- Murphy, K.T.; Ryall, J.G.; Snell, S.M.; Nair, L.; Koopman, R.; Krasney, P.A.; Ibebunjo, C.; Holden, K.S.; Loria, P.M.; Salatto, C.T.; et al. Antibody-directed myostatin inhibition improves diaphragm pathology in young but not adult dystrophic mdx mice. Am. J. Pathol. 2010, 176, 2425–2434. [Google Scholar] [CrossRef] [PubMed]
- Forcina, L.; Miano, C.; Pelosi, L.; Musarò, A. An Overview about the Biology of Skeletal Muscle Satellite Cells. Curr. Genom. 2019, 20, 24–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Datzkiw, D.; Rudnicki, M.A. Satellite cells in ageing: Use it or lose it. Open Biol. 2020, 10, 200048. [Google Scholar] [CrossRef] [PubMed]
- Mouly, V.; Aamiri, A.; Bigot, A.; Cooper, R.; Di Donna, S.; Furling, D.; Gidaro, T.; Jacquemin, V.; Mamchaoui, K.; Negroni, E. The mitotic clock in skeletal muscle regeneration, disease and cell mediated gene therapy. Acta Physiol. Scand. 2005, 184, 3–15. [Google Scholar] [CrossRef]
- Kipling, D.; Cooke, H.J. Hypervariable ultra-long telomeres in mice. Nature 1990, 347, 400–402. [Google Scholar] [CrossRef]
- Yucel, N.; Chang, A.C.; Day, J.W.; Rosenthal, N.; Blau, H.M. Humanizing the mdx mouse model of DMD: The long and the short of it. NPJ Regen. Med. 2018, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Van Pelt, D.W.; Kharaz, Y.A.; Sarver, D.C.; Eckhardt, L.R.; Dzierzawski, J.T.; Disser, N.P.; Piacentini, A.N.; Comerford, E.; McDonagh, B.; Mendias, C.L. Multiomics Analysis of the mdx/mTR Mouse Model of Duchenne Muscular Dystrophy. Connect. Tissue Res. 2020, 62. [Google Scholar] [CrossRef]
- Van Pelt, D.W.; Kharaz, Y.A.; Sarver, D.C.; Eckhardt, L.R.; Dzierzawski, J.T.; Disser, N.P.; Piacentini, A.N.; Comerford, E.; McDonagh, B.; Mendias, C.L. Evaluating Muscle Fiber Contractility and the Transcriptome, Proteome, Metabolome, and Lipidome of the mdx/mTR Mouse Model of Duchenne Muscular Dystrophy. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Meijer, J.P.; Jaspers, R.T.; Rittweger, J.; Seynnes, O.R.; Kamandulis, S.; Brazaitis, M.; Skurvydas, A.; Pišot, R.; Šimunič, B.; Narici, M.V.; et al. Single muscle fibre contractile properties differ between body-builders, power athletes and control subjects. Exp. Physiol. 2015, 100, 1331–1341. [Google Scholar] [CrossRef]
- Ahtiainen, J.P.; Walker, S.; Peltonen, H.; Holviala, J.; Sillanpää, E.; Karavirta, L.; Sallinen, J.; Mikkola, J.; Valkeinen, H.; Mero, A.; et al. Heterogeneity in resistance training-induced muscle strength and mass responses in men and women of different ages. AGE 2016, 38, 10. [Google Scholar] [CrossRef] [Green Version]
- Manini, T.M.; Clark, B.C. Dynapenia and Aging: An Update. J. Gerontol. Ser. A 2011, 67A, 28–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcotte, G.R.; West, D.W.D.; Baar, K. The molecular basis for load-induced skeletal muscle hypertrophy. Calcif. Tissue Int. 2015, 96, 196–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malisoux, L.; Francaux, M.; Nielens, H.; Theisen, D. Stretch-shortening cycle exercises: An effective training paradigm to enhance power output of human single muscle fibers. J. Appl. Physiol. 2006, 100, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, D.L.; Leinwand, L.A. Intracellular Calcium and Myosin Isoform Transitions: CALCINEURIN AND CALCIUM-CALMODULIN KINASE PATHWAYS REGULATE PREFERENTIAL ACTIVATION OF THE IIa MYOSIN HEAVY CHAIN PROMOTER. J. Biol. Chem. 2002, 277, 45323–45330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonanno, A.; Fields, R.D. Gene regulation by patterned electrical activity during neural and skeletal muscle development. Curr. Opin. Neurobiol. 1999, 9, 110–120. [Google Scholar] [CrossRef]
- Xuan, M.-F.; Luo, Z.-B.; Wang, J.-X.; Guo, Q.; Han, S.-Z.; Jin, S.-S.; Kang, J.-D.; Yin, X.-J. Shift from slow-to fast-twitch muscle fibres in skeletal muscle of newborn heterozygous and homozygous myostatin-knockout piglets. Reprod. Fertil. Dev. 2019, 31, 1628–1636. [Google Scholar] [CrossRef]
- Marini, J.-F.; Pons, F.; Leger, J.; Loffreda, N.; Anoal, M.; Chevallay, M.; Fardeau, M.; Leger, J.J. Expression of myosin heavy chain isoforms in Duchenne muscular dystrophy patients and carriers. Neuromuscul. Disord. 1991, 1, 397–409. [Google Scholar] [CrossRef]
- Pedemonte, M.; Sandri, C.; Schiaffino, S.; Minetti, C. Early decrease of IIx myosin heavy chain transcripts in Duchenne muscular dystrophy. Biochem. Biophys. Res. Commun. 1999, 255, 466–469. [Google Scholar] [CrossRef]
- Webster, C.; Silberstein, L.; Hays, A.P.; Blau, H.M. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 1988, 52, 503–513. [Google Scholar] [CrossRef]
- Anderson, E.J.; Neufer, P.D. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H2O2 generation. Am. J. Physiol. Cell Physiol. 2006, 290, C844–C851. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.L.; Fu, R.; Mitchell, E.W. Glutathione and antioxidant enzymes in skeletal muscle: Effects of fiber type and exercise intensity. J. Appl. Physiol. 1992, 73, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne Muscular Dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timpani, C.A.; Goodman, C.A.; Stathis, C.G.; White, J.D.; Mamchaoui, K.; Butler-Browne, G.; Gueven, N.; Hayes, A.; Rybalka, E. Adenylosuccinic acid therapy ameliorates murine Duchenne Muscular Dystrophy. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Timpani, C.A.; Trewin, A.J.; Stojanovska, V.; Robinson, A.; Goodman, C.A.; Nurgali, K.; Betik, A.C.; Stepto, N.; Hayes, A.; McConell, G.K. Attempting to compensate for reduced neuronal nitric oxide synthase protein with nitrate supplementation cannot overcome metabolic dysfunction but rather has detrimental effects in dystrophin-deficient mdx muscle. Neurotherapeutics 2017, 14, 429–446. [Google Scholar] [CrossRef] [Green Version]
- Disatnik, M.-H.; Dhawan, J.; Yu, Y.; Beal, M.F.; Whirl, M.M.; Franco, A.A.; Rando, T.A. Evidence of oxidative stress in mdx mouse muscle: Studies of the pre-necrotic state. J. Neurol. Sci. 1998, 161, 77–84. [Google Scholar] [CrossRef]
- Rando, T.A.; Disatnik, M.-H.; Yu, Y.; Franco, A. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul. Disord. 1998, 8, 14–21. [Google Scholar] [CrossRef]
- Talbot, J.; Maves, L. Skeletal muscle fiber type: Using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 518–534. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, I.; Pawlak, S.; Marraffino, S.; Christensen, J.; Sherlock, S.P.; Alvey, C.; Morris, C.; Arkin, S.; Binks, M. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Domagrozumab (PF-06252616), an Antimyostatin Monoclonal Antibody, in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2018, 7, 484–497. [Google Scholar] [CrossRef]
- Wagner, K.R.; Abdel-Hamid, H.Z.; Mah, J.K.; Campbell, C.; Guglieri, M.; Muntoni, F.; Takeshima, Y.; McDonald, C.M.; Kostera-Pruszczyk, A.; Karachunski, P.; et al. Randomized phase 2 trial and open-label extension of domagrozumab in Duchenne muscular dystrophy. Neuromuscul. Disord. 2020, 30, 492–502. [Google Scholar] [CrossRef]
- Bhasin, S.; Travison, T.G.; Manini, T.M.; Patel, S.; Pencina, K.M.; Fielding, R.A.; Magaziner, J.M.; Newman, A.B.; Kiel, D.P.; Cooper, C.; et al. Sarcopenia Definition: The Position Statements of the Sarcopenia Definition and Outcomes Consortium. J. Am. Geriatr. Soc. 2020, 68, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, B.J.; Ogborn, D.; Krieger, J.W. Effects of Resistance Training Frequency on Measures of Muscle Hypertrophy: A Systematic Review and Meta-Analysis. Sports Med. 2016, 46, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, H.; Loenneke, J.P.; Buckner, S.L.; Abe, T. Muscle growth across a variety of exercise modalities and intensities: Contributions of mechanical and metabolic stimuli. Med. Hypotheses 2016, 88, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Rindom, E.; Herskind, J.; Blaauw, B.; Overgaard, K.; Vissing, K.; de Paoli, F.V. Concomitant excitation and tension development are required for myocellular gene expression and protein synthesis in rat skeletal muscle. Acta Physiol. 2020, e13540. [Google Scholar] [CrossRef] [PubMed]
- Wackerhage, H.; Schoenfeld, B.J.; Hamilton, D.L.; Lehti, M.; Hulmi, J.J. Stimuli and sensors that initiate skeletal muscle hypertrophy following resistance exercise. J. Appl. Physiol. 2019, 126, 30–43. [Google Scholar] [CrossRef]
- Chan, S.; Head, S.I. The role of branched fibres in the pathogenesis of Duchenne muscular dystrophy. Exp. Physiol. 2011, 96, 564–571. [Google Scholar] [CrossRef]
- Murach, K.A.; Dungan, C.M.; Peterson, C.A.; McCarthy, J.J. Muscle fiber splitting is a physiological response to extreme loading in animals. Exerc. Sport Sci. Rev. 2019, 47, 108. [Google Scholar] [CrossRef]
- Lu, B.; Je, H.-S. Neurotrophic regulation of the development and function of the neuromuscular synapses. J. Neurocytol. 2003, 32, 931–941. [Google Scholar] [CrossRef]
- Mills, R.; Taylor-Weiner, H.; Correia, J.C.; Agudelo, L.Z.; Allodi, I.; Kolonelou, C.; Martinez-Redondo, V.; Ferreira, D.M.S.; Nichterwitz, S.; Comley, L.H.; et al. Neurturin is a PGC-1α1-controlled myokine that promotes motor neuron recruitment and neuromuscular junction formation. Mol. Metab. 2018, 7, 12–22. [Google Scholar] [CrossRef]
- Delezie, J.; Weihrauch, M.; Maier, G.; Tejero, R.; Ham, D.J.; Gill, J.F.; Karrer-Cardel, B.; Rüegg, M.A.; Tabares, L.; Handschin, C. BDNF is a mediator of glycolytic fiber-type specification in mouse skeletal muscle. Proc. Natl. Acad. Sci. USA 2019, 116, 16111–16120. [Google Scholar] [CrossRef] [Green Version]
- Lovering, R.M.; Iyer, S.R.; Edwards, B.; Davies, K.E. Alterations of neuromuscular junctions in Duchenne muscular dystrophy. Neurosci. Lett. 2020, 737, 135304. [Google Scholar] [CrossRef] [PubMed]
- Willmann, R.; Possekel, S.; Dubach-Powell, J.; Meier, T.; Ruegg, M.A. Mammalian animal models for Duchenne muscular dystrophy. Neuromuscul. Disord. 2009, 19, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Qiao, C.; Li, J.; Xiao, B.; Li, J.; Xiao, X. A GDF11/myostatin inhibitor, GDF11 propeptide-Fc, increases skeletal muscle mass and improves muscle strength in dystrophic mdx mice. Skelet. Muscle 2019, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Béchir, N.; Pecchi, E.; Vilmen, C.; Le Fur, Y.; Amthor, H.; Bernard, M.; Bendahan, D.; Giannesini, B. ActRIIB blockade increases force-generating capacity and preserves energy supply in exercising mdx mouse muscle in vivo. FASEB J. 2016, 30, 3551–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boido, M.; Butenko, O.; Filippo, C.; Schellino, R.; Vrijbloed, J.W.; Fariello, R.G.; Vercelli, A. A new protein curbs the hypertrophic effect of myostatin inhibition, adding remarkable endurance to motor performance in mice. PLoS ONE 2020, 15, e0228653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Ye, J.; Cao, L.; Zhang, Y.; Xia, W.; Zhu, D. Myostatin regulates glucose metabolism via the AMP-activated protein kinase pathway in skeletal muscle cells. Int. J. Biochem. Cell Biol. 2010, 42, 2072–2081. [Google Scholar] [CrossRef]
- Mouisel, E.; Relizani, K.; Mille-Hamard, L.; Denis, R.; Hourdé, C.; Agbulut, O.; Patel, K.; Arandel, L.; Morales-Gonzalez, S.; Vignaud, A.; et al. Myostatin is a key mediator between energy metabolism and endurance capacity of skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R444–R454. [Google Scholar] [CrossRef]
- Guo, T.; Jou, W.; Chanturiya, T.; Portas, J.; Gavrilova, O.; McPherron, A.C. Myostatin Inhibition in Muscle, but Not Adipose Tissue, Decreases Fat Mass and Improves Insulin Sensitivity. PLoS ONE 2009, 4, e4937. [Google Scholar] [CrossRef] [Green Version]
- McPherron, A.C.; Lee, S.-J. Suppression of body fat accumulation in myostatin-deficient mice. J. Clin. Investig. 2002, 109, 595–601. [Google Scholar] [CrossRef]
- Zhang, C.; McFarlane, C.; Lokireddy, S.; Masuda, S.; Ge, X.; Gluckman, P.D.; Sharma, M.; Kambadur, R. Inhibition of myostatin protects against diet-induced obesity by enhancing fatty acid oxidation and promoting a brown adipose phenotype in mice. Diabetologia 2012, 55, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Romani, P.; Valcarcel-Jimenez, L.; Frezza, C.; Dupont, S. Crosstalk between mechanotransduction and metabolism. Nat. Rev. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lebrasseur, N.K. Building muscle, browning fat and preventing obesity by inhibiting myostatin. Diabetologia 2012, 55, 13–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyer, A.L.; Wagner, K.R. Mammalian Mss51 is a skeletal muscle-specific gene modulating cellular metabolism. J. Neuromuscul. Dis. 2015, 2, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Rovira Gonzalez, Y.I.; Moyer, A.L.; LeTexier, N.J.; Bratti, A.D.; Feng, S.; Sun, C.; Liu, T.; Mula, J.; Jha, P.; Iyer, S.R.; et al. Mss51 deletion enhances muscle metabolism and glucose homeostasis in mice. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Ploquin, C.; Chabi, B.; Fouret, G.; Vernus, B.; Feillet-Coudray, C.; Coudray, C.; Bonnieu, A.; Ramonatxo, C. Lack of myostatin alters intermyofibrillar mitochondria activity, unbalances redox status, and impairs tolerance to chronic repetitive contractions in muscle. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1000–E1008. [Google Scholar] [CrossRef] [PubMed]
- Giannesini, B.; Vilmen, C.; Amthor, H.; Bernard, M.; Bendahan, D. Lack of myostatin impairs mechanical performance and ATP cost of contraction in exercising mouse gastrocnemius muscle in vivo. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E33–E40. [Google Scholar] [CrossRef] [Green Version]
- Baati, N.; Feillet-Coudray, C.; Fouret, G.; Vernus, B.; Goustard, B.; Coudray, C.; Lecomte, J.; Blanquet, V.; Magnol, L.; Bonnieu, A.; et al. Myostatin deficiency is associated with lipidomic abnormalities in skeletal muscles. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1044–1055. [Google Scholar] [CrossRef] [Green Version]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- Pauly, M.; Chabi, B.; Favier, F.B.; Vanterpool, F.; Matecki, S.; Fouret, G.; Bonafos, B.; Vernus, B.; Feillet-Coudray, C.; Coudray, C. Combined strategies for maintaining skeletal muscle mass and function in aging: Myostatin inactivation and AICAR-associated oxidative metabolism induction. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2015, 70, 1077–1087. [Google Scholar] [CrossRef] [Green Version]
- Kainulainen, H.; Papaioannou, K.G.; Silvennoinen, M.; Autio, R.; Saarela, J.; Oliveira, B.M.; Nyqvist, M.; Pasternack, A.; AC’t Hoen, P.A.; Kujala, U.M.; et al. Myostatin/activin blocking combined with exercise reconditions skeletal muscle expression profile of mdx mice. Mol. Cell Endocrinol. 2015, 399, 131–142. [Google Scholar] [CrossRef]
- Baati, N.; Feillet-Coudray, C.; Fouret, G.; Vernus, B.; Goustard, B.; Jollet, M.; Bertrand-Gaday, C.; Coudray, C.; Lecomte, J.; Bonnieu, A.; et al. New evidence of exercise training benefits in myostatin-deficient mice: Effect on lipidomic abnormalities. Biochem. Biophys. Res. Commun. 2019, 516, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Matsakas, A.; Macharia, R.; Otto, A.; Elashry, M.I.; Mouisel, E.; Romanello, V.; Sartori, R.; Amthor, H.; Sandri, M.; Narkar, V.; et al. Exercise training attenuates the hypermuscular phenotype and restores skeletal muscle function in the myostatin null mouse. Exp. Physiol. 2012, 97, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Matsakas, A.; Mouisel, E.; Amthor, H.; Patel, K. Myostatin knockout mice increase oxidative muscle phenotype as an adaptive response to exercise. J. Muscle Res. Cell Motil. 2010, 31, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Hulmi, J.J.; Hentilä, J.; DeRuisseau, K.C.; Oliveira, B.M.; Papaioannou, K.G.; Autio, R.; Kujala, U.M.; Ritvos, O.; Kainulainen, H.; Korkmaz, A.; et al. Effects of muscular dystrophy, exercise and blocking activin receptor IIB ligands on the unfolded protein response and oxidative stress. Free Radic. Biol. Med. 2016, 99, 308–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulmi, J.J.; Oliveira, B.M.; Silvennoinen, M.; Hoogaars, W.M.; Pasternack, A.; Kainulainen, H.; Ritvos, O. Exercise restores decreased physical activity levels and increases markers of autophagy and oxidative capacity in myostatin/activin-blocked mdx mice. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E171–E182. [Google Scholar] [CrossRef]
- Chinet, A.; Even, P.; Decrouy, A. Dystrophin-dependent efficiency of metabolic pathways in mouse skeletal muscles. Cell. Mol. Life Sci. 1994, 50, 602–605. [Google Scholar] [CrossRef]
- Onopiuk, M.; Brutkowski, W.; Wierzbicka, K.; Wojciechowska, S.; Szczepanowska, J.; Fronk, J.; Lochmüller, H.; Górecki, D.C.; Zabłocki, K. Mutation in dystrophin-encoding gene affects energy metabolism in mouse myoblasts. Biochem. Biophys. Res. Commun. 2009, 386, 463–466. [Google Scholar] [CrossRef]
- Passaquin, A.-C.; Renard, M.; Kay, L.; Challet, C.; Mokhtarian, A.; Wallimann, T.; Ruegg, U.T. Creatine supplementation reduces skeletal muscle degeneration and enhances mitochondrial function in mdx mice. Neuromuscul. Disord. 2002, 12, 174–182. [Google Scholar] [CrossRef]
- Percival, J.M.; Siegel, M.P.; Knowels, G.; Marcinek, D.J. Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Genet. 2013, 22, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in mdx Mice. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Ramos, S.V.; Hughes, M.C.; Delfinis, L.J.; Bellissimo, C.A.; Perry, C.G.R. Mitochondrial bioenergetic dysfunction in the D2.mdx model of Duchenne muscular dystrophy is associated with microtubule disorganization in skeletal muscle. PLoS ONE 2020, 15, e0237138. [Google Scholar] [CrossRef] [PubMed]
- Barbiroli, B.; Funicello, R.; Ferlini, A.; Montagna, P.; Zaniol, P. Muscle energy metabolism in female DMD/BMD carriers: A 31P-MR spectroscopy study. Muscle Nerve 1992, 15, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Barbiroli, B.; McCully, K.K.; Iotti, S.; Lodi, R.; Zaniol, P.; Chance, B. Further impairment of muscle phosphate kinetics by lengthening exercise in DMD/BMD carriers: An in vivo 31P-NMR spectroscopy study. J. Neurol. Sci. 1993, 119, 65–73. [Google Scholar] [CrossRef]
- Ljubicic, V.; Jasmin, B.J. Metformin increases peroxisome proliferator–activated receptor γ Co-activator-1α and utrophin a expression in dystrophic skeletal muscle. Muscle Nerve 2015, 52, 139–142. [Google Scholar] [CrossRef]
- Bell, E.L.; Shine, R.W.; Dwyer, P.; Olson, L.; Truong, J.; Fredenburg, R.; Goddeeris, M.; Stickens, D.; Tozzo, E. PPARδ modulation rescues mitochondrial fatty acid oxidation defects in the mdx model of muscular dystrophy. Mitochondrion 2019, 46, 51–58. [Google Scholar] [CrossRef]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. J. Pharmacol. Exp. Ther. 2011, 338, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Santhera Pharmaceuticals. Santhera to Discontinue Phase 3 SIDEROS Study and Development of Puldysa®®® in Duchenne Muscular Dystrophy (DMD) and Focus on Vamorolone. Press Release 6 October 2020. Available online: http://www.santhera.com/investors-and-media/news-and-media-center/press-releases (accessed on 19 November 2020).
- Grounds, M.D.; Radley, H.G.; Lynch, G.S.; Nagaraju, K.; De Luca, A. Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol. Dis. 2008, 31, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Kramerova, I.; Marinov, M.; Owens, J.; Lee, S.-J.; Becerra, D.; Spencer, M.J. Myostatin inhibition promotes fast fibre hypertrophy but causes loss of AMP-activated protein kinase signalling and poor exercise tolerance in a model of limb-girdle muscular dystrophy R1/2A. J. Physiol. 2020, 598, 3927–3939. [Google Scholar] [CrossRef]
- Glenn, N.O.; Henry, C.A. How muscle contraction strengthens tendons. eLife 2019, 8, e44149. [Google Scholar] [CrossRef]
- Li, Z.B.; Kollias, H.D.; Wagner, K.R. Myostatin directly regulates skeletal muscle fibrosis. J. Biol. Chem. 2008, 283, 19371–19378. [Google Scholar] [CrossRef] [Green Version]
- Kornegay, J.N.; Bogan, D.J.; Bogan, J.R.; Dow, J.L.; Wang, J.; Fan, Z.; Liu, N.; Warsing, L.C.; Grange, R.W.; Ahn, M.; et al. Dystrophin-deficient dogs with reduced myostatin have unequal muscle growth and greater joint contractures. Skelet. Muscle 2016, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansay, M.; Hanset, R. Anatomical, physiological and biochemical differences between conventional and double-muscled cattle in the Belgian blue and white breed. Livest. Prod. Sci. 1979, 6, 5–13. [Google Scholar] [CrossRef]
- Guo, W.; Miller, A.D.; Pencina, K.; Wong, S.; Lee, A.; Yee, M.; Toraldo, G.; Jasuja, R.; Bhasin, S. Joint dysfunction and functional decline in middle age myostatin null mice. Bone 2016, 83, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar] [PubMed]
- Zhou, L.; Lu, H. Targeting Fibrosis in Duchenne Muscular Dystrophy. J. Neuropathol. Exp. Neurol. 2010, 69, 771–776. [Google Scholar] [CrossRef]
- Desguerre, I.; Mayer, M.; Leturcq, F.; Barbet, J.P.; Gherardi, R.K.; Christov, C. Endomysial fibrosis in Duchenne muscular dystrophy: A marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 2009, 68, 762–773. [Google Scholar] [CrossRef] [Green Version]
- Stedman, H.; Sweeney, H.; Shrager, J.; Maguire, H.; Panettieri, R.; Petrof, B.a.; Narusawa, M.; Leferovich, J.; Sladky, J.; Kelly, A. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef]
- Nakatani, M.; Takehara, Y.; Sugino, H.; Matsumoto, M.; Hashimoto, O.; Hasegawa, Y.; Murakami, T.; Uezumi, A.; Takeda, S.i.; Noji, S.; et al. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J. 2008, 22, 477–487. [Google Scholar] [CrossRef]
- Qiao, C.; Li, J.; Jiang, J.; Zhu, X.; Wang, B.; Li, J.; Xiao, X. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum. Gene Ther 2008, 19, 241–254. [Google Scholar] [CrossRef]
- Bettica, P.; Petrini, S.; D’Oria, V.; D’Amico, A.; Catteruccia, M.; Pane, M.; Sivo, S.; Magri, F.; Brajkovic, S.; Messina, S.; et al. Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul. Disord. 2016, 26, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Gloss, D.; Moxley, R.T.; Ashwal, S.; Oskoui, M. Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy. Rep. Guidel. Dev. Subcomm. Am. Acad. Neurol. 2016, 86, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Hammers, D.W.; Hart, C.C.; Patsalos, A.; Matheny, M.K.; Wright, L.A.; Nagy, L.; Sweeney, H.L. Glucocorticoids counteract hypertrophic effects of myostatin inhibition in dystrophic muscle. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, D.L.; Loh, A.S. Posttranscriptional mechanisms involving microRNA-27a and b contribute to fast-specific and glucocorticoid-mediated myostatin expression in skeletal muscle. Am. J. Physiol. Cell Physiol. 2011, 300, C124–C137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.; Mallidis, C.; Bhasin, S.; Mahabadi, V.; Artaza, J.; Gonzalez-Cadavid, N.; Arias, J.; Salehian, B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E363–E371. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jiao, H.; Zhao, J.; Wang, X.; Lin, H. Glucocorticoids enhance muscle proteolysis through a myostatin-dependent pathway at the early stage. PLoS ONE 2016, 11, e0156225. [Google Scholar] [CrossRef] [PubMed]
- Consitt, L.A.; Clark, B.C. The Vicious Cycle of Myostatin Signaling in Sarcopenic Obesity: Myostatin Role in Skeletal Muscle Growth, Insulin Signaling and Implications for Clinical Trials. J. Frailty Aging 2018, 7, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Rodino-Klapac, L.R.; Janssen, P.M.; Shontz, K.M.; Canan, B.; Montgomery, C.L.; Griffin, D.; Heller, K.; Schmelzer, L.; Handy, C.; Clark, K.R.; et al. Micro-dystrophin and follistatin co-delivery restores muscle function in aged DMD model. Hum. Mol. Genet. 2013, 22, 4929–4937. [Google Scholar] [CrossRef] [PubMed]
- Hoogaars, W.M.; Mouisel, E.; Pasternack, A.; Hulmi, J.J.; Relizani, K.; Schuelke, M.; Schirwis, E.; Garcia, L.; Ritvos, O.; Ferry, A.; et al. Combined effect of AAV-U7-induced dystrophin exon skipping and soluble activin Type IIB receptor in mdx mice. Hum. Gene. Ther. 2012, 23, 1269–1279. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rybalka, E.; Timpani, C.A.; Debruin, D.A.; Bagaric, R.M.; Campelj, D.G.; Hayes, A. The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells 2020, 9, 2657. https://doi.org/10.3390/cells9122657
Rybalka E, Timpani CA, Debruin DA, Bagaric RM, Campelj DG, Hayes A. The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells. 2020; 9(12):2657. https://doi.org/10.3390/cells9122657
Chicago/Turabian StyleRybalka, Emma, Cara A. Timpani, Danielle A. Debruin, Ryan M. Bagaric, Dean G. Campelj, and Alan Hayes. 2020. "The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle" Cells 9, no. 12: 2657. https://doi.org/10.3390/cells9122657
APA StyleRybalka, E., Timpani, C. A., Debruin, D. A., Bagaric, R. M., Campelj, D. G., & Hayes, A. (2020). The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells, 9(12), 2657. https://doi.org/10.3390/cells9122657