
Antimyostatin Treatment in Health and Disease: The Story of Great Expectations and Limited Success


Abstract
:1. Introduction
2. Molecular Involvement of Myostatin in Mice and Humans
3. Myostatin in Healthy Humans and in Relation to Clinical Manifestations of Cachexia and Muscular Wasting
4. Myostatin in Response to Exercise
5. Preclinical Studies of Myostatin Inhibition in Animal Models of Neuromuscular Disorders
5.1. Antibodies against Myostatin
5.2. Myostatin Propeptide Administration or Overexpression
5.3. Systemic Administration of the Soluble Receptor ActRIIB
5.4. Administration of Antibodies Directed against ActRIIB
5.5. Follistatin Administration or Overexpression
5.6. Liver-Mediated Overexpression of Dominant-Negative Myostatin (dnMSTN), sActRIIB and Myostatin Propeptide
5.7. RNA Interference and Antioligonucleotides against Myostatin or ActRIIB
5.8. AAV-Cas9-Mediated Myostatin Gene Knock-Down
5.9. Crossbreeding Transgenic Myostatin Knock-Out Animals
6. Common Denominators in Animal Studies
7. Clinical Trials in Myostatin Inhibition
7.1. Clinical Trials in Muscular Dystrophy
7.2. Clinical Trials of Other Applications of Myostatin Inhibition
8. The Lack of Effect of Myostatin Inhibition in Clinical Trials of Muscular Dystrophy
9. Future Use of Myostatin Inhibition
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- McPherron, A.C.; Lawler, A.M.; Lee, S.-J. Regulation of Skeletal Muscle Mass in Mice by a New TGF-p Superfamily Member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Grobet, L.; Martin, L.J.; Poncelet, D.; Pirottin, D.; Brouwers, B.; Riquet, J.; Schoeberlein, A.; Dunner, S.; Ménissier, F.; Massabanda, J.; et al. A Deletion in the Bovine Myostatin Gene Causes the Double-Muscled Phenotype in Cattle. Nat. Genet. 1997, 17, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Kambadur, R.; Sharma, M.; Smith, T.P.; Bass, J.J. Mutations in Myostatin (GDF8) in Double-Muscled Belgian Blue and Piedmontese Cattle. Genome Res. 1997, 7, 910–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherron, A.C.; Lee, S.J. Double Muscling in Cattle Due to Mutations in the Myostatin Gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosher, D.S.; Quignon, P.; Bustamante, C.D.; Sutter, N.B.; Mellersh, C.S.; Parker, H.G.; Ostrander, E.A. A Mutation in the Myostatin Gene Increases Muscle Mass and Enhances Racing Performance in Heterozygote Dogs. PLoS Genet. 2007, 3. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M.; Wagner, K.R.; Stolz, L.E.; Hübner, C.; Riebel, T.; Kömen, W.; Braun, T.; Tobin, J.F.; Lee, S.-J. Myostatin Mutation Associated with Gross Muscle Hypertrophy in a Child. N. Engl. J. Med. 2004, 350, 2682–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendias, C.L.; Bakhurin, K.I.; Gumucio, J.P.; Shallal-Ayzin, M.V.; Davis, C.S.; Faulkner, J.A. Haploinsufficiency of Myostatin Protects against Aging-Related Declines in Muscle Function and Enhances the Longevity of Mice. Aging Cell 2015, 14, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.R.; Stricker, J.C.; Rosenberg, M.A.; Buranasombati, C.; Levitan, E.B.; Mittleman, M.A.; Rosenzweig, A. Effects of Myostatin Deletion in Aging Mice. Aging Cell 2009, 8, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogaars, W.M.H.; Jaspers, R.T. Past, Present, and Future Perspective of Targeting Myostatin and Related Signaling Pathways to Counteract Muscle Atrophy. In Muscle Atrophy; Xiao, J., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2018; pp. 153–206. ISBN 9789811314353. [Google Scholar]
- Sharma, M.; McFarlane, C.; Kambadur, R.; Kukreti, H.; Bonala, S.; Srinivasan, S. Myostatin: Expanding Horizons: Myostatin. Iubmb Life 2015, 67, 589–600. [Google Scholar] [CrossRef]
- Amthor, H.; Huang, R.; McKinnell, I.; Christ, B.; Kambadur, R.; Sharma, M.; Patel, K. The Regulation and Action of Myostatin as a Negative Regulator of Muscle Development during Avian Embryogenesis. Dev. Biol. 2002, 251, 241–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manceau, M.; Gros, J.; Savage, K.; Thomé, V.; McPherron, A.; Paterson, B.; Marcelle, C. Myostatin Promotes the Terminal Differentiation of Embryonic Muscle Progenitors. Genes Dev. 2008, 22, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.J.; Davies, M.V.; Pearson, A.A.; Wang, J.H.; Hewick, R.M.; Wolfman, N.M.; Qiu, Y. The Myostatin Propeptide and the Follistatin-Related Gene Are Inhibitory Binding Proteins of Myostatin in Normal Serum. J. Biol. Chem. 2002, 277, 40735–40741. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Kambadur, R.; Matthews, K.G.; Somers, W.G.; Devlin, G.P.; Conaglen, J.V.; Fowke, P.J.; Bass, J.J. Myostatin, a Transforming Growth Factor-β Superfamily Member, Is Expressed in Heart Muscle and Is Upregulated in Cardiomyocytes after Infarct. J. Cell. Physiol. 1999, 180, 1–9. [Google Scholar] [CrossRef]
- Gonzalez-Cadavid, N.F.; Taylor, W.E.; Yarasheski, K.; Sinha-Hikim, I.; Ma, K.; Ezzat, S.; Shen, R.; Lalani, R.; Asa, S.; Mamita, M.; et al. Organization of the Human Myostatin Gene and Expression in Healthy Men and HIV-Infected Men with Muscle Wasting. Proc. Natl. Acad. Sci. USA 1998, 95, 14938–14943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, B.D.; Weber, G.M. Sequence Conservation among Fish Myostatin Orthologues and the Characterization of Two Additional CDNA Clones from Morone Saxatilis and Morone Americana. Comp. Biochem. Physiol. Bbiochem. Mol. Biol. 2001, 129, 597–603. [Google Scholar] [CrossRef]
- Smith, T.P.; Lopez-Corrales, N.L.; Kappes, S.M.; Sonstegard, T.S. Myostatin Maps to the Interval Containing the Bovine Mh Locus. Mamm. Genome 1997, 8, 742–744. [Google Scholar] [CrossRef] [PubMed]
- Stavaux, D.; Art, T.; McEntee, K.; Reznick, M.; Lekeux, P. Muscle Fibre Type and Size, and Muscle Capillary Density in Young Double-Muscled Blue Belgian Cattle. Zent. Vet. A 1994, 41, 229–236. [Google Scholar] [CrossRef]
- Lee, S.-J.; McPherron, A.C. Regulation of Myostatin Activity and Muscle Growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thies, R.S.; Chen, T.; Davies, M.V.; Tomkinson, K.N.; Pearson, A.A.; Shakey, Q.A.; Wolfman, N.M. GDF-8 Propeptide Binds to GDF-8 and Antagonizes Biological Activity by Inhibiting GDF-8 Receptor Binding. Growth Factors 2001, 18, 251–259. [Google Scholar] [CrossRef]
- Wolfman, N.M.; McPherron, A.C.; Pappano, W.N.; Davies, M.V.; Song, K.; Tomkinson, K.N.; Wright, J.F.; Zhao, L.; Sebald, S.M.; Greenspan, D.S.; et al. Activation of Latent Myostatin by the BMP-1/Tolloid Family of Metalloproteinases. Proc. Natl. Acad. Sci. USA 2003, 100, 15842–15846. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-J. Genetic Analysis of the Role of Proteolysis in the Activation of Latent Myostatin. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amthor, H.; Nicholas, G.; McKinnell, I.; Kemp, C.F.; Sharma, M.; Kambadur, R.; Patel, K. Follistatin Complexes Myostatin and Antagonises Myostatin-Mediated Inhibition of Myogenesis. Dev. Biol. 2004, 270, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.J.; Qiu, Y.; Hewick, R.M.; Wolfman, N.M. Regulation of Myostatin in Vivo by Growth and Differentiation Factor-Associated Serum Protein-1: A Novel Protein with Protease Inhibitor and Follistatin Domains. Mol. Endocrinol. 2003, 17, 1144–1154. [Google Scholar] [CrossRef] [Green Version]
- Miura, T.; Kishioka, Y.; Wakamatsu, J.; Hattori, A.; Hennebry, A.; Berry, C.J.; Sharma, M.; Kambadur, R.; Nishimura, T. Decorin Binds Myostatin and Modulates Its Activity to Muscle Cells. Biochem. Biophys. Res. Commun. 2006, 340, 675–680. [Google Scholar] [CrossRef]
- Zhu, J.; Li, Y.; Shen, W.; Qiao, C.; Ambrosio, F.; Lavasani, M.; Nozaki, M.; Branca, M.F.; Huard, J. Relationships between Transforming Growth Factor-Β1, Myostatin, and Decorin Implications for Skeletal Muscle Fibrosis. J. Biol. Chem. 2007, 282, 25852–25863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemaladewi, D.U.; de Gorter, D.J.J.; Aartsma-Rus, A.; van Ommen, G.-J.; ten Dijke, P.; ’t Hoen, P.A.C.; Hoogaars, W.M. Cell-Type Specific Regulation of Myostatin Signaling. FASEB J. 2012, 26, 1462–1472. [Google Scholar] [CrossRef]
- Rebbapragada, A.; Benchabane, H.; Wrana, J.L.; Celeste, A.J.; Attisano, L. Myostatin Signals through a Transforming Growth Factor β-Like Signaling Pathway To Block Adipogenesis. Mol. Cell Biol. 2003, 23, 7230–7242. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-Dependent and Smad-Independent Pathways in TGF-Beta Family Signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Langley, B.; Thomas, M.; Bishop, A.; Sharma, M.; Gilmour, S.; Kambadur, R. Myostatin Inhibits Myoblast Differentiation by Down-Regulating MyoD Expression. J. Biol. Chem. 2002, 277, 49831–49840. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Langley, B.; Berry, C.; Sharma, M.; Kirk, S.; Bass, J.; Kambadur, R. Myostatin, a Negative Regulator of Muscle Growth, Functions by Inhibiting Myoblast Proliferation. J. Biol. Chem. 2000, 275, 40235–40243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartori, R.; Schirwis, E.; Blaauw, B.; Bortolanza, S.; Zhao, J.; Enzo, E.; Stantzou, A.; Mouisel, E.; Toniolo, L.; Ferry, A.; et al. BMP Signaling Controls Muscle Mass. Nat. Genet. 2013, 45, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Biesemann, N.; Mendler, L.; Wietelmann, A.; Hermann, S.; Schäfers, M.; Krüger, M.; Boettger, T.; Borchardt, T.; Braun, T. Myostatin Regulates Energy Homeostasis in the Heart and Prevents Heart Failure. Circ. Res. 2014, 115, 296–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip, B.; Lu, Z.; Gao, Y. Regulation of GDF-8 Signaling by the P38 MAPK. Cell Signal. 2005, 17, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Ratkevicius, A.; Joyson, A.; Selmer, I.; Dhanani, T.; Grierson, C.; Tommasi, A.M.; DeVries, A.; Rauchhaus, P.; Crowther, D.; Alesci, S.; et al. Serum Concentrations of Myostatin and Myostatin-Interacting Proteins Do Not Differ between Young and Sarcopenic Elderly Men. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Burch, P.M.; Pogoryelova, O.; Palandra, J.; Goldstein, R.; Bennett, D.; Fitz, L.; Guglieri, M.; Bettolo, C.M.; Straub, V.; Evangelista, T.; et al. Reduced Serum Myostatin Concentrations Associated with Genetic Muscle Disease Progression. J. Neurol. 2017, 264, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Szulc, P.; Schoppet, M.; Goettsch, C.; Rauner, M.; Dschietzig, T.; Chapurlat, R.; Hofbauer, L.C. Endocrine and Clinical Correlates of Myostatin Serum Concentration in Men--the STRAMBO Study. J. Clin. Endocrinol Metab. 2012, 97, 3700–3708. [Google Scholar] [CrossRef]
- Lakshman, K.M.; Bhasin, S.; Corcoran, C.; Collins-Racie, L.A.; Tchistiakova, L.; Forlow, S.B.; St Ledger, K.; Burczynski, M.E.; Dorner, A.J.; Lavallie, E.R. Measurement of Myostatin Concentrations in Human Serum: Circulating Concentrations in Young and Older Men and Effects of Testosterone Administration. Mol. Cell Endocrinol. 2009, 302, 26–32. [Google Scholar] [CrossRef]
- Schafer, M.J.; Atkinson, E.J.; Vanderboom, P.M.; Kotajarvi, B.; White, T.A.; Moore, M.M.; Bruce, C.J.; Greason, K.L.; Suri, R.M.; Khosla, S.; et al. Quantification of GDF11 and Myostatin in Human Aging and Cardiovascular Disease. Cell Metab. 2016, 23, 1207–1215. [Google Scholar] [CrossRef] [Green Version]
- Raue, U.; Slivka, D.; Jemiolo, B.; Hollon, C.; Trappe, S. Myogenic Gene Expression at Rest and after a Bout of Resistance Exercise in Young (18–30 Yr) and Old (80–89 Yr) Women. J. Appl. Physiol. 2006, 101, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Chew, J.; Tay, L.; Lim, J.P.; Leung, B.P.; Yeo, A.; Yew, S.; Ding, Y.Y.; Lim, W.S. Serum Myostatin and IGF-1 as Gender-Specific Biomarkers of Frailty and Low Muscle Mass in Community-Dwelling Older Adults. J. Nutr. Health Aging 2019, 23, 979–986. [Google Scholar] [CrossRef]
- Christensen, H.M.; Kistorp, C.; Schou, M.; Keller, N.; Zerahn, B.; Frystyk, J.; Schwarz, P.; Faber, J. Prevalence of Cachexia in Chronic Heart Failure and Characteristics of Body Composition and Metabolic Status. Endocrine 2013, 43, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.-N.; Lee, W.-J.; Liu, L.-K.; Lin, M.-H.; Chen, L.-K. Healthy Community-Living Older Men Differ from Women in Associations between Myostatin Levels and Skeletal Muscle Mass. J. Cachexia Sarcopenia Muscle 2018, 9, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, R.; Moghadam, B.H.; Church, D.D.; Tinsley, G.M.; Eskandari, M.; Moghadam, B.H.; Motevalli, M.S.; Baker, J.S.; Robergs, R.A.; Wong, A. The Effects of Concurrent Training Order on Body Composition and Serum Concentrations of Follistatin, Myostatin and GDF11 in Sarcopenic Elderly Men. Exp. Gerontol. 2020, 133, 110869. [Google Scholar] [CrossRef]
- Negaresh, R.; Ranjbar, R.; Baker, J.; Habibi, A.; Mokhtarzade, M.; Gharibvand, M.; Fokin, A. Skeletal Muscle Hypertrophy, Insulin-like Growth Factor 1, Myostatin and Follistatin in Healthy and Sarcopenic Elderly Men: The Effect of Whole-Body Resistance Training. Int. J. Prev. Med. 2019, 10, 29. [Google Scholar] [CrossRef]
- Zimmers, T.A.; Davies, M.V.; Koniaris, L.G.; Haynes, P.; Esquela, A.F.; Tomkinson, K.N.; McPherron, A.C.; Wolfman, N.M.; Lee, S.-J. Induction of Cachexia in Mice by Systemically Administered Myostatin. Science 2002, 296, 1486–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenk, K.; Schuler, G.; Adams, V. Skeletal Muscle Wasting in Cachexia and Sarcopenia: Molecular Pathophysiology and Impact of Exercise Training. J. Cachexia Sarcopenia Muscle 2010, 1, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Shyu, K.G.; Lu, M.J.; Wang, B.W.; Sun, H.Y.; Chang, H. Myostatin Expression in Ventricular Myocardium in a Rat Model of Volume-Overload Heart Failure. Eur. J. Clin. Investig. 2006, 36, 713–719. [Google Scholar] [CrossRef]
- Lenk, K.; Schur, R.; Linke, A.; Erbs, S.; Matsumoto, Y.; Adams, V.; Schuler, G. Impact of Exercise Training on Myostatin Expression in the Myocardium and Skeletal Muscle in a Chronic Heart Failure Model. Eur. J. Heart Fail. 2009, 11, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Lenk, K.; Erbs, S.; Höllriegel, R.; Beck, E.; Linke, A.; Gielen, S.; Winkler, S.M.; Sandri, M.; Hambrecht, R.; Schuler, G.; et al. Exercise Training Leads to a Reduction of Elevated Myostatin Levels in Patients with Chronic Heart Failure. Eur. J. Prev. Cardiol. 2012, 19, 404–411. [Google Scholar] [CrossRef]
- George, I.; Bish, L.T.; Kamalakkannan, G.; Petrilli, C.M.; Oz, M.C.; Naka, Y.; Lee Sweeney, H.; Maybaum, S. Myostatin Activation in Patients with Advanced Heart Failure and after Mechanical Unloading. Eur. J. Heart Fail. 2010, 12, 444–453. [Google Scholar] [CrossRef]
- Gruson, D.; Ahn, S.A.; Ketelslegers, J.-M.; Rousseau, M.F. Increased Plasma Myostatin in Heart Failure. Eur. J. Heart Fail. 2011, 13, 734–736. [Google Scholar] [CrossRef]
- Zamora, E.; Simó, R.; Lupón, J.; Galán, A.; Urrutia, A.; González, B.; Mas, D.; Valle, V. Serum Myostatin Levels in Chronic Heart Failure. Rev. Esp. Cardiol. 2010, 63, 992–996. [Google Scholar] [CrossRef]
- Furihata, T.; Kinugawa, S.; Fukushima, A.; Takada, S.; Homma, T.; Masaki, Y.; Abe, T.; Yokota, T.; Oba, K.; Okita, K.; et al. Serum Myostatin Levels Are Independently Associated with Skeletal Muscle Wasting in Patients with Heart Failure. Int. J. Cardiol. 2016, 220, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Wintgens, K.F.; Dschietzig, T.; Stoeva, S.; Paulsson, M.; Armbruster, F.P. Plasma Myostatin Measured by a Competitive ELISA Using a Highly Specific Antiserum. Clin. Chim. Acta 2012, 413, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Costelli, P.; Muscaritoli, M.; Bonetto, A.; Penna, F.; Reffo, P.; Bossola, M.; Bonelli, G.; Doglietto, G.B.; Baccino, F.M.; Fanelli, F.R. Muscle Myostatin Signalling Is Enhanced in Experimental Cancer Cachexia. Eur. J. Clin. Investig. 2008, 38, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of Cancer Cachexia and Muscle Wasting by ActRIIB Antagonism Leads to Prolonged Survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.T.; Chee, A.; Gleeson, B.G.; Naim, T.; Swiderski, K.; Koopman, R.; Lynch, G.S. Antibody-Directed Myostatin Inhibition Enhances Muscle Mass and Function in Tumor-Bearing Mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R716–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayot, M.; Rodriguez, J.; Vernus, B.; Carnac, G.; Jean, E.; Allen, D.; Goret, L.; Obert, P.; Candau, R.; Bonnieu, A. Myostatin Up-Regulation Is Associated with the Skeletal Muscle Response to Hypoxic Stimuli. Mol. Cell. Endocrinol. 2011, 332, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plant, P.J.; Brooks, D.; Faughnan, M.; Bayley, T.; Bain, J.; Singer, L.; Correa, J.; Pearce, D.; Binnie, M.; Batt, J. Cellular Markers of Muscle Atrophy in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2010, 42, 461–471. [Google Scholar] [CrossRef]
- Ju, C.-R.; Chen, R.-C. Serum Myostatin Levels and Skeletal Muscle Wasting in Chronic Obstructive Pulmonary Disease. Respir. Med. 2012, 106, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Avin, K.G.; Chen, N.X.; Organ, J.M.; Zarse, C.; O’Neill, K.; Conway, R.G.; Konrad, R.J.; Bacallao, R.L.; Allen, M.R.; Moe, S.M. Skeletal Muscle Regeneration and Oxidative Stress Are Altered in Chronic Kidney Disease. PLoS ONE 2016, 11, e0159411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.W.; Hill, R.J.; Krasney, P.A.; O’Conner, B.; Peirce, N.; Greenhaff, P.L. Disuse Atrophy and Exercise Rehabilitation in Humans Profoundly Affects the Expression of Genes Associated with the Regulation of Skeletal Muscle Mass. FASEB J. 2004, 18, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cross, J.M.; Bamman, M.M. Impact of Resistance Loading on Myostatin Expression and Cell Cycle Regulation in Young and Older Men and Women. Am. J. Physiol.-Endocrinol. Metab. 2005, 288, E1110–E1119. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.; Raue, U.; Yang, Y.; Jemiolo, B.; Trappe, S. Time Course of Proteolytic, Cytokine, and Myostatin Gene Expression after Acute Exercise in Human Skeletal Muscle. J. Appl. Physiol. 2007, 103, 1744–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hittel, D.S.; Axelson, M.; Sarna, N.; Shearer, J.; Huffman, K.M.; Kraus, W.E. Myostatin Decreases with Aerobic Exercise and Associates with Insulin Resistance. Med. Sci. Sports Exerc. 2010, 42, 2023–2029. [Google Scholar] [CrossRef] [Green Version]
- Whittemore, L.-A.; Song, K.; Li, X.; Aghajanian, J.; Davies, M.; Girgenrath, S.; Hill, J.J.; Jalenak, M.; Kelley, P.; Knight, A.; et al. Inhibition of Myostatin in Adult Mice Increases Skeletal Muscle Mass and Strength. Biochem. Biophys. Res. Commun. 2003, 300, 965–971. [Google Scholar] [CrossRef]
- Smith, R.C.; Cramer, M.S.; Mitchell, P.J.; Lucchesi, J.; Ortega, A.M.; Livingston, E.W.; Ballard, D.; Zhang, L.; Hanson, J.; Barton, K.; et al. Inhibition of Myostatin Prevents Microgravity-Induced Loss of Skeletal Muscle Mass and Strength. PLoS ONE 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Pirruccello-Straub, M.; Jackson, J.; Wawersik, S.; Webster, M.T.; Salta, L.; Long, K.; McConaughy, W.; Capili, A.; Boston, C.; Carven, G.J.; et al. Blocking Extracellular Activation of Myostatin as a Strategy for Treating Muscle Wasting. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.-P.G.; Petersen, M.C.; Abudukadier, A.; Moreira, G.V.; Jurczak, M.J.; Friedman, G.; Haqq, C.M.; Petersen, K.F.; Shulman, G.I. Anti-Myostatin Antibody Increases Muscle Mass and Strength and Improves Insulin Sensitivity in Old Mice. Proc. Natl. Acad. Sci. USA 2016, 113, 2212–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, H.; Kuramochi, T.; Katada, H.; Ueyama, A.; Ruike, Y.; Ohmine, K.; Shida-Kawazoe, M.; Miyano-Nishizawa, R.; Shimizu, Y.; Okuda, M.; et al. Novel Myostatin-Specific Antibody Enhances Muscle Strength in Muscle Disease Models. Sci. Rep. 2021, 11, 2160. [Google Scholar] [CrossRef]
- St. Andre, M.; Johnson, M.; Bansal, P.N.; Wellen, J.; Robertson, A.; Opsahl, A.; Burch, P.M.; Bialek, P.; Morris, C.; Owens, J. A Mouse Anti-Myostatin Antibody Increases Muscle Mass and Improves Muscle Strength and Contractility in the Mdx Mouse Model of Duchenne Muscular Dystrophy and Its Humanized Equivalent, Domagrozumab (PF-06252616), Increases Muscle Volume in Cynomolgus Monkeys. Skelet. Muscle 2017, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovich, S.; Krag, T.O.B.; Barton, E.R.; Morris, L.D.; Whittemore, L.-A.; Ahima, R.S.; Khurana, T.S. Functional Improvement of Dystrophic Muscle by Myostatin Blockade. Nature 2002, 420, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovich, S.; McNally, E.M.; Khurana, T.S. Myostatin Blockade Improves Function but Not Histopathology in a Murine Model of Limb-Girdle Muscular Dystrophy 2C. Muscle Nerve 2008, 37, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.A.; Millay, D.P.; Sargent, M.A.; McNally, E.M.; Molkentin, J.D. Age-Dependent Effect of Myostatin Blockade on Disease Severity in a Murine Model of Limb-Girdle Muscular Dystrophy. Am. J. Pathol. 2006, 168, 1975–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.T.; Ryall, J.G.; Snell, S.M.; Nair, L.; Koopman, R.; Krasney, P.A.; Ibebunjo, C.; Holden, K.S.; Loria, P.M.; Salatto, C.T.; et al. Antibody-Directed Myostatin Inhibition Improves Diaphragm Pathology in Young but Not Adult Dystrophic Mdx Mice. Am. J. Pathol. 2010, 176, 2425–2434. [Google Scholar] [CrossRef] [PubMed]
- Harish, P.; Malerba, A.; Lu-Nguyen, N.; Forrest, L.; Cappellari, O.; Roth, F.; Trollet, C.; Popplewell, L.; Dickson, G. Inhibition of Myostatin Improves Muscle Atrophy in Oculopharyngeal Muscular Dystrophy (OPMD). J. Cachexia Sarcopenia Muscle 2019, 10, 1016–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinklenberg, J.A.; Siebers, E.M.; Beatka, M.J.; Meng, H.; Yang, L.; Zhang, Z.; Ross, J.A.; Ochala, J.; Morris, C.; Owens, J.M.; et al. Myostatin Inhibition Using MRK35 Produces Skeletal Muscle Growth and Tubular Aggregate Formation in Wild Type and TgACTA1D286G Nemaline Myopathy Mice. Hum. Mol. Genet. 2018, 27, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.; Gao, F.; Wang, J.; Li, X.; Chu, B.; Wang, J.; Yang, G. Improvement of Muscular Atrophy by AAV-SaCas9-Mediated Myostatin Gene Editing in Aged Mice. Cancer Gene 2020, 27, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Holzbaur, E.L.F.; Howland, D.S.; Weber, N.; Wallace, K.; She, Y.; Kwak, S.; Tchistiakova, L.A.; Murphy, E.; Hinson, J.; Karim, R.; et al. Myostatin Inhibition Slows Muscle Atrophy in Rodent Models of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2006, 23, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Harish, P.; Forrest, L.; Herath, S.; Dickson, G.; Malerba, A.; Popplewell, L. Inhibition of Myostatin Reduces Collagen Deposition in a Mouse Model of Oculopharyngeal Muscular Dystrophy (OPMD) With Established Disease. Front. Physiol. 2020, 11, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latres, E.; Pangilinan, J.; Miloscio, L.; Bauerlein, R.; Na, E.; Potocky, T.B.; Huang, Y.; Eckersdorff, M.; Rafique, A.; Mastaitis, J.; et al. Myostatin Blockade with a Fully Human Monoclonal Antibody Induces Muscle Hypertrophy and Reverses Muscle Atrophy in Young and Aged Mice. Skelet Muscle 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Rong, H.; Gordi, T.; Bosley, J.; Bhattacharya, I. Translational Pharmacokinetic/Pharmacodynamic Analysis of MYO-029 Antibody for Muscular Dystrophy. Clin. Transl. Sci. 2016, 9, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovich, S.; Perkins, K.J.; Krag, T.O.B.; Whittemore, L.-A.; Khurana, T.S. Myostatin Propeptide-Mediated Amelioration of Dystrophic Pathophysiology. FASEB J. 2005, 19, 543–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, C.; Li, J.; Jiang, J.; Zhu, X.; Wang, B.; Li, J.; Xiao, X. Myostatin Propeptide Gene Delivery by Adeno-Associated Virus Serotype 8 Vectors Enhances Muscle Growth and Ameliorates Dystrophic Phenotypes in Mdx Mice. Hum. Gene Ther. 2008, 19, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.; Poupiot, J.; Vulin, A.; Fougerousse, F.; Arandel, L.; Daniele, N.; Roudaut, C.; Noulet, F.; Garcia, L.; Danos, O.; et al. AAV-Mediated Delivery of a Mutated Myostatin Propeptide Ameliorates Calpain 3 but Not α -Sarcoglycan Deficiency. Gene Ther. 2007, 14, 733–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akpan, I.; Goncalves, M.D.; Dhir, R.; Yin, X.; Pistilli, E.E.; Bogdanovich, S.; Khurana, T.S.; Ucran, J.; Lachey, J.; Ahima, R.S. The Effects of a Soluble Activin Type IIB Receptor on Obesity and Insulin Sensitivity. Int. J. Obes. 2009, 33, 1265–1273. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-S.; Peekhaus, N.; Weber, H.; Adamski, S.; Murray, E.M.; Zhang, H.Z.; Zhao, J.Z.; Ernst, R.; Lineberger, J.; Huang, L.; et al. Increased Muscle Force Production and Bone Mineral Density in ActRIIB-Fc-Treated Mature Rodents. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2013, 68, 1181–1192. [Google Scholar] [CrossRef] [Green Version]
- Cadena, S.M.; Tomkinson, K.N.; Monnell, T.E.; Spaits, M.S.; Kumar, R.; Underwood, K.W.; Pearsall, R.S.; Lachey, J.L. Administration of a Soluble Activin Type IIB Receptor Promotes Skeletal Muscle Growth Independent of Fiber Type. J. Appl. Physiol. 2010, 109, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, M.W.; Read, B.P.; Edelstein, R.; Yang, N.; Pierson, C.R.; Stein, M.J.; Wermer-Colan, A.; Buj-Bello, A.; Lachey, J.L.; Seehra, J.S.; et al. Inhibition of Activin Receptor Type IIB Increases Strength and Lifespan in Myotubularin-Deficient Mice. Am. J. Pathol. 2011, 178, 784–793. [Google Scholar] [CrossRef]
- Béchir, N.; Pecchi, É.; Relizani, K.; Vilmen, C.; Le Fur, Y.; Bernard, M.; Amthor, H.; Bendahan, D.; Giannesini, B. Mitochondrial Impairment Induced by Postnatal ActRIIB Blockade Does Not Alter Function and Energy Status in Exercising Mouse Glycolytic Muscle in Vivo. Am. J. Physiol.-Endocrinol. Metab. 2016, 310, E539–E549. [Google Scholar] [CrossRef] [Green Version]
- Relizani, K.; Mouisel, E.; Giannesini, B.; Hourdé, C.; Patel, K.; Gonzalez, S.M.; Jülich, K.; Vignaud, A.; Piétri-Rouxel, F.; Fortin, D.; et al. Blockade of ActRIIB Signaling Triggers Muscle Fatigability and Metabolic Myopathy. Mol. Ther. 2014, 22, 1423–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, J.A.; Kapur, K.; Taylor, R.S.; Sanchez, B.; Rutkove, S.B. Electrical Impedance Myography as a Biomarker of Myostatin Inhibition with ActRIIB-MFc: A Study in Wild-Type Mice. Future Sci. OA 2018, 4, FSO308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogaars, W.M.H.; Mouisel, E.; Pasternack, A.; Hulmi, J.J.; Relizani, K.; Schuelke, M.; Schirwis, E.; Garcia, L.; Ritvos, O.; Ferry, A.; et al. Combined Effect of AAV-U7-Induced Dystrophin Exon Skipping and Soluble Activin Type IIB Receptor in Mdx Mice. Hum. Gene Ther. 2012, 23, 1269–1279. [Google Scholar] [CrossRef]
- Béchir, N.; Pecchi, É.; Vilmen, C.; Bernard, M.; Bendahan, D.; Giannesini, B. Activin Type IIB Receptor Blockade Does Not Limit Adenosine Triphosphate Supply in Mouse Skeletal Muscle in Vivo. Muscle Nerve 2018, 58, 834–842. [Google Scholar] [CrossRef]
- Tauer, J.T.; Rauch, F. Novel ActRIIB Ligand Trap Increases Muscle Mass and Improves Bone Geometry in a Mouse Model of Severe Osteogenesis Imperfecta. Bone 2019, 128, 115036. [Google Scholar] [CrossRef]
- Li, Z.B.; Zhang, J.; Wagner, K.R. Inhibition of Myostatin Reverses Muscle Fibrosis through Apoptosis. J. Cell Sci. 2012, 125, 3957–3965. [Google Scholar] [CrossRef] [Green Version]
- Pistilli, E.E.; Bogdanovich, S.; Goncalves, M.D.; Ahima, R.S.; Lachey, J.; Seehra, J.; Khurana, T. Targeting the Activin Type IIB Receptor to Improve Muscle Mass and Function in the Mdx Mouse Model of Duchenne Muscular Dystrophy. Am. J. Pathol. 2011, 178, 1287–1297. [Google Scholar] [CrossRef]
- Iskenderian, A.; Liu, N.; Deng, Q.; Huang, Y.; Shen, C.; Palmieri, K.; Crooker, R.; Lundberg, D.; Kastrapeli, N.; Pescatore, B.; et al. Myostatin and Activin Blockade by Engineered Follistatin Results in Hypertrophy and Improves Dystrophic Pathology in Mdx Mouse More than Myostatin Blockade Alone. Skelet. Muscle 2018, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Hulmi, J.J.; Oliveira, B.M.; Silvennoinen, M.; Hoogaars, W.M.H.; Pasternack, A.; Kainulainen, H.; Ritvos, O. Exercise Restores Decreased Physical Activity Levels and Increases Markers of Autophagy and Oxidative Capacity in Myostatin/Activin-Blocked Mdx Mice. Am. J. Physiol.-Endocrinol. Metab. 2013, 305, E171–E182. [Google Scholar] [CrossRef] [PubMed]
- Tinklenberg, J.; Meng, H.; Yang, L.; Liu, F.; Hoffmann, R.G.; Dasgupta, M.; Allen, K.P.; Beggs, A.H.; Hardeman, E.C.; Pearsall, R.S.; et al. Treatment with ActRIIB-MFc Produces Myofiber Growth and Improves Lifespan in the Acta1 H40Y Murine Model of Nemaline Myopathy. Am. J. Pathol. 2016, 186, 1568–1581. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, M.W.; Viola, M.G.; Meng, H.; Edelstein, R.V.; Liu, F.; Yan, K.; Luna, E.J.; Lerch-Gaggl, A.; Hoffmann, R.G.; Pierson, C.R.; et al. Differential Muscle Hypertrophy Is Associated with Satellite Cell Numbers and Akt Pathway Activation Following Activin Type IIB Receptor Inhibition in Mtm1 p.R69C Mice. Am. J. Pathol. 2014, 184, 1831–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondulich, M.K.; Jolinon, N.; Osborne, G.F.; Smith, E.J.; Rattray, I.; Neueder, A.; Sathasivam, K.; Ahmed, M.; Ali, N.; Benjamin, A.C.; et al. Myostatin Inhibition Prevents Skeletal Muscle Pathophysiology in Huntington’s Disease Mice. Sci. Rep. 2017, 7, 14275. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-S.; Lehar, A.; Sebald, S.; Liu, M.; Swaggart, K.A.; Talbot, C.C.; Pytel, P.; Barton, E.R.; McNally, E.M.; Lee, S.-J. Muscle Hypertrophy Induced by Myostatin Inhibition Accelerates Degeneration in Dysferlinopathy. Hum. Mol. Genet. 2015, 24, 5711–5719. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, Y.; Hagiwara, H.; Nakatani, M.; Yasue, A.; Moriyama, K.; Murakami, T.; Tsuchida, K.; Noji, S.; Sunada, Y. Muscular Atrophy of Caveolin-3–Deficient Mice Is Rescued by Myostatin Inhibition. J. Clin. Investig. 2006, 116, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Lach-Trifilieff, E.; Minetti, G.C.; Sheppard, K.; Ibebunjo, C.; Feige, J.N.; Hartmann, S.; Brachat, S.; Rivet, H.; Koelbing, C.; Morvan, F.; et al. An Antibody Blocking Activin Type II Receptors Induces Strong Skeletal Muscle Hypertrophy and Protects from Atrophy. Mol. Cell Biol. 2014, 34, 606–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morvan, F.; Rondeau, J.-M.; Zou, C.; Minetti, G.; Scheufler, C.; Scharenberg, M.; Jacobi, C.; Brebbia, P.; Ritter, V.; Toussaint, G.; et al. Blockade of Activin Type II Receptors with a Dual Anti-ActRIIA/IIB Antibody Is Critical to Promote Maximal Skeletal Muscle Hypertrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 12448–12453. [Google Scholar] [CrossRef] [Green Version]
- Haidet, A.M.; Rizo, L.; Handy, C.; Umapathi, P.; Eagle, A.; Shilling, C.; Boue, D.; Martin, P.T.; Sahenk, Z.; Mendell, J.R.; et al. Long-Term Enhancement of Skeletal Muscle Mass and Strength by Single Gene Administration of Myostatin Inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 4318–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.M.; Kim, S.H.; Yamanaka, K.; Hester, M.; Umapathi, P.; Arnson, H.; Rizo, L.; Mendell, J.R.; Gage, F.H.; Cleveland, D.W.; et al. Gene Transfer Demonstrates That Muscle Is Not a Primary Target for Non-Cell-Autonomous Toxicity in Familial Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 19546–19551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kota, J.; Handy, C.R.; Haidet, A.M.; Montgomery, C.L.; Eagle, A.; Rodino-Klapac, L.R.; Tucker, D.; Shilling, C.J.; Therlfall, W.R.; Walker, C.M.; et al. Follistatin Gene Delivery Enhances Muscle Growth and Strength in Nonhuman Primates. Sci. Transl. Med. 2009, 1, 6ra15. [Google Scholar] [CrossRef] [Green Version]
- Pearsall, R.S.; Davies, M.V.; Cannell, M.; Li, J.; Widrick, J.; Mulivor, A.W.; Wallner, S.; Troy, M.E.; Spaits, M.; Liharska, K.; et al. Follistatin-Based Ligand Trap ACE-083 Induces Localized Hypertrophy of Skeletal Muscle with Functional Improvement in Models of Neuromuscular Disease. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castonguay, R.; Lachey, J.; Wallner, S.; Strand, J.; Liharska, K.; Watanabe, A.E.; Cannell, M.; Davies, M.V.; Sako, D.; Troy, M.E.; et al. Follistatin-288-Fc Fusion Protein Promotes Localized Growth of Skeletal Muscle. J. Pharm. Exp. 2018, 365, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, K.; Graham, I.R.; Otto, A.; Foster, H.; Trollet, C.; Yaworsky, P.J.; Walsh, F.S.; Bickham, D.; Curtin, N.A.; Kawar, S.L.; et al. Adeno-Associated Virus-8-Mediated Intravenous Transfer of Myostatin Propeptide Leads to Systemic Functional Improvements of Slow but Not Fast Muscle. Rejuvenation Res. 2009, 12, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hammers, D.W.; Barton, E.R.; Sweeney, H.L. Activin Receptor Type IIB Inhibition Improves Muscle Phenotype and Function in a Mouse Model of Spinal Muscular Atrophy. PLoS ONE 2016, 11, e0166803. [Google Scholar] [CrossRef] [PubMed]
- Morine, K.J.; Bish, L.T.; Selsby, J.T.; Gazzara, J.A.; Pendrak, K.; Sleeper, M.M.; Barton, E.R.; Lee, S.-J.; Sweeney, H.L. Activin IIB Receptor Blockade Attenuates Dystrophic Pathology in a Mouse Model of Duchenne Muscular Dystrophy. Muscle Nerve 2010, 42, 722–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morine, K.J.; Bish, L.T.; Pendrak, K.; Sleeper, M.M.; Barton, E.R.; Sweeney, H.L. Systemic Myostatin Inhibition via Liver-Targeted Gene Transfer in Normal and Dystrophic Mice. PLoS ONE 2010, 5, e9176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammers, D.W.; Hart, C.C.; Patsalos, A.; Matheny, M.K.; Wright, L.A.; Nagy, L.; Sweeney, H.L. Glucocorticoids Counteract Hypertrophic Effects of Myostatin Inhibition in Dystrophic Muscle. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Bish, L.T.; Sleeper, M.M.; Forbes, S.C.; Morine, K.J.; Reynolds, C.; Singletary, G.E.; Trafny, D.; Pham, J.; Bogan, J.; Kornegay, J.N.; et al. Long-Term Systemic Myostatin Inhibition via Liver-Targeted Gene Transfer in Golden Retriever Muscular Dystrophy. Hum. Gene Ther. 2011, 22, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Lu-Nguyen, N.; Malerba, A.; Popplewell, L.; Schnell, F.; Hanson, G.; Dickson, G. Systemic Antisense Therapeutics for Dystrophin and Myostatin Exon Splice Modulation Improve Muscle Pathology of Adult Mdx Mice. Mol. Nucleic Acids 2017, 6, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumonceaux, J.; Marie, S.; Beley, C.; Trollet, C.; Vignaud, A.; Ferry, A.; Butler-Browne, G.; Garcia, L. Combination of Myostatin Pathway Interference and Dystrophin Rescue Enhances Tetanic and Specific Force in Dystrophic Mdx Mice. Mol. Ther. 2010, 18, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Mendias, C.L.; Marcin, J.E.; Calerdon, D.R.; Faulkner, J.A. Contractile Properties of EDL and Soleus Muscles of Myostatin-Deficient Mice. J. Appl. Physiol. (1985) 2006, 101, 898–905. [Google Scholar] [CrossRef] [Green Version]
- Qaisar, R.; Renaud, G.; Morine, K.; Barton, E.R.; Sweeney, H.L.; Larsson, L. Is Functional Hypertrophy and Specific Force Coupled with the Addition of Myonuclei at the Single Muscle Fiber Level? FASEB J. 2011, 26, 1077–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amthor, H.; Macharia, R.; Navarrete, R.; Schuelke, M.; Brown, S.C.; Otto, A.; Voit, T.; Muntoni, F.; Vrbóva, G.; Partridge, T.; et al. Lack of Myostatin Results in Excessive Muscle Growth but Impaired Force Generation. Proc. Natl. Acad. Sci. USA 2007, 104, 1835–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girgenrath, S.; Song, K.; Whittemore, L.-A. Loss of Myostatin Expression Alters Fiber-Type Distribution and Expression of Myosin Heavy Chain Isoforms in Slow- and Fast-Type Skeletal Muscle. Muscle Nerve 2005, 31, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Hennebry, A.; Berry, C.; Siriett, V.; O’Callaghan, P.; Chau, L.; Watson, T.; Sharma, M.; Kambadur, R. Myostatin Regulates Fiber-Type Composition of Skeletal Muscle by Regulating MEF2 and MyoD Gene Expression. Am. J. Physiol.-Cell Physiol. 2009, 296, C525–C534. [Google Scholar] [CrossRef]
- Hennebry, A.; Oldham, J.; Shavlakadze, T.; Grounds, M.D.; Sheard, P.; Fiorotto, M.L.; Falconer, S.; Smith, H.K.; Berry, C.; Jeanplong, F.; et al. IGF1 Stimulates Greater Muscle Hypertrophy in the Absence of Myostatin in Male Mice. J. Endocrinol. 2017, 234, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Matsakas, A.; Mouisel, E.; Amthor, H.; Patel, K. Myostatin Knockout Mice Increase Oxidative Muscle Phenotype as an Adaptive Response to Exercise. J. Muscle Res. Cell Motil. 2010, 31, 111–125. [Google Scholar] [CrossRef]
- Kocsis, T.; Trencsenyi, G.; Szabo, K.; Baan, J.A.; Muller, G.; Mendler, L.; Garai, I.; Reinauer, H.; Deak, F.; Dux, L.; et al. Myostatin Propeptide Mutation of the Hypermuscular Compact Mice Decreases the Formation of Myostatin and Improves Insulin Sensitivity. Am. J. Physiol.-Endocrinol. Metab. 2016, 312, E150–E160. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R.; McPherron, A.C.; Winik, N.; Lee, S.-J. Loss of Myostatin Attenuates Severity of Muscular Dystrophy in Mdx Mice. Ann. Neurol. 2002, 52, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Liu, X.; Chang, X.; Allen, R.E. Muscle Regeneration in the Prolonged Absence of Myostatin. Proc. Natl. Acad. Sci. USA 2005, 102, 2519–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Shelton, G.D.; Engvall, E. Elimination of Myostatin Does Not Combat Muscular Dystrophy in Dy Mice but Increases Postnatal Lethality. Am. J. Pathol. 2005, 166, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Stedman, H.H.; Sweeney, H.L.; Shrager, J.B.; Maguire, H.C.; Panettieri, R.A.; Petrof, B.; Narusawa, M.; Leferovich, J.M.; Sladky, J.T.; Kelly, A.M. The Mdx Mouse Diaphragm Reproduces the Degenerative Changes of Duchenne Muscular Dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific Inhibition of Myostatin Activation Is Beneficial in Mouse Models of SMA Therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef] [Green Version]
- Graham, Z.A.; Collier, L.; Peng, Y.; Saéz, J.C.; Bauman, W.A.; Qin, W.; Cardozo, C.P. A Soluble Activin Receptor IIB Fails to Prevent Muscle Atrophy in a Mouse Model of Spinal Cord Injury. J. Neurotrauma 2016, 33, 1128–1135. [Google Scholar] [CrossRef]
- Pistilli, E.E.; Bogdanovich, S.; Mosqueira, M.; Lachey, J.; Seehra, J.; Khurana, T.S. Pretreatment with a Soluble Activin Type IIB Receptor/Fc Fusion Protein Improves Hypoxia-Induced Muscle Dysfunction. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2010, 298, R96–R103. [Google Scholar] [CrossRef] [Green Version]
- Tortoriello, D.V.; Sidis, Y.; Holtzman, D.A.; Holmes, W.E.; Schneyer, A.L. Human Follistatin-Related Protein: A Structural Homologue of Follistatin with Nuclear Localization. Endocrinology 2001, 142, 3426–3434. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, M.; Takehara, Y.; Sugino, H.; Matsumoto, M.; Hashimoto, O.; Hasegawa, Y.; Murakami, T.; Uezumi, A.; Takeda, S.; Noji, S.; et al. Transgenic Expression of a Myostatin Inhibitor Derived from Follistatin Increases Skeletal Muscle Mass and Ameliorates Dystrophic Pathology in Mdx Mice. FASEB J. 2008, 22, 477–487. [Google Scholar] [CrossRef]
- Shen, C.; Iskenderian, A.; Lundberg, D.; He, T.; Palmieri, K.; Crooker, R.; Deng, Q.; Traylor, M.; Gu, S.; Rong, H.; et al. Protein Engineering on Human Recombinant Follistatin: Enhancing Pharmacokinetic Characteristics for Therapeutic Application. J. Pharm. Exp. 2018, 366, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Malerba, A.; Kang, J.K.; McClorey, G.; Saleh, A.F.; Popplewell, L.; Gait, M.J.; Wood, M.J.; Dickson, G. Dual Myostatin and Dystrophin Exon Skipping by Morpholino Nucleic Acid Oligomers Conjugated to a Cell-Penetrating Peptide Is a Promising Therapeutic Strategy for the Treatment of Duchenne Muscular Dystrophy. Mol. Nucleic. Acids 2012, 1, e62. [Google Scholar] [CrossRef]
- Varga, L.; Szabo, G.; Darvasi, A.; Muller, G.; Sass, M.; Soller, M. Inheritance and Mapping of Compact (Cmpt), a New Mutation Causing Hypermuscularity in Mice. Genetics 1997, 147, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, J.J.; Lloyd, D.J.; Gekakis, N. Loss-of-Function Mutation in Myostatin Reduces Tumor Necrosis Factor Alpha Production and Protects Liver against Obesity-Induced Insulin Resistance. Diabetes 2009, 58, 1133–1143. [Google Scholar] [CrossRef] [Green Version]
- Kramerova, I.; Marinov, M.; Owens, J.; Lee, S.-J.; Becerra, D.; Spencer, M.J. Myostatin Inhibition Promotes Fast Fibre Hypertrophy but Causes Loss of AMP-Activated Protein Kinase Signalling and Poor Exercise Tolerance in a Model of Limb-Girdle Muscular Dystrophy R1/2A. J. Physiol. 2020, 598, 3927–3939. [Google Scholar] [CrossRef] [PubMed]
- Béchir, N.; Pecchi, E.; Vilmen, C.; Le Fur, Y.; Amthor, H.; Bernard, M.; Bendahan, D.; Giannesini, B. ActRIIB Blockade Increases Force-Generating Capacity and Preserves Energy Supply in Exercising Mdx Mouse Muscle in Vivo. FASEB J. 2016, 30, 3551–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, C.G.; Bruemmer, K.; Sesti, J.; Stefanski, C.; Curtis, H.; Ucran, J.; Lachey, J.; Seehra, J.S. Soluble Activin Receptor Type IIB Increased Forward Pulling Tension in the Mdx Mouse. Muscle Nerve 2011, 43, 694–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, C.J.; Booth, F.W.; Gordon, S.E. Skeletal Muscle Myostatin mRNA Expression Is Fiber-Type Specific and Increases during Hindlimb Unloading. Am. J. Physiol. 1999, 277, R601–R606. [Google Scholar] [CrossRef] [Green Version]
- Carnwath, J.W.; Shotton, D.M. Muscular Dystrophy in the Mdx Mouse: Histopathology of the Soleus and Extensor Digitorum Longus Muscles. J. Neurol. Sci. 1987, 80, 39–54. [Google Scholar] [CrossRef]
- Bloemberg, D.; Quadrilatero, J. Rapid Determination of Myosin Heavy Chain Expression in Rat, Mouse, and Human Skeletal Muscle Using Multicolor Immunofluorescence Analysis. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Mendias, C.L.; Lynch, E.B.; Gumucio, J.P.; Flood, M.D.; Rittman, D.S.; Van Pelt, D.W.; Roche, S.M.; Davis, C.S. Changes in Skeletal Muscle and Tendon Structure and Function Following Genetic Inactivation of Myostatin in Rats. J. Physiol. 2015, 593, 2037–2052. [Google Scholar] [CrossRef] [Green Version]
- Karpati, G.; Carpenter, S. Small-Caliber Skeletal Muscle Fibers Do Not Suffer Deleterious Consequences of Dystrophic Gene Expression. Am. J. Med. Genet. 1986, 25, 653–658. [Google Scholar] [CrossRef]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin Protects the Sarcolemma from Stresses Developed during Muscle Contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Jaswal, J.S. Energy Metabolic Phenotype of the Cardiomyocyte during Development, Differentiation, and Postnatal Maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Polla, B.; D’Antona, G.; Bottinelli, R.; Reggiani, C. Respiratory Muscle Fibres: Specialisation and Plasticity. Thorax 2004, 59, 808–817. [Google Scholar] [CrossRef] [Green Version]
- Greising, S.M.; Medina-Martínez, J.S.; Vasdev, A.K.; Sieck, G.C.; Mantilla, C.B. Analysis of Muscle Fiber Clustering in the Diaphragm Muscle of Sarcopenic Mice. Muscle Nerve 2015, 52, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A Phase I/IItrial of MYO-029 in Adult Subjects with Muscular Dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, I.; Pawlak, S.; Marraffino, S.; Christensen, J.; Sherlock, S.P.; Alvey, C.; Morris, C.; Arkin, S.; Binks, M. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Domagrozumab (PF-06252616), an Antimyostatin Monoclonal Antibody, in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2018, 7, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Abdel-Hamid, H.Z.; Mah, J.K.; Campbell, C.; Guglieri, M.; Muntoni, F.; Takeshima, Y.; McDonald, C.M.; Kostera-Pruszczyk, A.; Karachunski, P.; et al. Randomized Phase 2 Trial and Open-Label Extension of Domagrozumab in Duchenne Muscular Dystrophy. Neuromuscul Disord 2020, 30, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Jameson, G.S.; Von Hoff, D.D.; Weiss, G.J.; Richards, D.A.; Smith, D.A.; Becerra, C.; Benson, M.C.; Yuan, Z.; Robins, D.A.; Turik, M.; et al. Safety of the Antimyostatin Monoclonal Antibody LY2495655 in Healthy Subjects and Patients with Advanced Cancer. JCO 2012, 30, 2516. [Google Scholar] [CrossRef]
- Golan, T.; Geva, R.; Richards, D.; Madhusudan, S.; Lin, B.K.; Wang, H.T.; Walgren, R.A.; Stemmer, S.M. LY2495655, an Antimyostatin Antibody, in Pancreatic Cancer: A Randomized, Phase 2 Trial: LY2495655 in Patients with Stage II-IV Pancreatic Cancer. J. Cachexiasarcopenia Muscle 2018, 9, 871–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, C.; Lord, S.R.; Studenski, S.A.; Warden, S.J.; Fielding, R.A.; Recknor, C.P.; Hochberg, M.C.; Ferrari, S.L.; Blain, H.; Binder, E.F.; et al. Myostatin Antibody (LY2495655) in Older Weak Fallers: A Proof-of-Concept, Randomised, Phase 2 Trial. Lancet Diabetes Endocrinol. 2015, 3, 948–957. [Google Scholar] [CrossRef]
- Woodhouse, L.; Gandhi, R.; Warden, S.J.; Poiraudeau, S.; Myers, S.L.; Benson, C.T.; Hu, L.; Ahmad, Q.I.; Linnemeier, P.; Gomez, E.V.; et al. A Phase 2 Randomized Study Investigating the Efficacy and Safety of Myostatin Antibody LY2495655 versus Placebo in Patients Undergoing Elective Total Hip Arthroplasty. J. Frailty Aging 2016, 5, 62–70. [Google Scholar] [CrossRef]
- Attie, K.M.; Borgstein, N.G.; Yang, Y.; Condon, C.H.; Wilson, D.M.; Pearsall, A.E.; Kumar, R.; Willins, D.A.; Seehra, J.S.; Sherman, M.L. A Single Ascending-Dose Study of Muscle Regulator Ace-031 in Healthy Volunteers. Muscle Nerve 2013, 47, 416–423. [Google Scholar] [CrossRef]
- Smith, R.C.; Lin, B.K. Myostatin Inhibitors as Therapies for Muscle Wasting Associated with Cancer and Other Disorders. Curr. Opin. Support. Palliat Care 2013, 7, 352–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, C.; McMillan, H.J.; Mah, J.K.; Tarnopolsky, M.; Selby, K.; McClure, T.; Wilson, D.M.; Sherman, M.L.; Escolar, D.; Attie, K.M. Myostatin Inhibitor ACE-031 Treatment of Ambulatory Boys with Duchenne Muscular Dystrophy: Results of a Randomized, Placebo-Controlled Clinical Trial. Muscle Nerve 2017, 55, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Acceleron Discontinues Development of Phase 1 Molecule ACE-2494. 4 April 2019. Available online: http://investor.acceleronpharma.com/news-releases/news-release-details/acceleron-discontinues-development-phase-1-molecule-ace-2494 (accessed on 20 October 2019).
- Glasser, C.E.; Gartner, M.R.; Wilson, D.; Miller, B.; Sherman, M.L.; Attie, K.M. Locally Acting ACE-083 Increases Muscle Volume in Healthy Volunteers. Muscle Nerve 2018, 57, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Wong, B.L.; Byrne, B.J.; Tian, C.; Jacobsen, L.K.; Tirucherai, G.S.; Rabbia, M.; Kletzl, H.; Dukart, J.; Ong, R.; et al. A Phase 1b/2 Study of the Anti-Myostatin Adnectin RG6206 (BMS-986089) in Ambulatory Boys with Duchenne Muscular Dystrophy: A 72-Week Treatment Update (P1.6-062). Neurology 2018, 92, P1.6-062. [Google Scholar]
- Roche/Genentech Announces Decision to Discontinue Development of RG6206 (RO7239361). Available online: https://www.parentprojectmd.org/roche-genentech-announces-decision-to-discontinue-development-of-rg6206-ro7239361/ (accessed on 13 January 2021).
- Amato, A.A.; Sivakumar, K.; Goyal, N.; David, W.S.; Salajegheh, M.; Praestgaard, J.; Lach-Trifilieff, E.; Trendelenburg, A.-U.; Laurent, D.; Glass, D.J.; et al. Treatment of Sporadic Inclusion Body Myositis with Bimagrumab. Neurology 2014, 83, 2239–2246. [Google Scholar] [CrossRef] [Green Version]
- Amato, A.A.; Auberson, L.Z.; Hanna, M.G.; Badrising, U.A.; Needham, M.; Chinoy, H.; Aoki, M.; Koumaras, B.; Tanko, L.; Wu, M.; et al. Long-Term Efficacy and Safety of Bimagrumab in Inclusion Body Myositis: 2 Years Results (S38.003). Neurology 2018, 90, S38.003. [Google Scholar]
- Amato, A.A.; Hanna, M.G.; Machado, P.M.; Badrising, U.A.; Chinoy, H.; Benveniste, O.; Karanam, A.K.; Wu, M.; Tankó, L.B.; Schubert-Tennigkeit, A.A.; et al. Efficacy and Safety of Bimagrumab in Sporadic Inclusion Body Myositis: Long-Term Extension of RESILIENT. Neurology 2021. [Google Scholar] [CrossRef]
- Rooks, D.; Swan, T.; Goswami, B.; Filosa, L.A.; Bunte, O.; Panchaud, N.; Coleman, L.A.; Miller, R.R.; Garcia Garayoa, E.; Praestgaard, J.; et al. Bimagrumab vs Optimized Standard of Care for Treatment of Sarcopenia in Community-Dwelling Older Adults: A Randomized Clinical Trial. JAMA Netw. Open 2020, 3, e2020836. [Google Scholar] [CrossRef] [PubMed]
- Rooks, D.; Praestgaard, J.; Hariry, S.; Laurent, D.; Petricoul, O.; Perry, R.G.; Lach-Trifilieff, E.; Roubenoff, R. Treatment of Sarcopenia with Bimagrumab: Results from a Phase II, Randomized, Controlled, Proof-of-Concept Study. J. Am. Geriatr. Soc. 2017, 65, 1988–1995. [Google Scholar] [CrossRef]
- Rooks, D.S.; Laurent, D.; Praestgaard, J.; Rasmussen, S.; Bartlett, M.; Tankó, L.B. Effect of Bimagrumab on Thigh Muscle Volume and Composition in Men with Casting-induced Atrophy. J. Cachexia Sarcopenia Muscle 2017, 8, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Polkey, M.I.; Praestgaard, J.; Berwick, A.; Franssen, F.M.E.; Singh, D.; Steiner, M.C.; Casaburi, R.; Tillmann, H.-C.; Lach-Trifilieff, E.; Roubenoff, R.; et al. Activin Type II Receptor Blockade for Treatment of Muscle Depletion in Chronic Obstructive Pulmonary Disease. A Randomized Trial. Am. J. Respir. Crit. Care Med. 2019, 199, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Sahenk, Z.; Malik, V.; Gomez, A.M.; Flanigan, K.M.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Meadows, E.; Lewis, S.; et al. A Phase 1/2a Follistatin Gene Therapy Trial for Becker Muscular Dystrophy. Mol. Ther. 2015, 23, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Sahenk, Z.; Al-Zaidy, S.; Rodino-Klapac, L.R.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Miller, N.; Yalvac, M.; Dvorchik, I.; et al. Follistatin Gene Therapy for Sporadic Inclusion Body Myositis Improves Functional Outcomes. Mol. Ther. 2017, 25, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Padhi, D.; Higano, C.S.; Shore, N.D.; Sieber, P.; Rasmussen, E.; Smith, M.R. Pharmacological Inhibition of Myostatin and Changes in Lean Body Mass and Lower Extremity Muscle Size in Patients Receiving Androgen Deprivation Therapy for Prostate Cancer. J. Clin. Endocrinol. Metab. 2014, 99, E1967–E1975. [Google Scholar] [CrossRef] [PubMed]
- Mariot, V.; Joubert, R.; Hourdé, C.; Féasson, L.; Hanna, M.; Muntoni, F.; Maisonobe, T.; Servais, L.; Bogni, C.; Le Panse, R.; et al. Downregulation of Myostatin Pathway in Neuromuscular Diseases May Explain Challenges of Anti-Myostatin Therapeutic Approaches. Nat. Commun. 2017, 8, 1859. [Google Scholar] [CrossRef]
- Pedemonte, M.; Sandri, C.; Schiaffino, S.; Minetti, C. Early Decrease of IIx Myosin Heavy Chain Transcripts in Duchenne Muscular Dystrophy. Biochem. Biophys. Res. Commun. 1999, 255, 466–469. [Google Scholar] [CrossRef]
- Talbot, J.; Maves, L. Skeletal Muscle Fiber Type: Using Insights from Muscle Developmental Biology to Dissect Targets for Susceptibility and Resistance to Muscle Disease. Wiley Interdiscip Rev. Dev. Biol. 2016, 5, 518–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, C.; Silberstein, L.; Hays, A.P.; Blau, H.M. Fast Muscle Fibers Are Preferentially Affected in Duchenne Muscular Dystrophy. Cell 1988, 52, 503–513. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Debruin, D.A.; Bagaric, R.M.; Campelj, D.G.; Hayes, A. The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells 2020, 9, 2657. [Google Scholar] [CrossRef]
- Howard, J.F.; Utsugisawa, K.; Benatar, M.; Murai, H.; Barohn, R.J.; Illa, I.; Jacob, S.; Vissing, J.; Burns, T.M.; Kissel, J.T.; et al. Safety and Efficacy of Eculizumab in Anti-Acetylcholine Receptor Antibody-Positive Refractory Generalised Myasthenia Gravis (REGAIN): A Phase 3, Randomised, Double-Blind, Placebo-Controlled, Multicentre Study. Lancet Neurol. 2017, 16, 976–986. [Google Scholar] [CrossRef]
- Lu-Nguyen, N.; Ferry, A.; Schnell, F.J.; Hanson, G.J.; Popplewell, L.; Dickson, G.; Malerba, A. Functional Muscle Recovery Following Dystrophin and Myostatin Exon Splice Modulation in Aged Mdx Mice. Hum. Mol. Genet. 2019, 28, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Garito, T.; Roubenoff, R.; Hompesch, M.; Morrow, L.; Gomez, K.; Rooks, D.; Meyers, C.; Buchsbaum, M.S.; Neelakantham, S.; Swan, T.; et al. Bimagrumab Improves Body Composition and Insulin Sensitivity in Insulin-Resistant Individuals. Diabetes Obes Metab 2018, 20, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Heymsfield, S.B.; Coleman, L.A.; Miller, R.; Rooks, D.S.; Laurent, D.; Petricoul, O.; Praestgaard, J.; Swan, T.; Wade, T.; Perry, R.G.; et al. Effect of Bimagrumab vs Placebo on Body Fat Mass Among Adults With Type 2 Diabetes and Obesity: A Phase 2 Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e2033457. [Google Scholar] [CrossRef] [PubMed]


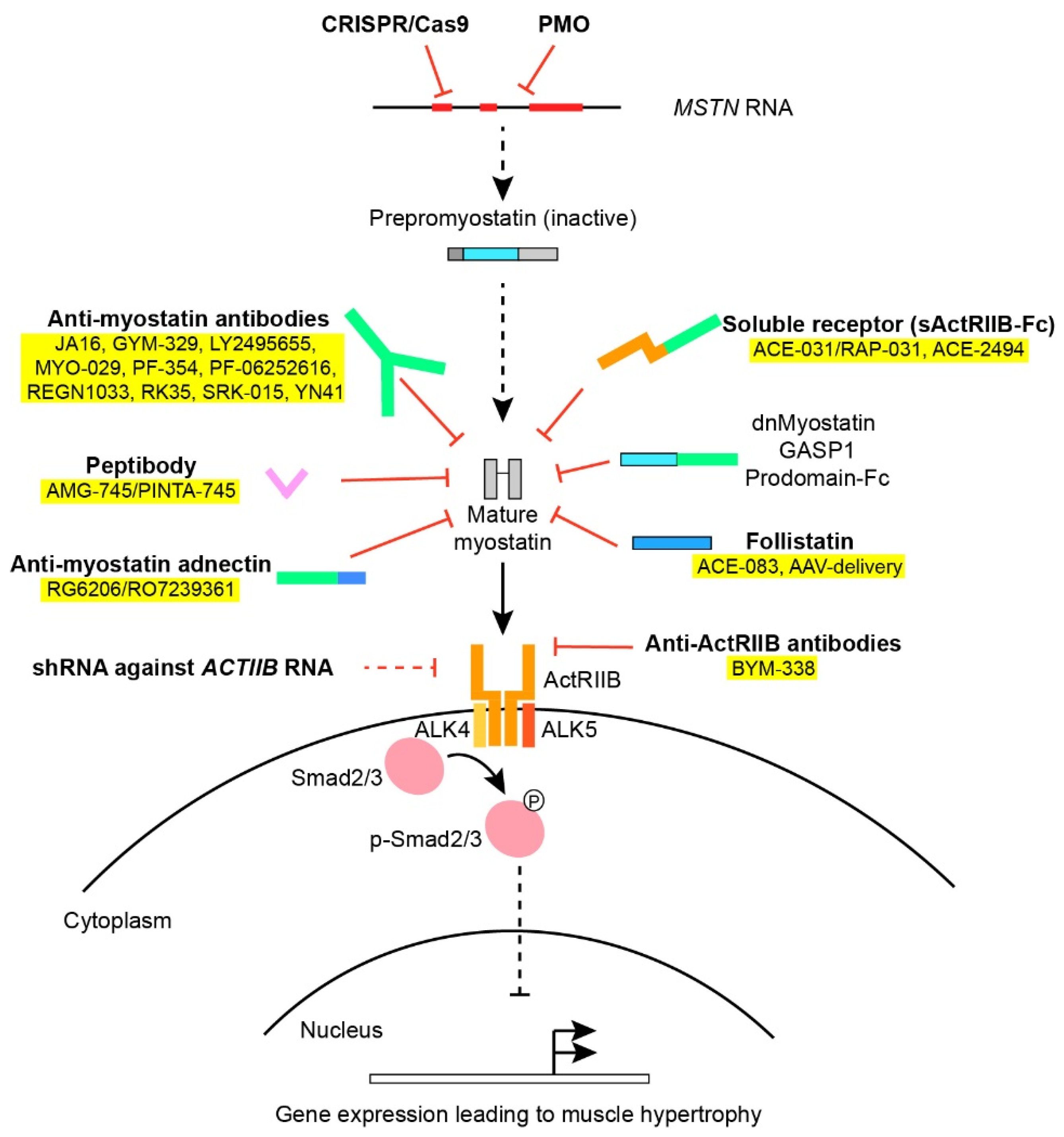{kind=link}
| Species/Model | Compound | Muscle Morphology | Fiber-Type Specific Changes | Absolute Force/ Glycolytic | Specific Force/Glycolytic | Absolute Force/ Oxidative | Specific Force/Oxidative | Stress-Induced Force Drop | Histopathological Effect of Myostatin Inhibition | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Antibodies Blocking Myostatin | ||||||||||
| Mouse/wild-type (BALB/c, C57BL/6) | JA16, ATA-842, mRK35, YN41, muSRK-015P, GYM-mFc | Fiber CSA increased in EDL [67] and Gas Increased weight of Gas, TA, Quad and TB, plantaris, Sol [68] | Increased IIB fiber CSA, no effect on overall composition [69] | Increased grip strength [68,70,71] | [67,68,69,70,71,72] | |||||
| Mouse/mdx, Mouse/Sgcd−/−, Sgcg−/− | JA16 | EDL: Increased weight and single fiber area [73,74]. Increase in TA, Quad, Gas [75] | EDL: increased force | EDL: No effect | No effect | Sgcd−/−: No improvement in histopathology of TA, EDL, Gas and diaphragm albeit hydroxyproline reduced in TA. Fibrosis in diaphragm increased (Sgcd−/− [75]) and decreased (mdx [73]) | [73,74,75] | |||
| Mouse/mdx | PF-354 | Increase in hindlimb muscle weight of 5 weeks treatment, no effect after 8 weeks. No effect/reduction in CSA | Diaphragm: No effect | Diaphragm increased (young)/no effect (old) | No effect | Diaphragm: Increased fiber size in young animals, decreased fiber size in old animals | [76] | |||
| Mouse/mdx, TgCTA1D286G, Sod1G93A, A17, Rat/Sod1G93A Mouse/SmnΔ7 | mRK35/RK35 muSRK-015P (in SmnΔ7) | TA, Gas, Quad, EDL, diaphragm weight increased. Increased CSA in TA, EDL. No effect on weight or CSA in soleus [77] | Quad: Increased proportion of IIB fibers [78] Increase in IIB fiber CSA, no effect in remaining fiber-types [79] | TA, EDL: Increased force Plantarflexor group increased torque [79] | TA, EDL: No effect effect on plantarflexor group [79] | Gas: reduced atrophy, preserved fiber diameter. Diaphragm integrity preserved [80]. Reduced collagen I, III, IV deposits. No effect on intranuclear inclusion bodies [77,81]. Increased number of tubular aggregates [78]. | [72,77,78,79,80,81] | |||
| Mouse/ CB17-SCID, C57BL/6, (Dexamethasone atrophy) | REGN1033 | Increased weight in Gas and TA. Fiber area increased in Gas | No effect on fiber type composition | TA: Increased force | TA: No effect | [82] | ||||
| Monkey/cynomolgus | MYO-029, Domagrozu-mab, GYM-cyfc | Increased muscular circumference | [71,72,83] | |||||||
| Myostatin Propeptide Administration or Overexpression | ||||||||||
| Mouse/mdx | Recombinant propeptide-Fc | EDL: weight, CSA, single fiber area increased | EDL: Increased force | EDL: Increased force | No effect | Decreased pathological changes | [84] | |||
| Mouse/mdx | AAV8- MPRO76AFc | TA, Quad, Gas, Diaphragm increased | TA: Increased force | TA: No effect | Larger fibers, less fibrosis | [85] | ||||
| Mouse/calpatin 3-null mice (LGMD2A), Sgca−/− (LGMD2D) | rAAV2/1mSeAP-propmyoD76A | Increased muscle mass in calpain-3-null mice, no effect in Sgca−/−-mice | EDL: increased force (calpain-3-null mice) | EDL: No effect (calpain-3-null mice) | Soleus: Increased force (calpain-3-null mice) | Soleus: No effect (calpain-3-null mice) | [86] | |||
| Soluble Receptor (sActRIIB-Fc) | ||||||||||
| Wild-type, C57BL/6 C57BL/10 | ACE-031, sActRIIB, RAP-031 ACE-2494 | Increased muscle weight. Fiber CSA increased in EDL [87] and in whole TA [88] | Soleus: Type I and II-fiber CSA increased [89]. Quad; increased size of I, IIA, IIB-fibers. No fiber-type switch [90] | EDL: twitch force increased, no effect on tetanic force [87]. Gas: no effect on max tetanic force [91] | EDL: no effect [92,93] Gas: decreased [91] | Soleus: increased force [92] | Soleus: no effect force [92] | [87,88,89,90,91,92,93,94,95,96] | ||
| Mouse/mdx | RAP-031, sActRIIB-Fc | Muscle weight increased Diaphragm and triceps myofiber increased [97]. EDL single fiber CSA increased [98]. | No fiber-type conversion [92] | EDL: increased force [98,99] | EDL: increased force [98], No effect [99]. EDL, TA decreased force in older animals | Soleus decreased force | Soleus decreased force | No effect | Diaphragm, TA: No effect on histopathology, hydroxyproline [94,98]. Fibrosis decreased [97]. No visible effects on H/E pathology. SDH stains without effect of treatment [100]. eMHC: no effect [94]. | [92,94,97,98,99,100] |
| Mouse/TgActa1H40Y, Mtm1R69C, Mtm1δ4, R6/2, Dysf−/−, Cav3P104L | RAP-031, sActRIIB-Fc | Increased muscle weight, increased fiber size | Quad: oxidative fiber diameter increased. Diaphragm: glycolytic myofibers hypertrophy [101]. IIB fiber hypertrophy, no fiber type switch [90,102] | No effect [101]. EDL, TA increased force [103] | No effect [101] | No effect [101] | No effect [101] | Nemaline rod structures unchanged [101]. Gross evaluation of diaphragm: unaffected by genotype or treatment [90]. Fibrotic changes improved | [90,101,102,103,104,105] | |
| Anti-ActRIIB Antibody | ||||||||||
| Mouse/SCID | BYM338 | Increased weight of TA, EDL, Gas. Soleus increased weight (in high dose) [106] | Gas: increased force [107] | [106,107] | ||||||
| Mouse/C57BL/6 (glucocorticoid-induced atrophy) | BYM338 | TA weight and CSA increased | TA increased force | [106] | ||||||
| Follistatin Administration or Overexpression | ||||||||||
| Mouse/ F66;Dysf−/−, F66;mdx | Follistatin overexpression | Muscle mass maintained in F66;mdx, decreased in F66;Dysf−/− | F66;Dysf−/−: EDL: Decreased force | F66;Dysf−/−: Exacerbation of dystrophic features. Increased Evans Blue Dye (EBD) uptake F66;mdx: Dystrophic features not exacerbated, mild improvement | [104] | |||||
| Mouse/mdx, Sod1G93A | AAV-delivered follistatin i.m. | Increased weight of TA, Gas, Quad, triceps | Increased grip strength | Young mdx: increased myofiber size. Satellite cell markers: no diff Old mdx: Fever necrotic fibers and mononuclear infiltrates | [108,109] | |||||
| Monkey/Cynomolgus | AAV-delivered follistatin i.m | Increased fiber size | Quad: Increased force | Myofiber hypertrophy | [110] | |||||
| Mouse/C57BL10, mdx, | ACE-083 | Increased CSA, weight | TA: increased force | TA: no effect | [111] | |||||
| Mouse/C57BL/6 | FS-EEE-mFc and FST288-Fc | Increased muscle weight | [99,112] | |||||||
| Mouse/mdx | FS-EEE-mFc | Increased weight in gas, Quad, triceps, TA | EDL: Increased force | EDL: No effect | Decreased necrosis and fibrosis in Quad, no effect in diaphragm | [99] | ||||
| Liver-mediated Overexpression of Dominant-negative Myostatin (dnMSTN), sActRIIB and Myostatin Propeptide | ||||||||||
| Mouse/MF-1 (wild-type) | AAV8 over-ekspression (propeptide) | Gas, TA increased mass. EDL and soleus increased CSA. | Increased CSA of type I, IIA and IIB-fibers | EDL: No effect | EDL: No effect | Soleus: increased force | Soleus: No effect | [113] | ||
| Mouse/SmaC/C | AAV-mediated systemic expression (dnMSTN and sActRIIB) | Increased weight in TA, Gas, Quad. dnMSTN-cohort: Increased CSA in EDL and TA but not in soleus | TA: Increased IIA size EDL: Increased IIA and IIB size and total fiber number. Soleus: No effect vs. controls. I-fibers generally unaffected | EDL increased vs. SMAC/C control | EDL; Decreased force | Soleus: Increased force | Soleus: No effect | [114] | ||
| Mouse/mdx | AAV-delivered liver-specific promoter: dnMSTN, sActRIIB | Increased weight in TA, Gas, Quad, EDL, Soleus EDL: increased CSA Soleus: No effect in weight [115] | EDL: IA + IIB increased fiber size. Increased proportion of IIB fibers in EDL and Soleus. Soleus: Increased size and proportion of IIA-fibers Diaphragm: IIX fibers proportion increased, IIA fibers proportion decrease [116] Diaphragm: No effect in specific fiber-type size [115] | EDL: increased force | No effect (decreased force by 10 months of treatment) | Soleus: increased force | Soleus increased force [116] Soleus no difference [115]. Diaphragm: no effect | [115,116,117] | ||
| Dog/GRMD | AAV-delivered liver-specific promoter (dnMSTN) | Increased weight in Tibialis cranialis, EDL, Gas, flexor digitorum superficialis | Increased size of IIA-fibers, no effect in I-fibers. No fiber type switch | [118] | ||||||
| RNA Interference and Anti-oligonucleotides against Myostatin or ActRIIB | ||||||||||
| Mouse/mdx | Antimyostatin PMO | No effect in weight of diaphragm, EDL, Gas, Soleus, TA | Diaphragm: no difference in fiber-type content (I, IIA, IIX, IIB) | Diaphragm and TA: no effect on fiber diameter and collagen IV content | [119] | |||||
| Mouse/mdx (female) | AAV-delivered shRNA, i.m. | TA: No effect on CSA, fiber number increased | TA: No effect | TA: No effect | [120] | |||||
| AAV-Cas9-mediated Myostatin Gene Editing | ||||||||||
| Mouse/C57/BL10 | rAAV-SaCas9 | Increased fiber area and number of fibers per area | [79] | |||||||
| Myostatin Knock-out/Crossbreeding | ||||||||||
| Mouse/Mstn−/− | Increased muscle weight vs. wild-type. Increased fiber number and CSA of EDL and soleus [121] | EDL fiber-type composition: IIA and IIX incidence decreased, IIB increased in EDL and TA. Soleus CSA increased only in IIA-fibers [122] | EDL: Increased [121]/no effect [122,123] | EDL: Decreased | Soleus: Increased | Soleus: No effect | EDL: Force deficit Soleus: No force deficit | Decreased hydroxyproline content in EDL, no effect in soleus [121]. Cytoplasmic inclusions of tubular aggregates in older mice [123] | [121,122,123,124,125,126,127] | |
| Mouse/BehC/C | Increased muscle weight | EDL: No effect [123] | EDL: Decreased force [123] | [123,128] | ||||||
| Mouse/ Mstn−/−;mdx Mstn−/−;Sgcd −/− MstnPro;Cav3P104L | Increased mean fiber diameter and muscle weight [105,129,130] | Mstn−/−;mdx: Reduced fibrosis [129] Mstn −/−;Sgcd −/−: Hydroxyproline content decreased in EDL [75] | [75,105,129,130] | |||||||
| Mouse/Mstn−/−; dyW/dyW | Increased muscle mass, muscle CSA and fiber CSA. (increased mortality) | Decreased type I fiber composition | No effect on necrosis, inflammation or infiltrating cells. Less fat tissue. | [131] | ||||||
| Treatment | Sponsor | Condition | Phase of Trial | Primary Outcome | Secondary Outcome | Result | Status | Reference |
|---|---|---|---|---|---|---|---|---|
| Neutralizing Monoclonal Antibodies | ||||||||
| MYO-029 (Stamulumab) | Wyeth | Healthy subjects | I | Safety, tolerability, PK/PD | N/A | Well tolerated | Completed | NCT# 00563810 |
| BMD, FSHD, LGMD (2A, 2B, 2C, 2D, 2E, 2I) | I/II | Safety | Biological activity (manual muscle test, QMT, TFT, pulmonary function test, subject-reported outcome, MRI, change in muscle mass, LBM) | Adverse effects, secondary outcome not reached | Completed | [154] EudraCT# 2004-000622-67 NCT# 00104078 | ||
| PF-06252616 (Domagrozumab) | Pfizer | Healthy subjects | I | Safety and tolerability | PK/PD, DXA evaluation | Well tolerated. LBM and muscle volume increased | Completed | [155] NCT# 01616277 |
| DMD | I | Safety and tolerability, mean change 4-stair climb | TFT, pulmonary function tests, muscle volume, PK/PD | No significant between-group differences in any secondary clinical endpoints, terminated. | Terminated | [156] NCT# 02310763 Extension: NCT# 02907619 | ||
| LGMD 2I (FKRP) | I/II | Safety and tolerability | Muscle strength, TFTs, pulmonary function, LBM, PK, PD. Exploratory outcome: muscle fat fraction | Preliminary results on clinicaltrials.gov per January 31, 2021 | Completed | NCT# 02841267 | ||
| LY2495655 (Landogrozumab) | Lilly | Healthy subjects | I | “Clinically significant effect” | PK, PD, thigh muscle volume | Well tolerated | Completed | [157] NCT# 01341470 |
| Advanced cancer | I | Safety and tolerability | PK | Well tolerated | Completed | [157] NCT# 01524224 | ||
| Pancreatic Cancer /cachexia | II | Overall survival | Progression-free survival, tumor response, duration of response, LBM, TFT, PRO, pain | Primary outcome not reached | Completed/Terminated | [158] NCT# 01505530 | ||
| Older, weak fallers | II | Change in appendicular LBM | TFTs, gait speed, QMT, body composition, rate of falls, myostatin serum concentration | Primary outcome reached | Completed | [159] NCT# 01604408 | ||
| Osteoarthritis undergoing total hip replacement | II | Change in appendicular LBM | Secondary: QMT, PRO, whole-body- composition | Primary outcome reached | Completed | [160] NCT# 01369511 | ||
| REGN1033 (Trevogrumab)/SAR391786 | Regeneron/ Sanofi | Healthy subjects | I | Assessment of safety, tolerability, administration | N/A | Results not reported (both studies) | Completed | NCT# 01507402, NCT# 01720576 |
| Healthy subjects | I | Change in total lean mass | Safety and tolerability, appendicular lean mass | Results not reported | Completed | NCT# 01910220 | ||
| Healthy subjects | I | PK in two different formulations of drug | Safety and tolerability | Results not reported | Completed | NCT# 02741739 | ||
| Sarcopenia | II | Change in total lean body mass | AE, appendicular lean mass, gait speed, SPPB, DXA-evaluated body composition, 6MWT, QMT, TFT | Results not reported | Completed | NCT# 01963598 | ||
| sIBM | II | Change in total lean mass | AE, TFT, 6MWT, 10MWT, QMT | N/A | Withdrawn | NCT# 03710941 | ||
| REGN2477 (Garetsomab, Activin A-antibody) alone and in combination with REGN1033 | Regeneron | Healthy subjects | I | Safety and tolerability | Thigh muscle volume, DXA-evaluated body composition, PK | Results not reported | Completed | NCT# 02943239 |
| SRK-015 (Apitegromab) | Scholar Rock | SMA 2, SMA 3 | II | Change from Baseline in the Revised Hammersmith Scale or Hammersmith Functional Motor Scale Expanded (HFMSE) | N/A | N/A | Active per January 31 2021 | NCT# 03921528 |
| GYM329/RG 6237 | Chugai Pharmaceutical/Roche | Healthy subjects (limb immobilization) | I | Thigh muscle strength | Safety and tolerability, PK, PD | Results not reported | Recruiting per January 31 2021 | NCT# 04708847 |
| Soluble ActRIIB | ||||||||
| ACE-031 (Ramatercept) | Acceleron | Healthy subjects | Ia | Safety and tolerability | PK/PD, body mass evaluation by DXA and MRI | Well tolerated | Completed | [161] NCT# 00755638 |
| Healthy subjects | Ib | Safety and tolerability | PK/PD | Adverse effect (epistaxis) Increased LBM and thigh muscle volume | Completed | [162] NCT# 00952887 | ||
| DMD | II | Safety and tolerability | PK/PD (MRI evaluation, bone mineral density, TFT) | Body mass, Bone mineral density MD improved vs. baseline (BL) No difference vs. placebo AE (telangiectasias, epistaxis) | Terminated | [163] NCT# 01099761 Extension: NCT# 01239758 | ||
| ACE-2494 | Healthy subjects | I | Safety and tolerability | PK/PD, DXA-evaluated body composition, thigh muscle volume evaluated by MRI | Development of antidrug antibodies | Terminated | [164] NCT# 03478319 | |
| Follistatin-Fc | ||||||||
| ACE-083 | Acceleron | Healthy subjects | I | Safety and tolerability | PK/PD, MRI/DXA evaluation, QMT | Well tolerated | Completed | [165] NCT# 02257489 |
| FSH | II | Safety and tolerability | PK, PD, QMT, TFT, QOL | Did not meet functional secondary endpoint | Terminated | NCT# 02927080 | ||
| Charcot–Marie–Tooth | II | Safety, tolerability, Muscle volume estimated by MRI | PK/PD, Muscular fat infiltration, QMT, TFT, QOL, Charcot–Marie–Tooth examination score) | Did not meet functional secondary endpoint | Terminated | NCT# 03124459 | ||
| Antimyostatin Adnectin | ||||||||
| BMS-986089 | Bristol-Meyers-Squibb/Hoffmann-La Roche/Roche/ Greentech | Healthy subjects | I | Safety and tolerability | Pharmacokinetics | Results not reported | Completed | NCT# 02145234 |
| RG6202/BMS-986089/ RO-7239361 | DMD | Ib/II | Safety and tolerability | Thigh contractive tissue, CSA, PK | No AE. Increased LBM | Terminated | [166] NCT# 02515669 | |
| RO-7239361/RG6206 | DMD | II/III | Changes in North Star Ambulatory Assessment score | TFT, QMT, 6MWT, walk, run and stride velocity | N/A | Discontinued | [167] NCT# 03039686 | |
| Anti-ActRIIB Antibody | ||||||||
| BYM-338 (Bimagrumab) | Novartis | sIBM | II | Change in muscle volume | Body composition, LBM, QMT, TFT, 6MWT | Primary outcome reached | Completed | [168] NCT# 01423110 Extension: NCT# 02250443 (terminated early) |
| sIBM | IIb/III | Change in 6MWT | LBM, QMT, sIBM functional assessment, rate of falls, SPPB | Primary outcome not reached | Completed | [169,170] NCT# 01925209 EudraCT# 2013-000705-23 Extension: NCT# 02573467 EudraCT# 2015-001411-12) | ||
| Sarcopenia | II | Change from baseline in SPPB | Safety, tolerability, 6MWT, gait speed, total LBM | Increased appendicular skeletal muscle index and LBM from baseline in 700 mg treatment cohort. No functional improvement [171] | Completed | [171] NCT# 02333331 EudraCT# 2014-003482-25 Extension: NCT# 02468674 Extension: 2015-000471-27 | ||
| Sarcopenia | II | Thigh muscle volume, intramuscular and subcutaneous fat tissue | Total LBM, QMT, TFT | Primary endpoint reached | Completed | [172] NCT# 01601600 | ||
| Patients undergoing surgical treatment of hip fracture | IIa/IIb | Change in total LBM | Gait speed, SPPB, safety and tolerability, rate of falls | Results not reported | Completed | NCT# 02152761 EudraCT# 2013-003439-31 | ||
| Casting-induced muscle atrophy (healthy) | N/A | Thigh muscle volume, change in intramuscular and subcutaneous adipose tissue | QMT, safety and tolerability | Primary endpoint reached (muscle volume) | [173] No clinical trial ID specified in article | |||
| COPD | II | Change in thigh muscle volume | 6MWT, PK | Primary endpoint reached | Completed | [174] NCT# 01669174 | ||
| Cancer cachexia (lung or pancreas) | II | Change in thigh muscle volume | Body weight, PK/PD, bone mineral density, LBM, physical activity levels | Results submitted, p-value not calculated | Completed | NCT# 01433263 | ||
| Type II diabetes | II | Change in body fat mass | HbA1c change, PK, body weight change, insulin resistance | Results not reported | Completed | NCT# 03005288 | ||
| Follistatin Gene Therapy | ||||||||
| AAV1.CMV.FS344 | Children’s Hospital/Milo Biotech | BMD | I/IIA (no placebo control) | 6MWT | QMT of quadriceps, muscle histology | Primary endpoint reached (in 4 of 6 subjects) | Completed | [175] NCT# 01519349 |
| sIBM | I/IIa | 6MWT | TFT, biopsy, Western blotting | Primary endpoint reached | Completed | [176] | ||
| rAAV1.CMV. huFollistatin344 | Jerry R. Mendell/Milo Therapeutics | DMD | I/II | AE | 6MWT, size of muscle fibers | Results not reported | Completed | NCT# 02354781 |
| Antimyostatin peptibody | ||||||||
| AMG-745/PINTA 745 | Amgen | Prostate cancer in patients treated with androgen deprivation therapy | I | AE, PK, DXA, QMT, SPPB, TFT | N/A | LBM increased, fat mass decreased. | Completed | [177] |
| Age-associated muscle loss | II | Thigh CSA | QMT, TFT, 6MWT, PK | N/A | Withdrawn | NCT# 00975104 | ||
| End stage renal disease, kidney disease, protein energy wasting | I/II | Safety and tolerability, LBM change | LBM, appendicular lean mass, mid upper arm muscle circumference, TFT, 6MWT | Results not reported | Completed | NCT# 01958970 | ||
| Myostatin Inhibition (Information on Myostatin Inhibition Strategy not Available) | ||||||||
| BLS-M22 | BioLeaders Corporation | Healthy subjects | I | Safety and tolerability | PK, immunogenicity, changes in muscle mass | Results not reported | Recruiting | NCT #03789734 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nielsen, T.L.; Vissing, J.; Krag, T.O. Antimyostatin Treatment in Health and Disease: The Story of Great Expectations and Limited Success. Cells 2021, 10, 533. https://doi.org/10.3390/cells10030533
Nielsen TL, Vissing J, Krag TO. Antimyostatin Treatment in Health and Disease: The Story of Great Expectations and Limited Success. Cells. 2021; 10(3):533. https://doi.org/10.3390/cells10030533
Chicago/Turabian StyleNielsen, Tue L., John Vissing, and Thomas O. Krag. 2021. "Antimyostatin Treatment in Health and Disease: The Story of Great Expectations and Limited Success" Cells 10, no. 3: 533. https://doi.org/10.3390/cells10030533
APA StyleNielsen, T. L., Vissing, J., & Krag, T. O. (2021). Antimyostatin Treatment in Health and Disease: The Story of Great Expectations and Limited Success. Cells, 10(3), 533. https://doi.org/10.3390/cells10030533